

Abstract
Retapamulin is a semisynthetic pleuromutilin derivative being developed as a topical antibiotic for treating bacterial infections of the skin. It is potent in vitro against susceptible and multidrug-resistant organisms commonly associated with bacterial skin infections. We report detailed mode of action studies demonstrating that retapamulin binds to the bacterial ribosome with high affinity, inhibits ribosomal peptidyl transferase activity, and partially inhibits the binding of the initiator tRNA substrate to the ribosomal P-site. Taken together, these data distinguish the mode of action of retapamulin from that of other classes of antibiotics. This unique mode of action may explain the lack of clinically relevant, target-specific cross-resistance of retapamulin with antibacterials in current use.
Pleuromutilin is a tricyclic, diterpene natural product first identified from the basidiomycete bacterial species Pleurotus mutilus (now referred to as Clitopilus scyphoides) that possesses modest antibacterial activity against primarily gram-positive bacterial organisms (8). Early studies on the mode of action of tiamulin and other semisynthetic pleuromutilin derivatives showed that it interfered with cell-free protein synthesis and formylmethionine-puromycin synthesis (5). Further work has shown that the binding of these compounds to the bacterial 50S ribosomal subunit is displaced by puromycin and chloramphenicol (6). Competitive rRNA footprinting experiments also indicated that tiamulin and another analogue, valnemulin, can bind concurrently with the macrolide antibiotic erythromycin. In contrast, these compounds compete with the peptidyl transferase inhibitor carbomycin (13). Additional evidence for the binding of pleuromutilins to the peptidyl transferase center has come from recent x-ray crystallographic data (17) which show tiamulin having interactions with both the ribosomal A- and P-sites. The interaction of pleuromutilins also appears to involve ribosomal protein L3 near the peptidyl transferase center, since Brachyspira sp. isolates with reduced tiamulin susceptibility have recently been identified with mutations in L3 (14). Thus, it is becoming clear that pleuromutilins have a mechanism of action which involves nucleotides within the peptidyl transferase center.
Despite the discovery and development of tiamulin and valnemulin, which have become important veterinary agents to treat swine disease, there has been little progress in the identification of pleuromutilin derivatives to treat bacterial infections in humans. Hence, there has been a renewed interest in the exploitation of novel pleuromutilin derivatives for deployment in human clinical practice (2, 7, 20). Retapamulin (Fig. 1A) is a selective, prokaryotic protein synthesis inhibitor being developed as a topical antibiotic for treating bacterial infections of the skin. This semisynthetic pleuromutilin has potent in vitro activity against susceptible and multidrug-resistant organisms commonly associated with bacterial skin infections (15, 16). Furthermore, in laboratory experiments, retapamulin shows a low propensity for development of resistance, indicating a low likelihood that resistance would develop via target mutation during therapy (9). In this paper, we present the mechanism of action of retapamulin and show that it is distinct from that of several other classes of antibiotics in current clinical use. Our data suggest a molecular mechanism which may help to explain why retapamulin shows no clinically relevant target-specific cross-resistance with other antibacterials.
FIG. 1.

(A) Structure of retapamulin. (B) Structures of other pleuromutilin derivatives used in this study. T indicates the positions of tritium atoms in SB-258781-AAA.
MATERIALS AND METHODS
Bacterial coupled transcription/translation (TnT) assay.
A luciferase reporter gene assay system was used to examine the effects of retapamulin on in vitro coupled transcription and translation of the plasmid pDNA-luciferase (pDNA-luc). The Escherichia coli S30 lysate was used as a source of transcription and translation factors in this assay. The LucLite luciferase reporter gene assay kit was obtained from Packard BioScience. The 2× TnT premix contained 625 μM concentrations of 20 natural l-amino acids, 700 mM potassium l-glutamate, 5 mM ATP, 1.25 mM (each) CTP, UTP, and GTP, 50 mM phosphoenol pyruvate, 44 mM HEPES, 44 mM Tris-acetate, 125 mM ammonium acetate, 2.5 mM cyclic AMP, 50 μg/ml folinic acid, 250 μg/ml E. coli MRE600 tRNA, 22.5 mM magnesium acetate, 2 mM isopropyl-β-d-thiogalactopyranoside (IPTG), 5 mM dithiothreitol, and 87.5 mg/ml polyethylene glycol 8000, pH 8.0. The final reaction conditions were 1% dimethyl sulfoxide (DMSO), 1× TnT premix, 4 mg/ml S30 lysate, 0.02 mg/ml pDNA-luc, and 0 to 50 μM test compound. Reaction mixtures (20 μl) containing test compound, TnT premix, and S30 lysate were preincubated for 15 min at 37°C. The reactions were started by the addition of 5 μl pDNA-luc and incubated at 37°C for 45 min, followed by incubation at ambient temperature for 10 min. Finally, 25 μl of LucLite luciferin substrate reagent was added to the reaction mixtures, and luminescence was measured on the LJL Biosystems Analyst HT. Background luminescence in the absence of template DNA was minimal. IC50s (50% inhibitory concentrations) were calculated using GraFit 4 (Erithacus Software).
