

Abstract
The pharmacokinetics of lopinavir were investigated by the use of a population approach performed with the nonlinear mixed effect modeling program NONMEM and 157 children ranging in age from 3 days to 18 years. The pharmacokinetics of lopinavir were well described by a one-compartment model in which the absorption and the elimination rate constants were equal. Typical population estimates of the apparent volume of distribution (V/F) and plasma clearance (CL/F) were 24.6 liters and 2.58 liters/h, respectively. The lopinavir V/F and CL/F were both related to body weight (BW), with an important increase in weight-normalized CL/F for the lowest BW. Combined treatment with lopinavir and nevirapine was found to increase the CL/F. The lopinavir CL/F was also age and sex related, as a 39% increase was observed after the age of 12 years for boys compared to the CL/F for girls. The consequences of these pharmacokinetic discrepancies and the necessity to modify the currently recommended dosage regimen should be further investigated.
Lopinavir is a protease inhibitor (PI) used for the treatment of human immunodeficiency virus (HIV) infection in children and adults. Lopinavir can be administered to children older than 6 months of age by the use of a liquid formulation as well as by the use of a solid oral formulation, both of which contain ritonavir for pharmacokinetic enhancement, at recommended lopinavir/ritonavir doses of 12/3 mg/kg of body weight (BW) twice daily (BID) for children with BWs between 7 and 14 kg and 10/2.5 mg/kg BID for children with BWs between 15 and 40 kg. For children with body weights greater than 40 kg, the 400/100-mg BID adult dosage regimen is recommended. Children younger than 6 months of age were not investigated in previously published studies (3, 27). This lack of data can explain to some extent why lopinavir is not recommended for use in children younger than age 6 months. Nevertheless, there are clinical situations, such as the presence of a viral strain less sensitive to the other antiretrovirals, which could require the administration of lopinavir to very young children. Another pediatric population for which pharmacokinetic data are lacking is children with BWs greater than 40 kg. However, this lack of data for adolescents is a very common phenomenon and is relevant not only to lopinavir. Frequently, adolescents are considered similar to adults from a pharmacokinetic point of view, which explains why adolescents often receive the recommended adult dosage regimen. However, adolescence is characterized by physiological and hormonal changes (26, 30), the possible consequences of which on the pharmacokinetics and metabolism of drugs are still unknown. Because pharmacokinetic data for children younger than age 6 months and adolescents are lacking, the relationships between lopinavir pharmacokinetic parameters and the individual characteristics of children are also not known. These relationships are nevertheless important for optimization of the lopinavir dosage regimen, as several studies performed with PI-experienced adults and children have identified relationships between the lopinavir concentration, the number of mutations in the gene coding for the viral protease that reduces susceptibility to lopinavir, and the virological response to lopinavir treatment (6, 11, 21). Therefore, to investigate the factors explaining the interindividual variability of lopinavir pharmacokinetics in children and to provide pharmacokinetic data for children younger than age 6 months and adolescents, the pharmacokinetics of lopinavir in a large population of children ranging in age from 3 days to 18 years were retrospectively investigated by the use of a population approach.
MATERIALS AND METHODS
Patients and treatment.
The population comprised pediatric patients receiving lopinavir for the treatment of HIV infection or the prevention of mother-to-child transmission. All the children were monitored by use of the plasma concentrations of antiretroviral drugs on a routine basis. For each patient, the time that elapsed between administration and sampling, gender, BW, and age were carefully recorded, as were combination treatments, particularly those with antiretroviral drugs. Body surface area (BSA) was calculated according to the following formula for the pediatric population: BSA = [(4 × BW) + 7]/(BW + 90). Additional information can be found at the Société Francophone de Médecine d'Urgence website (http://www.sfmu.org/calculateurs/SC_BB.htm). The number of protease mutations, the CD4 count, and the viral load at the baseline were obtained from the Virology and Immunology Departments of Necker-Enfants Malades Hospital.
Analytical methods.
The assay for lopinavir was performed by using high-pressure liquid chromatography with UV detection. Briefly, 500 μl of sodium carbonate (0.2 M) and 6 ml of ethyl acetate-hexane (50:50 vol/vol) was added to 200 μl of plasma. After 20 min of mixing, the supernatant was evaporated at 30°C under a stream of nitrogen. Dry residues were then reconstituted with 200 μl of the mobile phase (tetramethylammonium perchlorate [0.01 M] in trifluoroacetic acid [0.01%]-acetonitrile [39:61; vol/vol]), and 100 μl of this mixture was injected into the chromatographic system. The separation was performed on a Nucleosil C8 column (125 by 4.6 mm, 3 μm) at a flow rate of 1 ml/min. Detection was performed at 205 nm. The quantification limit of the method was 0.10 mg/liter, with the interassay precision and bias being less than 7% in the calibration range of 0.1 to 20 mg/liter.
Population pharmacokinetic modeling.
Concentration-time data were analyzed by use of the first-order conditional estimation method with interaction of the nonlinear mixed effects modeling program NONMEM (version V, level 1.1, double precision) (1). Several structural pharmacokinetic models were investigated. Classical one- and two-compartment models with first- and zero-order absorption were first evaluated. A one-compartment pharmacokinetic model in which a single rate constant (k) was used for both the absorption and the elimination processes was also tried (31). The equation for this model was as follows:
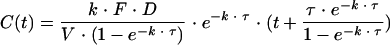 |
where τ is the time that elapsed between the administration of two doses (i.e., 12 h), C is concentration, t is time, F is bioavailability, D is lopinavir dose, and V is volume. The explicit solution for this pharmacokinetic model was coded in the $PRED section of the control stream, and its parameters were the apparent total clearance (CL/F) and the volume of distribution (V/F) of lopinavir.
