

Abstract
Perlecan, a heparan sulfate proteoglycan, is widely distributed in developing and adult tissues and plays multiple, important physiological roles. Studies with knockout mouse models indicate that expression of perlecan and heparan sulfate is critical for proper skeletal morphogenesis. Heparan sulfate chains bind and potentiate the activities of various growth factors such as fibroblast growth factor 2 (FGF-2). Previous studies indicate that important biological activities are associated with the heparan sulfate-bearing domain I of perlecan (PlnDI; French et al. J. Bone Miner. Res. 17, 48, 2002). In the present study, we have used recombinant, glycosaminoglycan-bearing PlnDI to reconstitute three-dimensional scaffolds of collagen I. Collagen I fibrils bound PlnDI much better than native collagen I monomers or heat-denatured collagen I preparations. Heparitinase digestion demonstrated that recombinant PlnDI was substituted with heparan sulfate and that these heparan sulfate chains were critically important not only for efficient integration of PlnDI into scaffolds, but also for FGF-2 binding and retention. PlnDI-containing collagen I scaffolds to which FGF-2 was bound sustained growth of both MG63, an osteoblastic cell line, and human bone marrow stromal cells (hBMSCs) significantly better than scaffolds lacking either PlnDI or FGF-2. Collectively, these studies demonstrate the utility of PlnDI in creating scaffolds that better mimic natural extracellular matrices and better support key biological activities.
INTRODUCTION
The extracellular matrix (ECM) consists of a complex mixture of proteins and glycoproteins serving multiple functions.1,2 During tissue repair the ECM serves as a platform to supply growth factors that modulate such diverse processes as angiogenesis, cell migration, cell proliferation, cell differentiation, and wound healing.1-3 These characteristics of ECM provide opportunities to improve biological activities of scaffolds used for tissue-engineering (TE) applications. The development of new classes of biomaterials for TE has focused on the design of biomimetic materials. In many cases, these materials utilize immobilized ECM components or peptide sequences derived from biologically active sequences within ECM proteins in concert with the basic materials.4,5 In addition, extensive studies have been performed to improve the design strategies for TE scaffolds.5-8
Heparan sulfate proteoglycans (HSPGs) in ECM have a wide range of biological activities and functions including modulation of cellular growth,2,3 development,9 angiogenesis,10,11 and tissue regeneration.2,3,12 Perlecan (Pln) is an important HSPG expressed in many extracellular matrices and basement membranes. Mutations in or genetic ablation of perlecan causes severe defects in various developmental systems including the skeleton.13 Pln has a protein core of approximately 400 kDa and consists of five distinct domains.14 Domain I is unique to Pln and contains three consensus SGD motifs that act as glycosaminoglycan (GAG) attachment sites.15 Through GAG chains attached to domain I, Pln can function as a ligand reservoir for storage and release of heparin-binding growth factors, such as fibroblast growth factor 2 (FGF-2) and vascular endothelial growth factor (VEGF), and also can protect these proteins from inactivation by proteolytic digestion.16-19 Pln also functions as a coreceptor for FGF-2,20,21 and regulates FGF activity in vitro and in vivo through its heparan sulfate (HS) chains.19,21-23 HS chains support growth factor binding to HSPGs.3,5,21-23 The benefits of including HS as a component of biomimetic materials are under study.4,5,7,8 These observations, along with previous studies mapping important biological activities of Pln to GAG-bearing domain I (PlnDI), suggest that inclusion of PlnDI is likely to improve biological activities of TE scaffolds.
Collagen I is a major structural protein in ECM. Collagen I is secreted as procollagen, which after proteolytic cleavage yields the triple-helical monomer composed of two α1 and one α2 chains.24,25 These monomers self-assemble in a regular staggered fashion into fibrils.24,25 It has been shown that HSPGs and HS can bind to natural fibrils, under physiological salt and pH conditions in vitro.26-28 In addition, HSPG and HS binding depends on the triple-helical and fibrillar conformation of collagen I fibrils.28,29 Furthermore, HS and HSPG binding requires a collagen structure that is optimal when collagen is in a fibrillar configuration.26,28 Thus, collagen I fibrils are ideal candidates with which to study the interaction with HS and HSPG. Collagen I has been used as a natural material for TE scaffolds30; however, it frequently has been used as a monomer8,31-34 and in denatured forms.35,36
Others have attempted to improve biological activity and growth factor delivery by TE scaffolds by covalently attaching GAG and heparin to collagen I scaffolds, using chemical procedures.31-34,37 We hypothesized that PlnDI would bind both FGF-2 and collagen I fibrils in a self-assembling fashion. These complexes were predicted to promote binding and better release of heparin-binding growth factors. In the present study, we used FGF-2 to assess such growth factor binding and biological activity of a scaffold including various forms of collagen type I and PlnDI. The results suggest that PlnDI used in conjunction with collagen I scaffolds improves FGF-2 binding and better sustains cell growth than do collagen I scaffolds alone.