Rabbit reticulocyte assay.
Inhibition of eukaryotic translation was evaluated with the rabbit reticulocyte lysate system (Promega) according to the manufacturer's instructions, utilizing luciferase mRNA as a control template. Background luminescence in the absence of mRNA was minimal. Inhibitors were tested from 2 nM to 100 μM, with a final DMSO concentration of 1%. Full-length luciferase was quantified using the Steady Glo luciferase assay system (Promega).
Measurement of pleuromutilin ligand displacement using fluorescence polarization.
Ribosomes were purified essentially as described by Staehelin and Maglott (21). Bacterial ribosomes were incubated at 37°C for 15 min before being diluted in binding buffer (20 mM HEPES, pH 7.5, 50 mM NH4Cl, 10 mM MgCl, 0.05% Tween 20). A fluorescently labeled pleuromutilin derivative (SB-452466) (Fig. 1B), labeled with 4,4-difluoro-3a,4a-diaza-s-indacene (BODIPY) by standard N-hydroxysuccinimidyl ester chemistry, was prepared, and its ribosome-binding properties were characterized kinetically to enable its use as a ligand in fluorescence polarization assays (23). The fluorescent pleuromutilin ligand was preincubated at a concentration of 5 nM with 30 nM E. coli erythromycin-susceptible (Erys) ribosomes for 30 min. Compounds were diluted in 10% DMSO and mixed with the above ligand-ribosome mixture in a 96-well plate in a final volume of 40 μl per well. The incubation continued at room temperature for 2 h. Fluorescence polarization values were measured using an LJL Analyst (excitation at 485 nm and emission at 530 nm.). IC50s were determined by fitting the binding data to a four-parameter IC50 equation (3).
Kd determination of retapamulin binding to ribosomes from E. coli and Staphylococcus aureus.
Bacterial ribosomes were incubated at 37°C for 15 min before dilution in binding buffer (20 mM HEPES, pH 7.5, 50 mM NH4Cl, 10 mM MgCl2, 0.05% Tween 20; for S. aureus ribosomes, this buffer contained 25 mM MgCl2). The on rate and off rate of retapamulin binding to both E. coli and S. aureus ribosomes was investigated in the presence of a radiolabeled pleuromutilin ([3H]SB-258781) (Fig. 1B) in a 96-well plate. In these experiments (n = 3), 20 nM retapamulin was mixed with ribosomes (5 to 15 nM) and [3H]SB-258781 (15 to 19 nM) in a final volume of 100 μl. The binding of [3H]SB-258781 to the ribosomes was competed with retapamulin at room temperature over a time course (2 h for E. coli ribosomes and 5 h for S. aureus ribosomes). The free and bound ligands were then separated through a filter plate (UniFilter GF/B; PerkinElmer) using a cell harvester. The plate was allowed to dry at 50°C for 30 min. After the addition of 50 μl of MicroScint-20 to the plate, the radioactivity in the plate was measured using a TopCount (PerkinElmer). Nonspecific binding was determined by displacement of [3H]SB-258781 with 10 μM SB-268091 (an additional semisynthetic pleuromutilin derivative) (Fig. 1B). The binding data were fit to the kinetics of competitive binding given by Motulsky and Mahan (11).
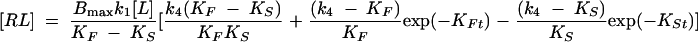 |
(1) |
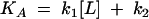 |
(2) |
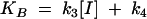 |
(3) |
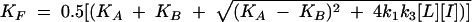 |
(4) |
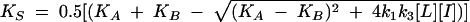 |
(5) |
where k1 and k2 are the on rate and off rate of the radio-ligand, respectively, and k3 and k4 are the on rate and off rate of the unlabeled compound, respectively.
Puromycin assay.