Several error models (i.e., proportional, exponential, and additive random effects model) were also investigated as means of describing interpatient and residual variabilities.
Systematic testing for the influence of continuous covariates on the pharmacokinetic parameters was done by use of a generalized model, according to the following equation, by using, for example, CL/F and BW: CL/F = TV(CL/F) · (BW/median BW)θBW, where TV(CL/F) is the typical value of the apparent clearance for a patient with the median covariate value, and θBW is the influential factor for body weight. Binary covariates (gender, combined treatment, dosage form) were investigated as follows: CL/F = TV(CL/F) · θSEX, where SEX is equal to 1 for males and 0 for females.
The possible effect of a covariate on lopinavir bioavailability was investigated as described above by estimation of the same influential factor on both CL/F and V/F.
The significance of a relationship between a pharmacokinetic parameter and a covariate was assessed by use of the chi-square test of the difference between the objective functions of the basic model (without the covariate) and the model with the covariate. A covariate was retained in the model if it produced a minimum decrease in the objective function of 4 units (P < 0.05, 1 degree of freedom) and if one of the following criteria was satisfied: (i) it led to a reduction of the interindividual variability (η) of the associated pharmacokinetic parameter or (ii) if its effect was biologically plausible. An intermediate multivariate model that included all selected covariates was then obtained. A covariate was retained in the final multivariate model if its deletion from the intermediate model led to a 7-point increase in the objective function (P < 0.01, 1 degree of freedom). At each step, the goodness of fit was evaluated by use of a graph of the weighted residuals versus time after administration of the dose (time) or by use of a graph of the weighted residuals versus the predicted concentration.
The accuracy and robustness of the final population model were assessed by a bootstrap method, which consisted of repeated random sampling with replacement from the original data set. This resampling was repeated 1,000 times, and the values of the parameters estimated from the bootstrap set were compared to the estimates obtained from the original data set. The entire procedure was performed in an automated fashion by using Wings for NONMEM (24).
Individual Bayesian estimates of the pharmacokinetic parameters were used to calculate the individual area under the concentration-time curve from time zero to 12 h (AUC0-12) and the trough concentration (Ctrough). The AUC0-12 achieved for the recommended pediatric dosage regimen was also calculated.
RESULTS
Demographic data.
One hundred fifty-seven children ranging in age from 3 days to 18 years were available for pharmacokinetic evaluation. Their main characteristics are listed in Table 1. The distribution of ages at the time of retrieval of the first sample is shown in Fig. 1. The number of protease mutations at the baseline, the viral load, and the CD4 count at the start of lopinavir treatment were known for 69 children, who were specifically described previously (11). Lopinavir was combined with at least one nucleoside reverse transcriptase inhibitor, one PI, or a nonnucleoside reverse transcriptase inhibitor in 90, 10, and 23% of the samples, respectively. More precisely, lopinavir was combined with one, two, or three nucleoside reverse transcriptase inhibitors in 16, 61, and 12% of the samples, respectively. Amprenavir was the PI that was the most frequently combined with lopinavir, but this association involved only 4% of the samples. Nevirapine was combined with lopinavir in 16% of the samples (whereas efavirenz was combined with lopinavir in 8% of the samples).
TABLE 1.
Characteristics of the 157 children (67 girls, 90 boys) at sampling time
| Characteristic | Mean | SD | Median | Range |
|---|---|---|---|---|
| Age (yra) | 9.1 | 4.8 | 10.2 | 3 days-18 |
| BW (kg) | 29.0 | 14.9 | 27.6 | 2-73 |
| Dose (mg) | 279 | 102 | 266 | 30-532 |
| Dose (mg/kg) | 109 | 3.7 | 10.4 | 4.4-29.4 |
| Dose (mg/m2) | 288 | 66 | 281 | 124-566 |
| No. of LPVb mutationsc | 2.6 | 2 | 2 | 0-8 |
| VL (copies/ml)d | 288,327 | 373,695 | 116,500 | 160-1,600,000 |
| CD4 cellsc (no./mm3) | 462 | 520 | 300 | 6-2,476 |
| % CD4 cellsc | 14.7 | 10 | 13.5 | 1-42 |
| Lopinavir plasma concn (mg/liter) | 8.99 | 4.89 | 8.25 | 0.33-29.7 |
| No. of samples | 541 | |||
| No. of samples per patient | 3.5 | 3 | 1-14 |
Unless indicated otherwise.
LPV, lopinavir.
n = 69 patients.
VL, viral load. The results are for the 68 patients whose viral loads were >50 copies/ml.
FIG. 1.

Age distribution at the time of retrieval of the first blood sample in the population of the study.
Population pharmacokinetics.