MATERIALS AND METHODS
Materials
Recombinant human FGF-basic (FGF-2) and biotinylated mouse anti-human FGF-2 antibody were obtained from & Systems (Minneapolis, MN). Biotinylated rabbit anti-human FGF-2 antibody was purchased from Research Diagnostics (Flanders, NJ). Rabbit anti-FGF-2 was obtained from Chemicon International (Temecula, CA). Rabbit anti-mouse PlnDI was a gift from R. Timpl (Max-Planck-Institut für Biochemie, Martinsried, Germany). Collagen type I solution (prepared from rat tail tendon, 3.6-10.9 mg/mL) in 0.02 N acetic acid was purchased from BD Biosciences (Bedford, MA). Heparinases I, II, and III, chondroitinase AC, dexamethasone, bovine serum albumin (BSA), Tween 20, and d-(+)-glucose were obtained from Sigma-Aldrich (St. Louis, MO). l-Ascorbic acid-2-phosphate was purchased from Wako Pure Chemical Industries (Japan). Protein quantitation assay reagent (Coomassie Plus-200 protein assay reagent), NeutrAvidin (horseradish peroxidase conjugated) (NA-HRP, NeutrAvidin-HRP), 3,3′,5,5′tetramethylbenzidine (TMB, 1-Step Ultra TMB-ELISA), Coomassie Plus-200 protein assay reagent, blocking buffer (SuperBlock blocking buffer), and chemiluminescence substrate (SuperSignal West Dura extended duration substrate) were purchased from Pierce Biotechnology (Rockford, IL).
Methods
Preparation of recombinant perlecan domain I. Recombinant perlecan domain I (PlnDI) was prepared as described previously,38 with some modification. Briefly, complementary DNA (cDNA) encoding domain I of perlecan (amino acid residues 22-194) was inserted in frame behind the leader sequence of the basement membrane protein, BM-40. This construct then was inserted into the pCEP-Pu vector and stably transfected into 293 EBNA cells, according to the manufacturer’s instructions (Invitrogen, Carlsbad, CA). Positive clones were selected with puromycin (100 μg/mL) and the production of PlnDI by stable transfectants was verified by indirect immunoflouresence employing perlecan domain I-specific antibodies, and by dot-blot analysis of conditioned medium (see below).
To generate PlnDI-containing conditioned medium, clones expressing PlnDI were cultured in Nunc cell factories (Nalge Nunc International, Naperville, IL). Serum-free conditioned medium was collected every other day for 10 days, and stored at −20°C. After the filtration of cell debris, the conditioned medium was concentrated and partially purified with a spiral-wound membrane cartridge with a 10,000 molecular weight cutoff (Amicon, Beverly, MA). The medium then was applied to a diethylaminoethyl (DEAE)-cellulose column (2.5 × 10 cm; Bio-Rad, Hercules, CA), equilibrated with 0.05 M Tris-HCl (pH 8.6), 2 M urea, 0.25 M NaCl, 2.5 mM EDTA, 0.5 mM phenylmethylsulfonyl fluoride (PMSF), and 5 mM benzamidine. After extensive washes with the equilibration buffer, PlnDI was eluted with 0.5 M NaCl, in 0.05 M Tris-HCl (pH 8.6), 2 M urea, 2.5 mM EDTA, 0.5 mM PMSF, and 5 mM benzamidine. PlnDI purity was assessed by NuPAGE gel electrophoretic analysis (4-12% [w/v] Bis-Tris with morpholinoethanesulfonic acid [MES; Invitrogen]) according to the manufacturer’s instructions, followed by Western blot analysis with PlnDI-specific antibodies.