Inhibition of ribosomal peptidyl transferase activity was measured in a scintillation proximity assay similar to that described by Polacek et al. (12). E. coli tRNAfmet (Sigma) was aminoacylated with [3H]methionine (Amersham) and formylated as described by Schwartz and Ofengand (19) using purified methionyl tRNA synthetase as well as E. coli lysate S150 as a source of methionyl tRNA formyltransferase. MVF mRNA of 120 nucleotides encoding methionine, valine, phenylalanine and a stop codon was transcribed from a linear plasmid and purified by standard methods. Biotin-puromycin was obtained from Dharmacon (Lafayette, CO) or Jena BioSciences (Jena, Germany).
Reaction mixtures containing P buffer [50 mM HEPES, pH 7.5, 100 mM NH4Cl, 15 mM Mg(OAc)2], 1 mM dithiothreitol, 50 nM 70S ribosomes, and 125 nM mRNA were incubated at 37°C for 10 min in the presence of compound (1% DMSO final concentration). [3H]fmet-tRNA (50 nM) was added, and the reaction mixture was incubated for 30 min at 37°C. Reaction mixtures were cooled to room temperature, 500 nM of biotin-puromycin was added, and the mixture was incubated for 3 h. Reactions were quenched with 4.1 mg/ml streptavidin-SPA beads (Amersham) in 1× phosphate-buffered saline-125 mM EDTA. The [3H]fmet-puromycin-biotin product was quantified in a Wallac Microbeta (Perkin Elmer). Background counts from reactions lacking biotin-puromycin were minimal and were subtracted from experimental samples.
P-Site binding.
[3H]fmet-tRNA, tRNAphe, and retapamulin were prepared in 10% DMSO, and 20 μl of this ligand-compound mixture was added to a 96-well plate. E. coli ribosomes (Erys) were incubated at 37°C for 15 min and then diluted in P buffer [50 mM HEPES, pH 7.5, 100 mM NH4Cl, 15 mM Mg(OAc)2]. One hundred eighty microliters of the ribosome mixture was added to a 96-well plate containing the above ligand and compound. The final assay mixture contained 50 nM ribosomes, 100 nM [3H]fmet-tRNA, and 50 nM tRNAphe. The binding reactions were incubated at 37°C for 30 min, and the plate was then placed on ice. The bound and free ligands were separated by filtration using a filter plate (MultiScreen HA; Millipore). The plate was then washed twice with P buffer and allowed to dry at 50°C for 30 min. Background counts, determined from binding reactions conducted in the absence of ribosomes, were subtracted from experimental binding reactions. After the addition of 60 μl of MicroScint-20 to the plate, the radioactivity was measured using a TopCount (PerkinElmer). To explore the mode of inhibition of retapamulin in P-site binding, a Schild-type analysis was performed by titrating retapamulin at four different concentrations of [3H]fmet-tRNA ranging from 10 nM to 200 nM. IC50s were determined from each concentration of the radioligand, and the bound ligand (%) was plotted as a function of retapamulin concentration.
Fluorescent chemical rRNA footprinting.
E. coli MRE600 ribosomes were prepared as described above. Dimethyl sulfate (DMS) was obtained from Acros Organics. ThermoScript RNase H-reverse transcriptase was obtained from Invitrogen. MicroSpin S-300 HR columns were obtained from Amersham Biosciences. Reversed-phase high-performance liquid chromatography-purified, 5,6-carboxyfluorescein-labeled DNA primers (sequences below) were supplied by Integrated DNA Technologies, Inc. All other chemical reagents were obtained from Sigma-Aldrich Chemical Co. Ribosomes were activated for the binding reaction by incubation at 42°C for 5 min. Activated E. coli MRE600 ribosomes (200 nM) were incubated with 200 μM compound at 37°C for 20 min and then at ambient temperature for 10 min in 40 mM HEPES (pH 7.4), 150 mM KCl, 10 mM MgCl2, and 6 mM β-mercaptoethanol.