The classical one-compartment model with first-order absorption (subroutines ADVAN2 and TRANS2) was first tried. For the basic model, estimates of the values for CL/F, V/F, and ka were 2.86 ± 0.15 liters/h, 24.8 ± 11.8 liters, and 0.155 ± 0.048 h−1, respectively. It can be seen that the estimate for ka was very close to the value of kel derived from CL/F and V/F (i.e., 0.115 h−1). Furthermore, the inclusion of covariates provided high standard deviations for estimates of the values of ka and 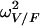 (where
(where 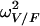 is the interindividual variability of V/F), suggesting that these parameters could not be estimated (data not shown). Because of these results, a limited form of this model that corresponded to a one-compartment model with first-order absorption when ka and kel are not distinguishable was used (31). This model provided the best fit, with a further 20-point decrease in the objective function compared to the value obtained by use of the classical one-compartment model. The one-compartment model with zero-order absorption led to pharmacokinetic parameter estimates that were in disagreement with previously published values (data not shown), and the two-compartment model seemed overparameterized for our data, as it systematically led to convergence failure. The graph of the observed concentrations as a function of time after dosing along with the typical pharmacokinetic curve is shown in Fig. 2. Interpatient variability was described by an exponential error model, whereas residual variability was described by a combined exponential and additive error model. A significant covariance term was found between the η values for CL/F and V/F.
is the interindividual variability of V/F), suggesting that these parameters could not be estimated (data not shown). Because of these results, a limited form of this model that corresponded to a one-compartment model with first-order absorption when ka and kel are not distinguishable was used (31). This model provided the best fit, with a further 20-point decrease in the objective function compared to the value obtained by use of the classical one-compartment model. The one-compartment model with zero-order absorption led to pharmacokinetic parameter estimates that were in disagreement with previously published values (data not shown), and the two-compartment model seemed overparameterized for our data, as it systematically led to convergence failure. The graph of the observed concentrations as a function of time after dosing along with the typical pharmacokinetic curve is shown in Fig. 2. Interpatient variability was described by an exponential error model, whereas residual variability was described by a combined exponential and additive error model. A significant covariance term was found between the η values for CL/F and V/F.
FIG. 2.

Observed lopinavir concentrations and typical pharmacokinetic curve.
The use of body weight and sex were found to improve the fit. However, the relationship between sex and CL/F was found to be age related. Indeed, for ages greater than 12 years, the lopinavir CL/F was increased in boys, whereas sex did not influence CL/F beyond the age of 12 years. Among the concomitantly administered antiretroviral agents, a significant interaction was found only with nevirapine, which increased the lopinavir CL/F. The effects of the different covariates tested on the objective function and the interindividual variability of the pharmacokinetic parameters are summarized in Table 2.
TABLE 2.
Effect of the tested covariates on the objective functiona
| Covariate tested | Pharmacokinetic parameter | ΔFobj1 | Δη1 (%) | ΔFobj2 | Δη2 (%) |
|---|---|---|---|---|---|
| BW | CL/F | −71 | −41 | +48 | +100 |
| BW | V/F | −13 | −63 | +18 | +165 |
| Age | CL/F | −42 | −16 | ↔ | |
| Age | V/F | −5 | −43 | ↔ | |
| Sex | CL/F | −33 | −17 | +18 | +15 |
| Sex | V/F | ↔ | |||
| Dosage form | F | ↔ | |||
| Nevirapine | CL/F | −7 | ↔ | +11 | +15 |
| Efavirenz | CL/F | ↔ | |||
| Amprenavir | CL/F | ↔ | |||
| Lopinavir | CL/F | ↔ |
ΔFobj, observed change in the objective function induced by the corresponding covariate after its addition to the base model (ΔFobj1) or its deletion from the intermediate model (ΔFobj2); Δη, percent change in the interindividual variability of the corresponding pharmacokinetic parameter provided by the addition of the tested covariate in the base model (Δη1) or by its deletion from the intermediate model (Δη2); ↔, no change.
The final covariate submodel was then V/F (liters) = 24.6 × (BW/27)0.72. If the child's age was <12 years, CL/F (liters/h) = 2.58 × (BW/27)0.46 × 1.34N, and if the child's age was >12 years, CL/F (liters/h) = 2.58 × (BW/27)0.46 × 1.34N × 1.39S, where N is equal to 1 if nevirapine was combined with lopinavir (and 0 if not) and S is equal to 1 for boys and 0 for girls.
Table 3 summarizes the population parameter estimates. The goodness of fit was also evaluated graphically from the distribution of the points on graphs of the weighted residuals versus time and weighted residuals predicted concentration (Fig. 3A and B).
TABLE 3.
Population pharmacokinetic parameters of lopinavir in 157 children and bootstrap validationa
| Parameter | TV (CL/F) (liters/h) | CL/F
|
TV (V/F) (liters) | V/F, θBW | 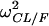 |
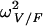 |
covCL,V | Proportional residual variability (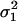 ) ) |
Additive residual variability (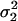 ) ) |
||
|---|---|---|---|---|---|---|---|---|---|---|---|
| θBW | θSEX (if age >12 yr) | θnevirapine | |||||||||
| Final model, original data set | |||||||||||
| Mean | 2.58 | 0.46 | 1.39 | 1.34 | 24.6 | 0.72 | 0.0957 | 0.180 | 0.131 | 0.138 | 1.83 |
| SE | 0.12 | 0.066 | 0.09 | 0.14 | 4.25 | 0.11 | 0.021 | 0.064 | 0.031 | 0.014 | 0.81 |
| Bootstrap valueb | |||||||||||
| Mean | 2.59 | 0.44 | 1.41 | 1.39 | 26.2 | 0.73 | 0.092 | 0.23 | 0.13 | 0.137 | 1.81 |
| SE | 0.13 | 0.074 | 0.13 | 0.19 | 10.5 | 0.17 | 0.024 | 0.10 | 0.040 | 0.07 | 0.86 |
SE, standard error of the estimate; TV, typical value of the corresponding pharmacokinetic parameter; θcovariate, influential factor for the covariate; ω2, interindividual variability; covCL,V: covariance between η values of CL/F and V/F.