FGF-2 binding to PlnDI. A dot-blotting format was used to determine whether PlnDI was functionally active in binding FGF-2. Briefly, PlnDI was digested with chondroitinase AC or heparinases I, II, and III for 4 h at 37°C. Three micrograms of undigested or digested PlnDI was blotted onto nitrocellulose, and subsequently blocked with 5% (w/v) fat-free milk powder in blocking buffer for 4 h at 4°C. After washing with blocking buffer, 100 ng of FGF-2 was added to each well of the blotting apparatus and incubated for 4 h at room temperature. The membrane then was removed from the blotting apparatus, and blocked with 3% (w/v) BSA in blocking buffer for 4 h at 4°C, before incubation in biotinylated rabbit anti-FGF-2 antibody (0.1 μg/mL) for 1 h at room temperature. After this incubation, the membrane was washed in 0.05% (v/v) Tween 20 in phosphate-buffered saline (PBS-T), and incubated with NA-HRP for 20 min at room temperature. After washing with PBS-T, the bound antibody was detected via enhanced chemiluminescence. The binding of FGF-2 to PlnDI was evaluated by densitometry and expressed as individual density values (IDVs).
PlnDl binding to collagen I fibrils. To monitor PlnDI binding to collagen I fibrils, the binding assay was performed essentially as described previously26,39 with some modification. Briefly, collagen I fibrils were generated by dialyzing a collagen type I solution (3.60 mg/mL) in 0.02 N acetic acid for 18 h against PBS at 4°C. PlnDI was biotinylated with sulfo-NHS-LC-biotin [sulfosuccinimidyl-6-(biotinamido) hexanoate], using an EZ-Link Sulfo-NHS-LC-Biotinylation kit (Pierce Biotechnology) according to the manufacturer’s instructions.
For the binding analysis, biotinylated PlnDI (3 μg), digested or undigested with heparinases I, II, and III and/or chondroitinase AC, was added to a 200- μL solution of PBS containing 3% (w/v) BSA and 20 μg of collagen I fibrils in a 500- μL polyethylene microcentrifuge tube and then blocked with 3% (w/v) BSA for 1 h at room temperature. After a 2-h incubation with biotinylated PlnDI at room temperature, collagen I fibrils were collected by centrifugation for 10 min at room temperature. To remove trapped biotinynated PlnDI from the collagen I fibrils, pellets were resuspended in 200 μL of PBS by brief sonication on ice, and then centrifuged for 5 min at 10,000 × g in a microcentrifuge. This wash step was repeated three times. Collagen I fibrils then were resuspended and incubated with NA-HRP (1 μg/mL) in 200 μL of blocking buffer for 40 min at room temperature. After washing with PBS, the pellets were reacted with 25 μL of TMB for 5-8 min. Two hundred microliters of solution was finally transferred to each well of 96-well plate for optical density (OD) measurement at 450 nm.
FGF-2 binding to PlnDI-collagen fibril complexes. PlnDI (3 μg), undigested or digested with heparinases I, II, and III, was bound to collagen I fibrils (20 μg) to form two different complexes: digested PlnDI/collagen I (D-PF) and undigested PlnDI/collagen (PF) fibrils. Collagen I fibrils alone (F), and collagen I fibrils digested (D-F) with heparinases I, II, and III, were used as controls. D-PF, PF, D-F, and F then were incubated with 50 ng of FGF-2 in 200 μL of 3% (w/v) BSA for 2 h at room temperature. After washing, these samples were reacted with biotinylated mouse anti-FGF-2 antibody (20 ng/mL) in 200 μL of 1% (w/v) BSA plus 2% (v/v) heat-inactivated normal goat serum for 2 h at room temperature. After washing with PBS, these samples then were incubated with NA-HRP (1 μg/mL) in 200 μL of blocking buffer for 40 min at room temperature, and pelleted by centrifugation. The pellets were reacted with TMB followed by washing with PBS as described above. Two hundred microliters of solution was transferred to each well of a 96-well plate for OD measurement at 450 nm.
Fabrication of collagen I scaffolds and binding of PlnDI to scaffolds. Collagen I scaffolds were prepared, employing three different forms of collagen I: collagen I monomer, denatured collagen I fibrils, and collagen I fibrils. Each scaffold was made as follows:
Collagen I fibrils: 8 volumes of collagen I fibril suspension (10 mg/mL) and 2 volumes of collagen I dispersion (10 mg/mL) in 0.02 N acetic acid were mixed on ice, and diluted with PBS to 7 mg/mL of collagen I fibril suspension.