The fluorescent chemical rRNA footprinting assay described herein is similar to conventional rRNA footprinting except that fluorescently labeled primers followed by automated sequencing are used to obtain data (4). Chemical modification of rRNA by methylation of adenine residues was performed by adding 4 μl DMS (1:12 in 100% ethanol) to each 100-μl reaction mixture and incubating the mixture at 37°C for 10 min. The reactions were stopped by ethanol precipitation, and samples were resuspended in 0.3 M sodium acetate (pH 5.4), 2.5 mM EDTA. Immediately prior to purification of the modified rRNA, samples were brought to 0.5% sodium dodecyl sulfate and then purified by phenol-CHCl3-isoamylalcohol (25:24:1, vol/vol) extraction and ethanol precipitation. The methylation pattern of the rRNA bases was monitored by primer extension using ThermoScript RNase H-reverse transcriptase as described by the manufacturer. Primer oLM7, 5′-5,6-carboxyfluorescein-CCT ACA CAT CAA GGC TC-3′ was used to screen A2058, A2059, and A2062. Excess primer was removed from the reactions using a MicroSpin S-300 HR column as described by the manufacturer. The samples were aliquoted into 1-μl fractions, dried under a vacuum, resuspended in loading dye solution, and separated by capillary gel electrophoresis.
Sequencing data was analyzed using GeneScan Analysis Software (ABI PRISM). The footprinting pattern of compound bound to the ribosome is obtained by comparing the methylation patterns at nucleotide bases within the peptidyl transferase center produced in the absence and presence of compound. The data were normalized by monitoring the change in methylation pattern at a base that lies outside of the peptidyl transferase center.
Compounds.
The following compounds were obtained from Medicinal Chemistry, GlaxoSmithKline, Tonbridge, United Kingdom: retapamulin (1), tiamulin, telithromycin, SB-268091, [3H]SB-258781-AAA, and SB-452466. Sparsomycin and erythromycin were purchased from Sigma Chemical, and clarithromycin and azithromycin were purchased from Apin Chemical Limited.
RESULTS
Selective inhibition of bacterial protein synthesis.
Retapamulin was tested in a bacterial coupled transcription/translation assay system which measures the expression of the luciferase gene in an E. coli S30 lysate. Using lysates prepared from erythromycin-susceptible E. coli cells, retapamulin was a potent inhibitor of protein synthesis, with an IC50 of 0.33 ± 0.02 μM (Fig. 2A). Erythromycin was used as a positive control in this experiment and gave an IC50 of 0.264 ± 0.02 μM (data not shown). In contrast, retapamulin was ineffective in inhibiting eukaryotic translation; when tested in a rabbit reticulocyte lysate system with the cellular components necessary for mammalian protein synthesis, retapamulin never achieved more than 20% inhibition at the highest concentration tested (100 μM) (Fig. 2B). Sparsomycin, a compound known to be a nonselective prokaryotic and eukaryotic protein synthesis inhibitor, showed complete inhibition of mammalian translation, with an IC50 of 73 ± 4 nM.
FIG. 2.

Selective inhibition of protein synthesis. (A) Effect of retapamulin on bacterial coupled transcription/translation in S30 lysates from Erys E. coli. Reaction mixtures containing increasing concentrations of retapamulin were incubated as described in Materials and Methods, and the luminescence resulting from the translation of luciferase was measured. (B) Effect of inhibitors on mammalian protein synthesis. Reaction mixtures were incubated with retapamulin (○) or sparsomysin (•) in a rabbit reticulocyte lysate as described in Materials and Methods, and the translation of luciferase was quantified.
Binding of retapamulin to bacterial ribosomes.
A competitive fluorescence polarization binding assay (23) using a fluorescently labeled pleuromutilin derivative (SB-452466) was developed to allow for the determination of pleuromutilin displacement by other compounds. Figure 3A shows that retapamulin binds to Erys ribosomes and fully displaces the labeled ligand with an IC50 of 26.1 ± 3.6 nM; tiamulin also displaced the labeled ligand with an IC50 of 148.8 ± 23.9 nM. In contrast, however, macrolides like erythromycin, azithromycin, and clarithromycin are not able to displace significantly the pleuromutilin ligand from E. coli Erys ribosomes (Fig. 3B). An additional competitive binding assay, using a radiolabeled pleuromutilin ([3H]SB-258781), was used to measure the kinetics of ribosome binding by unlabeled compounds. Our results from these experiments indicate that although retapamulin bound to both E. coli and S. aureus ribosomes with similar potencies (Kd, ∼3 nM), it associated with and dissociated from E. coli ribosomes faster than with S. aureus ribosomes. The kinetic rate constants are presented in Table 1. Representative time courses of binding to these ribosomes are shown in Fig. 4.
FIG. 3.

Binding of pleuromutilins and macrolides to E. coli Erys ribosomes as measured in a fluorescence polarization (FP) competitive binding assay. Ribosomes were incubated with a BODIPY-labeled pleuromutilin derivative for 30 min, inhibitors at various concentrations were added, and the fluorescence polarization values (mP) were measured. IC50 determinations were made by fitting the binding data to a four-parameter IC50 equation. Graphs indicate the binding of retapamulin (○) and tiamulin (•) (A) and erythromycin (•), clarithromycin (□), and azithromycin (▴) (B).