Mean of 1,000 bootstrap analyses.
FIG. 3.

Goodness of fit visualized on the weighted residuals (WRES) versus time after dose (A) and weighted residuals versus predicted plasma lopinavir concentrations (PRED) (B).
Bootstrap validation.
The final model obtained with the original data set was subjected to a bootstrap analysis. As shown in Table 3, the mean parameter estimates obtained from the bootstrap process (with 1,000 runs) were not statistically significantly different from the estimates previously obtained with the original data set.
Evolution of lopinavir exposure with respect to BW, age, and sex.
The different values of the influential factor of body weight on CL/F and V/F indicated the nonlinear pattern of the lopinavir half-life (t1/2) with respect to body weight. These nonlinear relationships between CL/F, weight-normalized CL/F, t1/2, and body weight are displayed in Fig. 4. As seen in Fig. 4, the weight-normalized CL/F was even higher in the neonatal subgroup (i.e., age <8 days), with a mean value of 0.44 liter/h/kg (six neonates). Eleven children, including the six neonates, had BWs less than 7 kg at the time of sampling and had a mean age and a mean BW of 2.3 months and 3.7 kg, respectively. The mean CL/F, V/F, and t1/2 values for these children were 1.15 liters/h (i.e., 0.35 liter/h/kg), 6.70 liters, and 3.9 h, respectively. In this subpopulation, the 12-mg/kg lopinavir dose that is recommended for children with BWs between 7 and 14 kg would provide mean AUC0-12, maximum concentration, and Ctrough values of 38.6 mg · h/liter, 3.68 mg/liter, and 2.16 mg/liter, respectively. If the pharmacokinetic target is the mean AUC0-12 calculated for our population (i.e., 108 mg · h/liter), a 34-mg/kg dose would be necessary. The corresponding calculated mean Ctrough would be 6.11 mg/liter.
FIG. 4.

Evolution of lopinavir plasma clearance (A), weight-normalized clearance (B), and terminal half-life (C) according to BW.
The lopinavir CL/F was also found to be related to sex in children whose ages were greater than 12 years. To illustrate this, the individual AUC0-12 and Ctrough values achieved for the currently recommended pediatric dose (12 mg/kg for children with BWs between 7 and 14 kg, 10 mg/kg for children with BWs between 14 and 40 kg, and 400 mg for children with BWs greater than 40 kg) were calculated. The mean curve representing those values with respect to BW and sex is displayed in Fig. 5. Twenty-nine girls and 35 boys were older than 12 years of age at the time of sampling. However, as the PI concentration is thought to be more especially important for pretreated patients, i.e., patients infected with viral strains less susceptible to PIs (21), we focused on the possible difference between the efficacy of lopinavir, expressed as the viral load decrease, after 1 year of treatment in pretreated children by age and sex. There was a trend for a decrease in treatment efficacy after the age of 12 years for boys compared to the treatment efficacy for girls, but because of the small number of children, no statistical analysis was performed with these data (Table 4).
FIG. 5.

Calculated (A) mean lopinavir AUC0-12 and (B) steady-state Ctrough values achieved for the recommended pediatric dosage regimen in boys and girls with respect to BW (calculated values and mean curves).
TABLE 4.
Lopinavir efficacy after 12 months of treatment in pretreated children by age and sexa
| Age (yr) | Characteristic | Girls | Boys |
|---|---|---|---|
| <12 | No. of children | 9 | 12 |
| Age | 8.6 | 6.4 | |
| Baseline VL (log10 no. of copies/ml) | 4.5 | 4.9 | |
| Baseline no. of LPV mutations | 3 | 3 | |
| Lopinavir dose (mg) | 266 | 266 | |
| AUC0-12 (mg · h/liter) | 108 | 116 | |
| VL decrease (log10 copies/ml) | −1.7 | −2.5 | |
| No. of children with VLs <50 copies/ml | 6 | 6 | |
| >12 | No. of children | 6 | 9 |
| Age | 14.8 | 15 | |
| Baseline VL (log10 copies/ml) | 4.9 | 5.2 | |
| Baseline no. of LPV mutations | 3 | 4 | |
| Lopinavir dose (mg) | 399 | 399 | |
| AUC0-12 (mg · h/liter) | 97 | 75 | |
| VL decrease (log10 no. of copies/ml) | −2.5 | −0.5 | |
| No. of children with VLs <50 copies/ml | 4 | 2 |
All values provided are median values. LPV, lopinavir; VL, viral load.