Denatured collagen I fibrils: Collagen I fibrils (10 mg/mL) were denatured at 56°C for 30 min, and then mixed with collagen I dispersion (10 mg/mL) in 0.02 N acetic acid on ice, and diluted with PBS to 7 mg/mL of collagen I fibril suspension.
Collagen I monomer: Collagen I dispersion in 0.02 N acetic acid (10.9 mg/mL) was diluted with 0.02 N acetic acid to 7.0 mg/mL of collagen I dispersion.
After addition of d-(+)-glucose (9 mM) and deaeration of the suspension under vacuum to remove entrapped air bubbles, 200 μL of each form of collagen I solution was poured into individual polyethylene molds (Nunc-Immuno Modules; Nalge Nunc International) at -40°C for 24 h and then lyophilized for 48 h. The scaffolds then were cross-linked as described previously.40,41 The scaffolds were placed inside an ultraviolet (UV) cross-linking chamber (Stratalinker 2400; Stratagene Cloning Systems, La Jolla, CA) for 3 h at a distance of 4.5 in. from a bank of five UV bulbs with a primary emission at 254 nm.
For the PlnDI-binding assay, biotinylated PlnDI (3 μg), undigested or digested with heparinases I, II, and III, was incubated with each scaffold in 200 μL of PBS for 2 h at room temperature, after blocking with 3% (w/v) BSA in PBS for 1 h at room temperature. After washing extensively with PBS, employing a Vortex mixer (Scientific Industries, Bohemia, NY), the scaffolds were incubated with NA-HRP (1 μg/mL) in 3% BSA (w/v) in PBS for 40 min at room temperature, and then reacted with TMB. The OD at 450 nm was determined as described above.
FGF-2 binding to collagen I scaffolds. Collagen I fibrils were incubated with PlnDI at a ratio of 30 μg of PlnDI to 10 mg of collagen I fibrils in 1 mL of PBS to form PF complex. After washing with PBS extensively, the PF complex was used to prepare scaffolds (PF-scaffolds) as described above. Natural fibrils of collagen I alone were used to prepare collagen I fibril scaffolds (F-scaffolds). After treatment (or no treatment) with heparinase I, II, and III for 4 h, each scaffold was incubated with 50 ng of FGF-2 in 500 μL of PBS for 2 h after blocking with 3% (w/v) BSA in PBS. The binding of FGF-2 to PF- and F-scaffolds was detected with a biotinylated anti-FGF-2 antibody (20 ng/mL) in 500 μL of 1% BSA (w/v) and 2% (v/v) heat-inactivated normal goat serum in PBS for 2 h at room temperature, and finally reacted with NA-HRP (1 μg/mL) in 3% (w/v) BSA in PBS for 40 min at room temperature, and then reacted with TMB. The OD at 450 nm was determined as described above.
Immunolocalization of PlnDI and FGF-2 in collagen I scaffolds. Immunostaining was used to demonstrate the distribution of PlnDI in PF-scaffolds, and the effect of PlnDI associated in PF-scaffolds on immobilization of FGF-2. PF-scaffolds and F-scaffolds were produced as described above. After blocking with 3% (w/v) BSA in PBS, each of the PF-scaffolds and F-scaffolds was incubated with 50 ng of FGF-2 in 500 μL of 3% (w/v) BSA in PBS for 2 h. After washing with PBS, the samples, PF-scaffolds and F-scaffolds, were fixed briefly in 4% (w/v) paraformaldehyde in PBS for 30 min. After washing with PBS, the samples were embedded in O.C.T. medium (Sakura Finetek, Torrance, CA), frozen on dry ice, and then sectioned in a cryostat. Sixty-micron sections of the samples were mounted on gelatin-coated coverslips blocked with DAKO serum-free protein block (DakoCytomation, Carpinteria, CA) and incubated with antibodies specific for PlnDI (diluted 1:200) or FGF-2 (diluted 1:200) for 1 h at 37°C. After washing with PBS, the sections were incubated with goat anti-rabbit IgG conjugated with Alex Fluor 568 (Molecular Probes, Eugene, OR), diluted 1:50 in PBS, for 40 min at 37°C, and subsequently observed by confocal microscopy.