TABLE 1.
Kinetics and thermodynamics of retapamulin binding to E. coli and S. aureus ribosomes
| Ribosome | kon (M−1 s−1)a | koff (s−1)a | Kd (koff/kon) (M) |
|---|---|---|---|
| E. coli | (3.0 ± 0.4) × 105 | (8.3 ± 1.0) × 10−4 | 2.8 × 10−9 |
| S. aureus | (6.3 ± 2.9) × 104 | (2.0 ± 0.2) × 10−4 | 3.2 × 10−9 |
Mean values ± standard errors from three experiments (n = 3). kon, on rate; koff, off rate.
FIG. 4.

A time course of retapamulin binding to E. coli (A) and S. aureus (B) ribosomes. The on rate and off rate of retapamulin binding to bacterial ribosomes were measured in a competitive filter-binding assay using a [3H]pleuromutilin derivative as a ligand. The binding data were fit to a kinetics of competitive binding model. Binding was conducted in the absence (•) and presence (▴) of 20 nM retapamulin.
Inhibition of bacterial peptidyl transferase and fmet-tRNA binding.
The effect of retapamulin on peptide bond formation was evaluated using a version of the puromycin reaction adapted to a higher-throughput scintillation proximity assay format (12). This assay measures the ability of the ribosome to catalyze peptide bond formation between the aminoacylated initiator fmet-tRNA substrate in the P-site and biotinylated puromycin (acceptor substrate) in the A-site of the ribosome. Figure 5A shows the concentration-response plots of retapamulin and tiamulin when titrated against peptidyl transferase activity reconstituted with E. coli ribosomes. The IC50 for both compounds was below the tight binding limit of the assay (∼100 nM). In addition, Fig. 5B shows that retapamulin partially inhibits the ability of charged, N-blocked tRNA to bind to the P-site of E. coli ribosomes, with an IC50 of 17.4 ± 2.1 nM (maximum inhibition = 80%). To confirm the specificity of this inhibition, we investigated whether other ribosome-targeting drugs were able to interfere with binding of the fmet-tRNA substrate in a filter binding assay. The antibiotics erythromycin, clarithromycin, azithromycin, telithromycin, and linezolid were tested. Unlike retapamulin, none of these compounds displaced binding of fmet-tRNA from ribosomes (data not shown) at concentrations up to 10 μM.
FIG. 5.

(A) Inhibition of ribosomal peptidyl transferase activity. Reaction mixtures containing ribosomes, [3H]fmet-tRNA, mRNA, and biotinylated puromycin were carried out in the presence of increasing concentrations of tiamulin (•) or retapamulin (○) as described in Materials and Methods, and the amount of [3H]fmet-biotin-puromycin was quantified using strepatividin SPA beads. IC50 determinations were made by fitting the data to a four-parameter IC50 equation. (B) Inhibition of fmet-tRNA binding to E. coli ribosomes. Ribosomes were incubated with [3H]fmet-tRNA (P-site substrate), tRNAphe (A-site substrate), mRNA, and increasing concentrations of retapamulin. The bound and free ligands were separated by filtration, and IC50 values were calculated. (C) Schild analysis of retapamulin displacement of fmet-tRNA binding. P-site binding reactions, as described above, were conducted by titrating retapamulin at 10 nM (▪), 20 nM (□), 60 nM (•), or 200 nM (○) concentrations of [3H]fmet-tRNA, and the bound ligand (%) was plotted as a function of the retapamulin concentration.
To address further how retapamulin interferes with binding of the P-site tRNA substrate, a Schild-type analysis was performed by titrating the compound with several concentrations of [3H]fmet-tRNA. As seen in Fig. 5C, the IC50s of retapamulin are independent of radioligand concentrations. These data are most consistent with a mechanism of inhibition for retapamulin that is antagonistic but not mutually exclusive, with fmet-tRNA binding to the ribosome (3).
rRNA footprinting by retapamulin.