DISCUSSION
The pharmacokinetics of lopinavir in plasma were well described by a simplified one-compartment model in which a single rate constant represented both the absorption and the elimination processes. This model could possibly be inaccurate for estimation of the specific absorption rate constant for lopinavir. However, this choice seemed relevant, as our model satisfyingly predicted the values of the lopinavir pharmacokinetic parameters in neonates and children. Indeed, the previously published pediatric pharmacokinetic study, which included 98 children ranging in age from 6 months to 12.6 years, reported mean values of the time to the maximum concentration, the maximum concentration, Ctrough, t1/2, and AUC0-12 of 4.0 h, 11.7 mg/liter, 6.90 mg/liter, 6.1 h, and 102.8 mg · h/liter, respectively (27), for a mean lopinavir/ritonavir dose of 300/75 mg/m2, whereas our model provided values for our population of 4.5 h, 9.60 mg/liter, 7.90 mg/liter, 6.6 h, and 108 mg · h/liter, respectively, for a mean dose of 288 mg/m2 dose. The previous pharmacokinetic study also reported an approximately 40% decrease in the lopinavir half-life when lopinavir was combined with nevirapine. This result is in agreement with the 34% increase in the lopinavir CL/F induced by nevirapine that was found in the present study. Similar pharmacokinetic interactions are known to occur when lopinavir is combined with efavirenz (3) and amprenavir (29), but these covariates did not provide a significant decrease in the objective function, probably because not enough samples were involved. Another important pharmacokinetic interaction is the increase in lopinavir exposure provided by ritonavir (15). However, the use of the ritonavir concentration as a covariate in the model was not investigated in the present study, as the study design did not allow it. Indeed, plasma samples were drawn at different times postdosing for the different children, and as the ritonavir concentration naturally evolves with the time postdosing, the ritonavir concentrations measured at different times were not comparable and therefore could not be used as a covariate. Besides, the purpose of our study was to develop a population model that could predict the values of lopinavir pharmacokinetic parameters for children, an objective not achievable with a covariate like the ritonavir concentration, which cannot be known a priori. The ritonavir dose could be a predictable covariate, but it was not investigated in the present study, as the lopinavir/ritonavir dose ratio remains constant, regardless of the lopinavir dose administered.
For neonates and infants, our model also showed the decrease in the lopinavir half-life in children with BWs of less than about 10 kg that was recently reported for 12 infants whose ages were between 14 days and 6 months (E. G. Chadwick, J. H. Rodman, P. Palumbo, D. Persaud, J. Chen, J. Gardella, K. Luzuriaga, R. Yogev, P. Emmanuel, M. Rathore, and the PACTG 1030 Study Team, Abstr. 12th Conf. Retrovir. Opportunistic Infect., abstr. 766, 2005). Indeed, for a median BW of 5.1 kg, that study reported median CL/F and V/F values of 1.48 liters/h and 7.74 liters, respectively. If a one-compartment model is assumed, these parameters allow calculation of an elimination half-life of 3.6 h, which is consistent with our own results (Fig. 4). Such a result is surprising, as lopinavir is extensively metabolized by cytochrome P450 3A4 (CYP 3A4) (15), the activity of which is known to be markedly reduced in the first weeks of life (18). A first possible explanation would be a decrease in lopinavir bioavailability secondary to absorption disorders. However, a change in bioavailability would have the same consequences on V/F and CL/F, and the elimination half-life would remain constant throughout the BW range of the study. Another possibility could be the presence, at least during the first weeks of life, of CYP 3A7 (4). This enzyme, which has a 90% amino acid sequence similarity with CYP 3A4 (16), could be involved in lopinavir metabolism; and the pharmacokinetic enhancement by ritonavir could be weaker with this cytochrome than with CYP 3A4. The pharmacokinetic enhancement by ritonavir could also be reduced by a decrease in ritonavir bioavailability in children younger than 6 months of age. Indeed, a previously published study investigating the pharmacokinetics of ritonavir in 48 children between the ages of 6 months and 14 years found that the two children who were 6 months of age had a ritonavir CL/F almost 10 times higher than the CL/F found in older children and imputed this result to a decrease in ritonavir bioavailability (23). Although this result was obtained with high ritonavir doses, this phenomenon could also be observed with the low ritonavir dose of the lopinavir/ritonavir combination and explain the increase in lopinavir CL/F that we observed for children younger than 6 months of age. The metabolic immaturity could also be balanced by an increase in the lopinavir free fraction (fu) in the youngest children. Indeed, lopinavir is known to be highly bound to α1-acid glycoprotein (AAG) (5), the concentration of which is markedly reduced at birth (about 0.2 to 0.3 g/liter) and gradually increases to reach adult values (0.7 to 1.0 g/liter) after 9 to 12 months of life (22). Ritonavir is also highly bound to plasma proteins, including AAG (14); but as the plasma ritonavir concentration is much lower than the lopinavir plasma concentration (most of the plasma ritonavir concentrations were less than 0.5 mg/liter in our population), a decrease in the AAG concentration could have more consequences on lopinavir binding than on ritonavir binding. Therefore, more free lopinavir than free ritonavir could be available for CYP 3A4, which could partially counteract the competitive inhibition by ritonavir.
So, it seems likely that this increased ability to eliminate lopinavir in these children is, in fact, a consequence of a decrease in the pharmacokinetic enhancement provided by ritonavir. Such a phenomenon could therefore be found for the pharmacokinetics of other boosted PIs in neonates and infants. Unfortunately, to our knowledge, no information about the pharmacokinetics of boosted PIs in this particular population is available. Such studies are therefore warranted to confirm this hypothesis. These changes in lopinavir pharmacokinetics in neonates and infants could have consequences on treatment efficacy in this subpopulation. Indeed, because of the decrease in the elimination half-life, it will be difficult to achieve the Ctroughs or genotypic inhibitory quotients that are usually accepted as target values (6, 17). The 12-mg/kg BID lopinavir dose that is recommended for children with BWs greater than 7 kg could be inappropriate for the youngest children, as this dose provides a mean Ctrough of 2.5 mg/liter, which was found to be the limit below which treatment failure is likely, whatever the lopinavir mutation score at the baseline is (6). Our results indicate that a 34-mg/kg BID dose would provide a lopinavir exposure close to the mean reported exposure in children. However, this dose is at least three times higher than the recommended pediatric dose and could therefore raise a safety issue. Furthermore, this theoretical 34-mg/kg dose was calculated by assuming that the pharmacokinetics of lopinavir are linear with the dose, which is probably not the case. Finally, because a change in fu is thought to have no clinically relevant consequence for metabolized and orally administered drugs, as the concentration of the free drug (i.e., the active form) remains constant (2), a dose increase could be irrelevant if the change in the pharmacokinetics of lopinavir are a result of modified protein binding, as suggested above. It therefore seems important to investigate the active form of lopinavir and to specifically determine the optimal lopinavir dosing regimen for neonates and infants in further studies. For example, an alternative, given the decrease in the lopinavir half-life, could be the administration of lopinavir three times a day instead of BID.