FGF-2 release. Dry collagen I scaffolds, PF-scaffolds, and F-scaffolds were loaded with 10 ng of FGF-2 in 5 μL of PBS and then incubated for 2 h at room temperature to form PF-scaffolds preloaded with FGF-2 (PF/FGF-scaffolds) and F-scaffolds preloaded with FGF-2 (F/FGF-scaffolds). Samples (PF/FGF-scaffolds and F/FGF-scaffolds) were transferred to release medium (Dulbecco’s modified Eagle’s medium [DMEM] plus penicillin [100 U/mL] and streptomycin [100 μg/mL]), and incubated in 4°C. The release medium was harvested at 1 h, 6 h, 12 h, 24 h, 3 days, 1 week, 2 weeks, and 4 weeks, and stored at -40°C. The content of FGF-2 in the release medium was determined in a sandwich ELISA (Quantikine bFGF ELISA; & Systems), according to the manufacturer’s instructions.
Digestion of collagen I scaffolds loaded with FGF-2. After blocking with 3% (w/v) BSA in PBS, PF-scaffolds and F-scaffolds were incubated with FGF-2 (50 ng/mL) in 500 μL of PBS to form PF/FGF-scaffolds and F/FGF-scaffolds, respectively. After washing extensively with PBS in a Vortex mixer, PF/FGF-scaffolds and F/FGF-scaffolds were treated with heparinases I, II, and III for 4 h at room temperature or left untreated. The content of FGF-2 in the supernatant was measured in a sandwich ELISA (Quantikine bFGF ELISA; & Systems) according to the manufacturer’s instructions.
MG63 and hBMSC proliferation on scaffolds. PF-scaffolds and F-scaffolds were incubated with recombinant human FGF-2 (rhFGF-2, 10 ng/mL) in DMEM supplemented with 5% (v/v) fetal calf serum (FCS) for 2 h at room temperature to form PF/FGF- and F/FGF-scaffolds, respectively. After washing with PBS extensively, the scaffolds, PF/FGF, F/FGF, PF, and F, were placed in 24-well tissue culture dishes containing an MG63 (American Type Culture Collection [ATCC], Manassas, VA) cell suspension (2 × 105/mL) in DMEM supplemented with 5% (v/v) FCS. The 24-well plates were shaken gently at intervals of 30-40 min for 4 h. Cell-seeded scaffolds then were transferred to new 24-well plates and incubated in DMEM containing 1% (v/v) FCS at 37°C in a humidified atmosphere of 95% air and 5% CO2. Culture medium containing 1% (v/v) FCS was changed every 3 days. Cultures were harvested on days 1, 3, 6, 9, and 12. Human bone marrow stromal cells (hBMSCs) were isolated and expanded as previously described.42,43 Briefly, human bone marrow aspirates were obtained from the iliac crest of health adult human donors after obtaining informed consent, and were resuspended in DMEM supplemented with 10% (v/v) FCS. The hBMSCs were selected on the basis of their ability to adhere to the tissue culture plastic; nonadherent hematopoietic cells were removed during medium replacement. The hBMSCs were expanded in DMEM supplemented with 20% (v/v) FCS and cultured at 37°C in a humidified atmosphere of 95% air and 5% CO2. These cells from 80-90% confluent monolayers were expanded through four passages. Using the same method applied for MG63 cells, hBMSCs (2 × 105/mL) in DMEM supplemented with 10% (v/v) FCS were seeded onto scaffolds (PF/FGF, F/FGF, PF, and F). The seeded scaffolds then were transferred to new 24-well plates and incubated in DMEM containing 10% (v/v) FCS, 10 nM dexamethasone, and ascorbate 2-phosphate (50 μg/mL) at 37°C in a humidified atmosphere of 95% air and 5% CO2 for 15 days. Culture medium was changed every 3 days. Cultures were terminated on days 1, 5, 10, and 15. Proliferation of MG63 cells and hBMSCs was determined by immunodetection of bromodeoxyuridine (BrdU) incorporation (Cell Proliferation ELISA Biotrak System; Amersham Biosciences, Piscataway, NJ), according to the manufacturer’s instructions with minor modifications. Cells seeded on scaffolds were labeled with BrdU, and then incubated with peroxidase-labeled anti-BrdU antibody. After washing extensively with washing buffer in a Vortex mixer to remove excess anti-BrdU antibody, the scaffolds were incubated with TMB substrate to produce a colored solution. In addition, the TMB substrate reaction was monitored so that the OD450 value remained in the linear range of detection. The same volume of TMB with the same reaction time was used for scaffolds obtained at all collection points. After 200 μL of colored solution was transferred to each well of a 96-well plate, BrdU incorporation was measured by absorbance at 450 nm.