Table 2 shows the extent of protection or enhancement of reactivity to DMS for A2058, A2059, and A2062 when ribosomes were incubated in the presence of saturating amounts of compound. In contrast to the protection of A2058 and A2059 reactivity afforded by telithromycin, both pleuromutilin derivatives enhanced the reactivity of these nucleotides to the chemical modification reagent; retapamulin showed an almost threefold enhancement of A2059 reactivity compared to tiamulin. Figure 6 shows the observed protection and enhancement patterns of these nucleotides mapped onto the secondary structure of the peptidyl transferase region of 23S rRNA.
TABLE 2.
Fluorescent chemical rRNA footprinting of retapamulin, tiamulin, and telithromycin
| Nucleotide | % P or E in the presence of compounda:
|
||
|---|---|---|---|
| Telithromycin | Tiamulin | Retapamulin | |
| A2058 | 89 (P) | 148 (E) | 147 (E) |
| A2059 | 88 (P) | 221 (E) | 635 (E) |
| A2062 | 63 (E) | 32 (E) | 44 (E) |
P, protection; E, enhancement. The terms protection and enhancement refer to the effect of compound binding on the susceptibility of the nucleotide to chemical modification by DMS.
FIG. 6.

Chemical footprint of retapamulin and telithromycin on E. coli 23S rRNA. The extent of protection or enhancement of chemical modification by DMS for rRNA was measured in the presence of retapamulin, tiamulin, or telithromycin. The resulting methylation pattern of rRNA bases was monitored by extension of fluorescently labeled primers and subsequent sequencing by capillary gel electrophoresis. ○, enhancement of reactivity by retapamulin; ▴, enhancement of reactivity by telithromycin; •, protection of reactivity by telithromycin.
DISCUSSION
Retapamulin was tested for its ability to inhibit bacterial protein synthesis in a coupled transcription/translation assay using lysate from erythromycin-sensitive E. coli. This compound was a potent inhibitor of this assay, with an IC50 of 0.33 μM (Fig. 2A). While these data do not necessarily differentiate an effect on transcription or translation, our data are consistent with a direct inhibitory effect on translation based on the known mechanism of action of pleuromutilins (7). When tested in a eukaryotic, nonorganelle protein synthesis assay using rabbit reticulocyte lysate, retapamulin was not effective in inhibiting eukaryotic translation (Fig. 2B). In contrast, sparsomycin, a known nonselective protein synthesis inhibitor, blocked eukaryotic translation with an IC50 of 73 nM. While retapamulin is a potent inhibitor of bacterial protein synthesis, its selectivity against mitochondrial protein synthesis remains unknown.
A method using fluorescence polarization to characterize drug interactions with the bacterial ribosome has recently been described (23). We took advantage of this technology to develop a robust and sensitive competitive binding assay using a fluorescently labeled pleuromutilin (SB-452466) to determine the IC50s of pleuromutilin displacement by other compounds. These competitive binding studies show that retapamulin efficiently binds to and completely displaces the labeled ligand from erythromycin-susceptible ribosomes, while drugs like erythromycin, azithromycin, and clarithromycin do not (Fig. 3). An additional competitive binding assay using a radiolabeled pleuromutilin was used to determine the kinetics of inhibitor binding to ribosomes (Fig. 4). Taken together, our data indicate that the potent antimicrobial activity of retapamulin is likely due to its high-affinity interaction with the ribosome (Kd, ∼3 nM). Further, although pleuromutilins and macrolides both inhibit bacterial growth by inhibition of protein synthesis, the present data indicate that the two classes of inhibitors bind to bacterial ribosomes at distinct sites.
Peptide bond formation is the principal reaction of protein synthesis and takes place in the peptidyl transferase center of the 50S ribosomal subunit. While some macrolide-lincosamide-streptogramin B (MLSB) antibiotics are known to be inhibitors of peptidyl transferase (e.g., carbomycin, tylosin, spiramycin, lincomycin), the majority of clinically relevant macrolides are not (e.g., erythromycin, telithromycin, clarithromycin, and azithromycin) (18, 22). Retapamulin is a potent inhibitor of ribosomal peptidyl transferase activity on erythromycin-susceptible ribosomes from E. coli (Fig. 5A) and thus inhibits protein synthesis by preventing peptide bond formation. Our data are consistent with earlier work showing the inhibition of peptidyl transferase by pleuromutilin derivatives (5, 13). Additionally, retapamulin was found to partially inhibit binding of fmet-tRNA to the P-site of the ribosome (Fig. 5B), suggesting that the compound may interfere with binding of the P-site tRNA substrate. Our inability to detect complete inhibition of P-site binding is particularly intriguing. The Schild analysis shown in Fig. 5C indicates that the binding of retapamulin is not competitive with P-site tRNA, suggesting that there is an allosteric component to the inhibition of tRNA-binding. This is not at all surprising given the recent structural data (17) indicating that tiamulin clearly interferes with the correct positioning of both A- and P-site tRNA substrates. Earlier biochemical footprinting data have shown that tiamulin and valnemulin protect U2584 and U2585 in the peptidyl transferase center (13). Taken together with the fact that these nucleotides are implicated in the binding of P-site tRNA (10), it is reasonable to suggest that pleuromutilins, including retapamulin, may destabilize tRNA binding to the P-site and/or cause the repositioning of tRNA on the ribosome so that the acceptor arm is no longer able to participate in peptidyl transfer. While its contribution to the overall inhibition of protein synthesis remains to be explored further, the inhibition of P-site binding by retapamulin suggests an effect on translation initiation which further differentiates this compound from macrolide and ketolide bacterial protein synthesis inhibitors in current human clinical use.