The sex-related change in the lopinavir CL/F that was found in adolescents has an important consequence on lopinavir exposure, as, for the current recommended lopinavir regimen, the AUC0-12 calculated for the oldest boys (i.e., 50 mg · h/liter) is approximately half the AUC0-12 calculated for the oldest girls (i.e., 100 mg · h/liter). Poorer compliance by boys than by girls could have been a possible bias that would explain our results. The omission of the previous doses is indeed impossible to investigate when a single blood sample is drawn after the drug was taken during the consultation. However, such a link between sex and compliance would have provided a relationship between sex and V/F similar to the one that was found between sex and CL/F, which was not the case (Table 2). The previously published pediatric pharmacokinetic studies did not find such a sex-related difference, probably because of the lack of children older than 12 years of age (27) or the small number of patients older than 12 years of age included (3). A previously published study performed with adults (18 men and 7 women) also investigated the sex-related differences in the pharmacokinetics of lopinavir (25). Even though the median lopinavir AUC0-12 seemed greater in women (90.6 mg · h/liter) than in men (74.1 mg · h/liter), the difference was not significant. As stated above, lopinavir is extensively metabolized at the hepatic level by CYP 3A4, a metabolic pathway that is not known to display clinically relevant sex-related differences in adults (8, 28). Our results could therefore indicate that adolescence could be characterized by pharmacokinetic specificities compared to the pharmacokinetics in both younger children and adults, so that the current pharmacokinetic assumption that places adults and adolescents in the same category should be reconsidered, at least for metabolized drugs. This was previously suggested for CYP 1A2, as the rate of demethylation of caffeine decreased in early puberty for girls and late puberty for boys (20). However, it must be noticed that similar sex-related differences in the pharmacokinetics of indinavir (10) and saquinavir (25), two other PIs metabolized by CYP 3A4, have been reported in adults. Unfortunately, the pharmacokinetic studies performed with children did not include enough adolescents to investigate the possible sex-based differences in indinavir and saquinavir pharmacokinetics in this population (7, 12, 13, 19). Furthermore, a result that could support a sex-related difference in the lopinavir CL/F in adults is that our model seemed to predict the values of the lopinavir pharmacokinetic parameters in adults quite accurately. Indeed, the study cited above reported median lopinavir AUC0-12s of 90.6 and 74.1 mg · h/liter for women and men, respectively (25). As the lopinavir dose was 400 mg BID, these AUC0-12 values corresponded to CL/F values of 4.41 and 5.40 liters/h for women and men, respectively. The inclusion of the mean BW of 70 kg that was reported in this study in our CL/F equations gave values of 4 liters/h for women and 5.6 liters/h for men. Another previously published pharmacokinetic study performed with adults reported mean CL/F and V/F values of 5.73 liters/h and 61.6 liters, respectively (9). As the population in that study was characterized by a mean BW of 72 kg and comprised 85% men, it was possible to calculate respective CL/F and V/F values of 5.6 liters/h (by using the CL/F equation for boys older than age 12 years) and 49.8 liters, respectively, by the use of our model. So, the consistency between the values calculated by the use of our model and the values reported in previous studies could suggest a sex-related difference in lopinavir pharmacokinetics in adults. Besides, it also indicates that our model accurately predicts the lopinavir CL/F and V/F over a very large BW range (i.e., from the BWs of neonates to the BWs adults), which supports the structural pharmacokinetic model used.
Our data did not allow us to investigate rigorously the possible consequence of this increase in the lopinavir CL/F on treatment efficacy, so the need to give higher lopinavir doses to boys than to girls should be evaluated in further studies. Such a study seems more especially important, as many countries do not practice therapeutic drug monitoring with antiretroviral drugs, which could balance the pharmacokinetic discrepancies between patients. However, the interest in therapeutic drug monitoring for children may be hampered by the high intraindividual variability that we observed. This variability is probably overestimated, as our study design (i.e., a single sample was obtained per occasion) did not allow us to estimate the interoccasion variability. This interoccasion variability could also be important, as the median delay between the first and the last occasion was 0.9 year (range, 0.03 to 2.4 years), a delay during which the pharmacokinetic parameters values naturally evolve, especially in young children.
In conclusion, this study has shown that the pharmacokinetics of lopinavir vary greatly during childhood and identified children younger than age 6 months and boys older than age 12 years as subpopulations in which the levels of exposure to lopinavir may be decreased. The virological and clinical relevance of this pharmacokinetic modification and the usefulness of a higher dosage in these children should be further evaluated.
Acknowledgments
This work was supported by l'Institut National de la Santé et de la Recherche Médicale (contrat de recherche stratégique 2002).
Footnotes
Published ahead of print on 28 August 2006.
REFERENCES
- 1.Beal, S. L., and L. B. Sheiner. 1991. NONMEM user's guide. NONMEM Project Group, University of California at San Francisco, San Francisco, Calif.