RESULTS
Production, purification, and characterization of PlnDI
For PlnDI production, 293 EBNA clones expressing high amounts of PlnDI were selected and seeded in Nunclon cell factories (Nalge Nunc International, Rochester, NY) under serum-free conditions. Conditioned medium was collected every other day for 10 days, yielding 10 L of medium. Finally, PlnDI (∼0.35 mg/L) was isolated by DEAE-cellulose chromatography and eluted with an NaCl gradient. Purity was assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by Coomassie blue staining and Western blot analyses (Fig. 1). Fully glycosylated PlnDI migrated as a diffuse band, with an Mr ranging from 35,000 to 90,000, that stained poorly with Commassie blue (not shown) and reacted poorly with antibodies directed against the core protein (Fig. 1, lane 1). In contrast, pretreatment with he-parinases I, II, and III, to selectively cleave HS chains yielded a product that migrated at approximately 35,000 with much improved immunoreactivity (Fig. 1, lane 2). Pretreatment with chondroitinase AC, to selectively cleave chondroitin 4- and 6-sulfate chains, yielded two major bands migrating at approximately 30,000 and 22,000 (Fig. 1, lane 3). Pretreatment with both heparinases and chondroitinase AC yielded bands with similar sizes as observed with either enzyme alone (Fig. 1, lane 4). To verify the biological activity of PlnDI in vitro, murine mesenchymal cells (C3H10T1/2 cell line) were plated on tissue culture surfaces coated with PlnDI as previously described.44,45 Within a few hours of plating, these cells began to aggregate and formed dense prechondrogenic nodules as observed previously with intact Pln and PlnDI obtained from other sources (data not shown). Thus, these PlnDI preparations were considered to be appropriately decorated with glycosaminoglycan chains and biologically active.
FIG. 1.
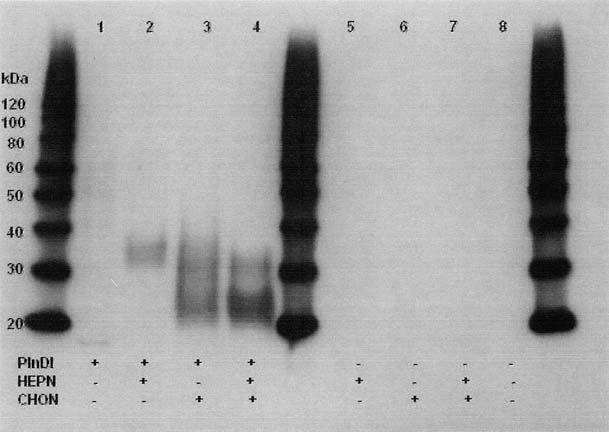
Western blot of PlnDI. Expression of PlnDI in EBNA cells, purification, Western blotting, and enzyme digestions were performed as described in Materials and Methods. Lane 1, undigested PlnDI; lane 2, PlnDI digested with heparinases I, II, and III (HEPN); lane 3, PlnDI digested with chondroitinase AC (CHON); lane 4, PlnDI digested with both HEPN and CHON; lanes 5-8, negative controls in which PlnDI was omitted. Numbers in the margin denote the migration position of protein molecular weight markers (in kDa).