The effect of retapamulin, tiamulin, and telithromycin (Ketek) on the peptidyl transferase region of 23S rRNA was measured by chemically probing the accessibility of the N1 position in adenosine residues in the putative compound binding sites. Consistent with earlier biochemical data for tiamulin and valnemulin (13), our results indicate that retapamulin results in an enhancement of reactivity and suggests that retapamulin may bind either allosterically or orthosterically to the rRNA and induce a different conformational change in the peptidyl transferase center such that the accessibility of these nucleotides is altered. This is in contrast to data we obtained for telithromycin, which showed a protection of A2058 and A2059. Furthermore, the effects of retapamulin on nucleotides in this region are entirely consistent with its activity as a potent inhibitor of ribosomal peptidyl transferase.
Overall, these studies demonstrate that retapamulin can be clearly differentiated from macrolide and ketolide bacterial protein synthesis inhibitors which are currently in clinical use. Retapamulin is fully active against clinical isolates carrying resistance determinants to these classes of protein synthesis inhibitors as well as isolates resistant to antibiotics that act by mechanisms other than inhibition of protein synthesis (e.g., β-lactams, quinolones). While more work remains to be done, these data suggest that retapamulin exhibits a complex mode of interaction with the ribosomal target which play an important role in retapamulin achieving antibacterial activity against susceptible and multidrug-resistant pathogens (15, 16).
Footnotes
Published ahead of print on 28 August 2006.
REFERENCES
- 1.Berry, V., S. Dabbs, H. C. Frydrych, E. Hunt, G. Woodnutt, and D. F. Sanderson. May 1999. Pleuromutilin derivatives as antimicrobials. International patent WO99/21855.
- 2.Brooks, G., W. Burgess, D. Colthurst, J. D. Hinks, E. Hunt, M. J. Pearson, B. Shea, A. K. Takle, J. M. Wilson, and G. Woodnutt. 2001. Pleuromutilins. Part 1. The identification of novel mutilin 14-carbamates. Bioorg. Med. Chem. 9:1221-1231. [DOI] [PubMed] [Google Scholar]
- 3.Copeland, R. A. 2005. Evaluation of enzyme inhibitors in drug discovery: a guide for medicinal chemists and pharmacologists. John Wiley & Sons, Inc., Hoboken, N.J. [PubMed]
- 4.Gould, P. S., H. Bird, and A. J. Easton. 2005. Translation toeprinting assays using fluorescently labeled primers and capillary electrophoresis. BioTechniques 38:397-400. [DOI] [PubMed] [Google Scholar]
- 5.Hodgin, L. A., and G. Hogenauer. 1974. The mode of action of pleuromutilin derivatives. Effect on cell-free polypeptide synthesis. Eur. J. Biochem. 47:527-533. [DOI] [PubMed] [Google Scholar]
- 6.Hogenauer, G. 1975. The mode of action of pleuromutilin derivatives. Location and properties of the pleuromutilin binding site on Escherichia coli ribosomes. Eur. J. Biochem. 52:93-98. [DOI] [PubMed] [Google Scholar]
- 7.Hunt, E. 2000. Pleuromutilin antibiotics. Drugs Future 25:1163-1168. [Google Scholar]
- 8.Kavanagh, F., A. Hervey, and W. J. Robbins. 1951. Antibiotic substances from Basidiomycetes. VIII. Pleurotus multilus (fr.) Sacc. and Pleurotus Passeckerianus Pilat. Proc. Natl. Acad. Sci. USA 37:570-574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kosowska-Shick, K., C. Clark, K. Credito, P. McGhee, B. Dewasse, T. Bogdanovich, and P. C. Appelbaum. 2006. Single- and multistep resistance selection studies on the activity of retapamulin compared to other agents against Staphylococcus aureus and Streptococcus pyogenes. Antimicrob. Agents Chemother. 50:765-769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moazed, D., and H. F. Noller. 1989. Interaction of tRNA with 23S rRNA in the ribosomal A, P, and E sites. Cell 57:585-597. [DOI] [PubMed] [Google Scholar]