- 2.Benet, L. Z., and B. A. Hoener. 2002. Changes in plasma protein binding have little clinical relevance. Clin. Pharmacol. Ther. 71:115-121. [DOI] [PubMed] [Google Scholar]
- 3.Bergshoeff, A. S., P. L. Fraaij, J. Ndagijimana, G. Verweel, N. G. Hartwig, T. Niehues, R. De Groot, and D. M. Burger. 2005. Increased dose of lopinavir/ritonavir compensates for efavirenz-induced drug-drug interaction in HIV-1-infected children. J. Acquir. Immune Defic. Syndr. 39:63-68. [DOI] [PubMed] [Google Scholar]
- 4.Blake, M. J., L. Castro, J. S. Leeder, and G. L. Kearns. 2005. Ontogeny of drug metabolizing enzymes in the neonate. Semin. Fetal Neonatal Med. 10:123-138. [DOI] [PubMed] [Google Scholar]
- 5.Boffito, M., P. G. Hoggard, W. E. Lindup, S. Bonora, A. Sinicco, S. H. Khoo, G. Di Perri, and D. J. Back. 2004. Lopinavir protein binding in vivo through the 12-hour dosing interval. Ther. Drug Monit. 26:35-39. [DOI] [PubMed] [Google Scholar]
- 6.Breilh, D., I. Pellegrin, A. Rouzes, K. Berthoin, F. Xuereb, H. Budzinski, M. Munck, H. J. Fleury, M. C. Saux, and J. L. Pellegrin. 2004. Virological, intracellular and plasma pharmacological parameters predicting response to lopinavir/ritonavir (KALEPHAR study). AIDS 18:1305-1310. [DOI] [PubMed] [Google Scholar]
- 7.Burger, D. M., A. M. van Rossum, P. W. Hugen, M. H. Suur, N. G. Hartwig, S. P. Geelen, H. J. Scherpbier, R. M. Hoetelmans, A. G. Vulto, and R. de Groot. 2001. Pharmacokinetics of the protease inhibitor indinavir in human immunodeficiency virus type 1-infected children. Antimicrob. Agents Chemother. 45:701-705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cotreau, M. M., L. L. von Moltke, and D. J. Greenblatt. 2005. The influence of age and sex on the clearance of cytochrome P450 3A substrates. Clin. Pharmacokinet 44:33-60. [DOI] [PubMed] [Google Scholar]
- 9.Crommentuyn, K. M., B. S. Kappelhoff, J. W. Mulder, A. T. Mairuhu, E. C. van Gorp, P. L. Meenhorst, A. D. Huitema, and J. H. Beijnen. 2005. Population pharmacokinetics of lopinavir in combination with ritonavir in HIV-1-infected patients. Br. J. Clin. Pharmacol. 60:378-389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Csajka, C., C. Marzolini, K. Fattinger, L. A. Decosterd, A. Telenti, J. Biollaz, and T. Buclin. 2004. Population pharmacokinetics of indinavir in patients infected with human immunodeficiency virus. Antimicrob. Agents Chemother. 48:3226-3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delaugerre, C., J. P. Teglas, J. M. Treluyer, P. Vaz, V. Jullien, F. Veber, C. Rouzioux, M. L. Chaix, and S. Blanche. 2004. Predictive factors of virologic success in HIV-1-infected children treated with lopinavir/ritonavir. J. Acquir. Immune Defic. Syndr. 37:1269-1275. [DOI] [PubMed] [Google Scholar]
- 12.Gatti, G., A. Vigano, N. Sala, S. Vella, M. Bassetti, D. Bassetti, and N. Principi. 2000. Indinavir pharmacokinetics and parmacodynamics in children with human immunodeficiency virus infection. Antimicrob. Agents Chemother. 44:752-755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grub, S., P. Delora, E. Ludin, F. Duff, C. V. Fletcher, R. C. Brundage, M. W. Kline, N. R. Calles, H. Schwarzwald, and K. Jorga. 2002. Pharmacokinetics and pharmacodynamics of saquinavir in pediatric patients with human immunodeficiency virus infection. Clin. Pharmacol. Ther. 71:122-130. [DOI] [PubMed] [Google Scholar]
- 14.Hsu, A., G. R. Granneman, and R. J. Bertz. 1998. Ritonavir. Clinical pharmacokinetics and interactions with other anti-HIV agents. Clin. Pharmacokinet. 35:275-291. [DOI] [PubMed] [Google Scholar]
- 15.Hurst, M., and D. Faulds. 2000. Lopinavir. Drugs 60:1371-1379. [DOI] [PubMed] [Google Scholar]
- 16.Itoh, S., T. Yanagimoto, S. Tagawa, H. Hashimoto, R. Kitamura, Y. Nakajima, T. Okochi, S. Fujimoto, J. Uchino, and T. Kamataki. 1992. Genomic organization of human fetal specific P-450IIIA7 (cytochrome P-450HFLa)-related gene(s) and interaction of transcriptional regulatory factor with its DNA element in the 5′ flanking region. Biochim. Biophys. Acta 1130:133-138. [DOI] [PubMed] [Google Scholar]
- 17.Kappelhoff, B. S., K. M. Crommentuyn, M. M. de Maat, J. W. Mulder, A. D. Huitema, and J. H. Beijnen. 2004. Practical guidelines to interpret plasma concentrations of antiretroviral drugs. Clin. Pharmacokinet. 43:845-853. [DOI] [PubMed] [Google Scholar]