FGF-2 binding to PlnDI
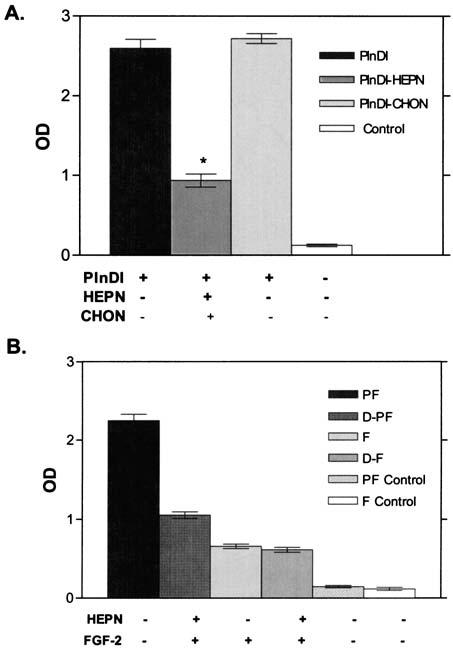
Dot-blot assays were employed to monitor FGF-2 binding to PlnDI. Briefly, PlnDI was immobilized on nitrocellulose in its native form or after digestion with heparinases I, II, and III or chondroitinase AC. Binding of FGF-2 to PlnDI was detected with FGF-2-specific antibodies. Figure 2A and B shows a representative dot blot depicting FGF-2 binding to PlnDI. No binding was observed to areas lacking PlnDI and digested with glycosaminoglycan lyases. In contrast, FGF-2 bound well to intact, chondroitinase AC-digested, but not heparinase-digested, PlnDI. Figure 2C summarizes the densitometric analyses of these data. These results demonstrate that FGF-2 binds well to PlnDI in an HS-dependent manner.
FIG. 2.

Dot-blot analyses of FGF-2 binding to PlnDI. (A) Undigested PlnDI, and PlnDI digested with heparinases I, II, and III (HEPN) or chondroitinase AC (CHON), were blotted in triplicate on nitrocellulose membrane. (B) PBS, HEPN, and CHON only served as negative controls. FGF-2 binding was determined as described in Materials and Methods. (C) Bar graph summarizes densitometric measurements performed on dot blot (A) above. Each bar indicates the mean ± SD (n = 3).
PlnDI binding to collagen I fibrils
Next, PlnDI binding to various collagen I forms was examined. The forms of collagen used were collagen I fibrils and heat-denatured collagen I fibrils. Biotinylated PlnDI was used to monitor binding in these assays. In suspension assays, biotinylated PlnDI bound well to collagen I fibrils (Fig. 3A). Binding was not affected by predigestion of PlnDI with chondroitinase AC (Fig. 3A), but was greatly reduced by predigestion with heparinases I, II, and III. In solid-phase assays, the results were similar; however, wells coated with heat-denatured collagen I fibrils did not bind biotinylated PlnDI (data not shown). Taken together, these findings suggest that PlnDI binding to collagen I fibrils is HS dependent, and that collagen conformation is also important.
FIG. 3.
(A) PlnDI binding to collagen I fibrils. Binding of biotinylated PlnDI was determined as described in Materials and Methods. The bar graph indicates means ± SD of triplicate determinations of PlnDI, PlnDI digested with heparinases I, II, and III (HEPN), and PlnDI digested with chondroitinase AC (CHON) to collagen I fibrils. Collagen I fibrils incubated in PBS without binding interaction with PlnDI served as control. *p ± 0.01 relative to undigested or chondroitinase-digested PlnDI. (B) FGF-2 binding to PlnDI associated with collagen I fibrils. PlnDI and PlnDI digested with heparinases I, II, and III (HEPN) interact with collagen I fibrils to form PF (PlnDI-fibrils) and D-PF (digested PlnDI-fibrils), respectively. These complexes were used as matrices for FGF-2 binding. FGF-2 binding was determined by ELISA as described in Materials and Methods. Each bar indicates the mean ± SD of triplicate determinations.
FGF-2 binding to PlnDI associated with collagen I fibrils
Because PlnDI binding to both FGF-2 and collagen I fibrils was HS dependent, we determined whether FGF-2 could bind to PlnDI when PlnDI was associated with collagen I fibrils. In this experiment, PlnDI, either undigested or predigested with heparinases, was bound to collagen I fibrils. Collagen I fibrils, undigested or digested with heparinases I, II, and III served as controls. After blocking nonspecific protein binding, FGF-2 was incubated with each of these preparations. Bound FGF-2 was detected with an anti-FGF-2 antibody. PlnDI-fibril complexes bound significantly more FGF-2 (2 to 3-fold) than any of the other complexes (Fig. 3B) (p < 0.01). Dose-response studies demonstrated a molar ratio of FGF-2 to PlnDI binding of 1:44 at saturating amounts of FGF-2 (data not shown). Similar to the results obtained for PlnDI binding to collagen I fibrils, heparinase digestion of PlnDI markedly reduced binding. Collagen fibrils alone bound significant amounts of FGF-2, but this did not appear to be HS dependent because heparinase digestion of collagen fibrils had no effect on FGF-2 binding. Collectively, these observations indicated that FGF-2 can bind to PlnDI-collagen I fibril complexes and that this association is greatly improved or stabilized by the presence of HS chains.