- 11.Motulsky, H. J., and L. C. Mahan. 1984. The kinetics of competitive radioligand binding predicted by the law of mass action. Mol. Pharmacol. 25:1-9. [PubMed] [Google Scholar]
- 12.Polacek, N., S. Swaney, D. Shinabarger, and A. S. Mankin. 2002. SPARK-a novel method to monitor ribosomal peptidyl transferase activity. Biochemistry 41:11602-11610. [DOI] [PubMed] [Google Scholar]
- 13.Poulsen, S. M., M. Karlsson, L. B. Johansson, and B. Vester. 2001. The pleuromutilin drugs tiamulin and valnemulin bind to the RNA at the peptidyl transferase centre on the ribosome. Mol. Microbiol. 41:1091-1099. [DOI] [PubMed] [Google Scholar]
- 14.Pringle, M., J. Poehlsgaard, B. Vester, and K. S. Long. 2004. Mutations in ribosomal protein L3 and 23S ribosomal RNA at the peptidyl transferase centre are associated with reduced susceptibility to tiamulin in Brachyspira spp. isolates. Mol. Microbiol. 54:1295-1306. [DOI] [PubMed] [Google Scholar]
- 15.Rittenhouse, S., C. Singley, J. Hoover, R. Page, and D. Payne. 2006. Use of the surgical wound infection model to determine the efficacious dosing regimen of retapamulin, a novel topical antibiotic. Antimicrob. Agents Chemother. 50.: 3886-3888. [DOI] [PMC free article] [PubMed]
- 16.Rittenhouse, S., S. Biswas, J. Broskey, L. McCloskey, T. Moore, S. Vasey, J. West, M. Zalacain, R. Zonis, and D. Payne. 2006. Selection of retapamulin, a novel pleuromutilin for topical use. Antimicrob. Agents Chemother. 50:3882-3885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schlunzen, F., E. Pyetan, P. Fucini, A. Yonath, and J. M. Harms. 2004. Inhibition of peptide bond formation by pleuromutilins: the structure of the 50S ribosomal subunit from Deinococcus radiodurans in complex with tiamulin. Mol. Microbiol. 54:1287-1294. [DOI] [PubMed] [Google Scholar]
- 18.Schlünzen, F., R. Zarivach, J. Harms, A. Bashan, A. Tocilj, R. Albrecht, A. Yonath, and F. Franceschi. 2001. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature 413:814-821. [DOI] [PubMed] [Google Scholar]
- 19.Schwartz, I., and J. Ofengand. 1978. Photochemical cross-linking of unmodified acetylvalyl-tRNA to 16S RNA at the ribosomal P site. Biochemistry 17:2524-2530. [DOI] [PubMed] [Google Scholar]
- 20.Springer, D. M., M. E. Sorenson, S. Huang, T. P. Connolly, J. J. Bronson, J. A. Matson, R. L. Hanson, D. B. Brzozowski, T. L. LaPorte, and R. N. Patel. 2003. Synthesis and activity of a C-8 keto pleuromutilin derivative. Bioorg. Med. Chem. Lett. 13:1751-1753. [DOI] [PubMed] [Google Scholar]
- 21.Staehelin, T., and D. T. Maglott. 1971. Preparation of Escherichia coli ribosomal subunits active in polypeptide synthesis. Methods Enzymol. 20:449-456. [Google Scholar]
- 22.Tenson, T., M. Lovmar, and M. Ehrenberg. 2003. The mechanism of action of macrolides, lincosamides and streptogramin B reveals the nascent peptide exit path in the ribosome. J. Mol. Biol. 330:1005-1014. [DOI] [PubMed] [Google Scholar]
- 23.Yan, K., E. Hunt, J. Berge, E. May, R. A. Copeland, and R. R. Gontarek. 2005. Fluorescence polarization method to characterize macrolide-ribosome interactions. Antimicrob. Agents Chemother. 49:3367-3372. [DOI] [PMC free article] [PubMed] [Google Scholar]