- 18.Kearns, G. L., S. M. Abdel-Rahman, S. W. Alander, D. L. Blowey, J. S. Leeder, and R. E. Kauffman. 2003. Developmental pharmacology—drug disposition, action, and therapy in infants and children. N. Engl. J. Med. 349:1157-1167. [DOI] [PubMed] [Google Scholar]
- 19.Kline, M. W., R. C. Brundage, C. V. Fletcher, H. Schwarzwald, N. R. Calles, N. E. Buss, P. Snell, P. DeLora, M. Eason, K. Jorga, C. Craig, and F. Duff. 2001. Combination therapy with saquinavir soft gelatin capsules in children with human immunodeficiency virus infection. Pediatr. Infect. Dis. J. 20:666-671. [DOI] [PubMed] [Google Scholar]
- 20.Lambert, G. H., D. A. Schoeller, A. N. Kotake, C. Flores, and D. Hay. 1986. The effect of age, gender, and sexual maturation on the caffeine breath test. Dev. Pharmacol. Ther. 9:375-388. [DOI] [PubMed] [Google Scholar]
- 21.Marcelin, A. G., I. Cohen-Codar, M. S. King, P. Colson, E. Guillevic, D. Descamps, C. Lamotte, V. Schneider, J. Ritter, M. Segondy, H. Peigue-Lafeuille, L. Morand-Joubert, A. Schmuck, A. Ruffault, P. Palmer, M. L. Chaix, V. Mackiewicz, V. Brodard, J. Izopet, J. Cottalorda, E. Kohli, J. P. Chauvin, D. J. Kempf, G. Peytavin, and V. Calvez. 2005. Virological and pharmacological parameters predicting the response to lopinavir-ritonavir in heavily protease inhibitor-experienced patients. Antimicrob. Agents Chemother. 49:1720-1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mazoit, J. X., and B. J. Dalens. 2004. Pharmacokinetics of local anaesthetics in infants and children. Clin. Pharmacokinet. 43:17-32. [DOI] [PubMed] [Google Scholar]
- 23.Mueller, B. U., R. P. Nelson, Jr., J. Sleasman, J. Zuckerman, M. Heath-Chiozzi, S. M. Steinberg, F. M. Balis, P. Brouwers, A. Hsu, R. Saulis, S. Sei, L. V. Wood, S. Zeichner, T. T. Katz, C. Higham, D. Aker, M. Edgerly, P. Jarosinski, L. Serchuck, S. M. Whitcup, D. Pizzuti, and P. A. Pizzo. 1998. A phase I/II study of the protease inhibitor ritonavir in children with human immunodeficiency virus infection. Pediatrics 101:335-343. [DOI] [PubMed] [Google Scholar]
- 24.Parke, J., N. H. Holford, and B. G. Charles. 1999. A procedure for generating bootstrap samples for the validation of nonlinear mixed-effects population models. Comput. Methods Programs Biomed. 59:19-29. [DOI] [PubMed] [Google Scholar]
- 25.Ribera, E., R. M. Lopez, M. Diaz, L. Pou, L. Ruiz, V. Falco, M. Crespo, C. Azuaje, I. Ruiz, I. Ocana, B. Clotet, and A. Pahissa. 2004. Steady-state pharmacokinetics of a double-boosting regimen of saquinavir soft gel plus lopinavir plus minidose ritonavir in human immunodeficiency virus-infected adults. Antimicrob. Agents Chemother. 48:4256-4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rogol, A. D. 2004. Gender and hormonal regulation of growth. J. Pediatr. Endocrinol. Metab. 17(Suppl. 4):1259-1265. [PubMed] [Google Scholar]
- 27.Saez-Llorens, X., A. Violari, C. O. Deetz, R. A. Rode, P. Gomez, E. Handelsman, S. Pelton, O. Ramilo, P. Cahn, E. Chadwick, U. Allen, S. Arpadi, M. M. Castrejon, R. S. Heuser, D. J. Kempf, R. J. Bertz, A. F. Hsu, B. Bernstein, C. L. Renz, and E. Sun. 2003. Forty-eight-week evaluation of lopinavir/ritonavir, a new protease inhibitor, in human immunodeficiency virus-infected children. Pediatr. Infect. Dis. J. 22:216-224. [DOI] [PubMed] [Google Scholar]
- 28.Schwartz, J. B. 2003. The influence of sex on pharmacokinetics. Clin. Pharmacokinet. 42:107-121. [DOI] [PubMed] [Google Scholar]
- 29.Taburet, A. M., G. Raguin, C. Le Tiec, C. Droz, A. Barrail, I. Vincent, L. Morand-Joubert, G. Chene, F. Clavel, and P. M. Girard. 2004. Interactions between amprenavir and the lopinavir-ritonavir combination in heavily pretreated patients infected with human immunodeficiency virus. Clin. Pharmacol. Ther. 75:310-323. [DOI] [PubMed] [Google Scholar]
- 30.Veldhuis, J. D., J. N. Roemmich, E. J. Richmond, A. D. Rogol, J. C. Lovejoy, M. Sheffield-Moore, N. Mauras, and C. Y. Bowers. 2005. Endocrine control of body composition in infancy, childhood, and puberty. Endocrine Rev. 26:114-146. [DOI] [PubMed] [Google Scholar]
- 31.Wijnand, H. P. 1988. Pharmacokinetic model equations for the one- and two-compartment models with first-order processes in which the absorption and exponential elimination or distribution rate constants are equal. J. Pharmacokinet. Biopharm. 16:109-128. [DOI] [PubMed] [Google Scholar]