Properties and binding capacity of collagen I scaffolds
Scaffolds prepared in these studies display a porous structure as described previously.46 An immunohistochemical analysis was employed to compare the distribution of FGF-2 and PlnDI binding in PF-scaffolds and F-scaffolds. As expected, PlnDI was not detected in F-scaffolds (Fig. 4A). In contrast, PlnDI was distributed throughout PF-scaffolds (Fig. 4B). Staining of PF-scaffolds and F-scaffolds incubated with FGF-2 with FGF-2 antibodies also suggested that PF-scaffolds contained significantly more FGF-2 than did F-scaffolds and that FGF-2 was distributed throughout the scaffold (Fig. 4C and D).
FIG. 4.
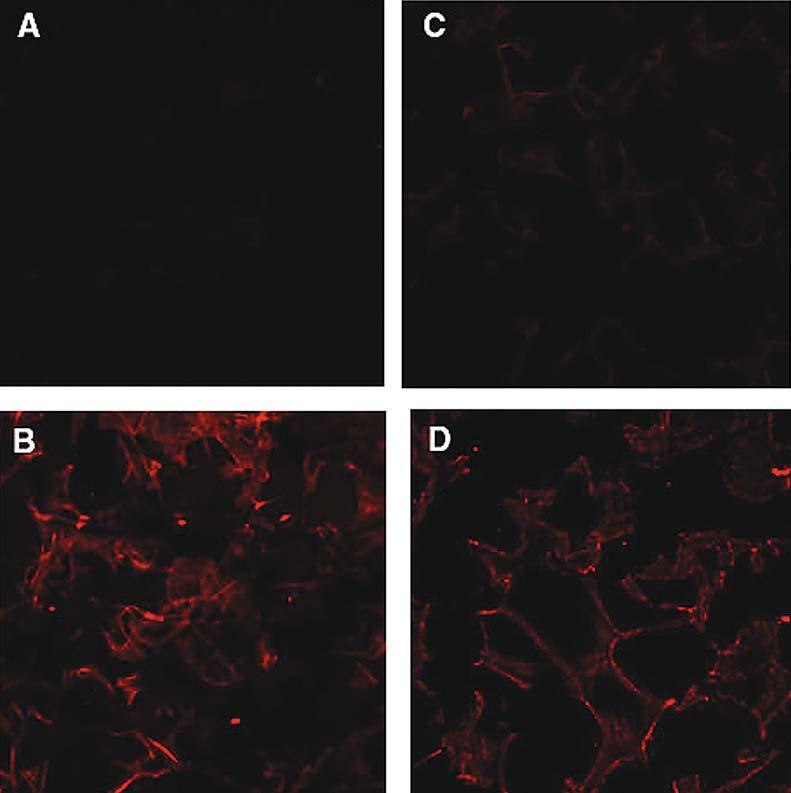
Immunofluorescence staining for PlnDI and FGF-2 located in collagen I scaffolds. PlnDI was detected in scaffolds prepared with collagen I fibrils only (A and C) or PlnDI-collagen I fibril complexes (B and D). Subsequently, these scaffolds were incubated with FGF-2, and binding was detected by immunostaining with anti-Pln antibodies (A and B) or anti-FGF-2 antibodies (C and D) as described in Materials and Methods.
Subsequent experiments employing similarly treated scaffolds were carried out to analyze the FGF-2-binding properties of PF-scaffolds after treatment with heparinases. As expected, untreated PF-scaffolds immobilized more FGF-2 than did F-scaffolds (Fig. 5A) (p < 0.01). Consistent with our previous observations, the binding of FGF-2 to PF-scaffolds after treatment with heparinases was reduced to a level similar to that observed with F-scaffolds (Fig. 5A) (p < 0.01). In contrast, heparinase digestion did not affect FGF-2 binding to F-scaffolds. Taken together, these findings indicate that PlnDI stably associates with UV cross-linked collagen I fibril scaffolds and greatly promotes HS-dependent FGF-2 binding.
FIG. 5.
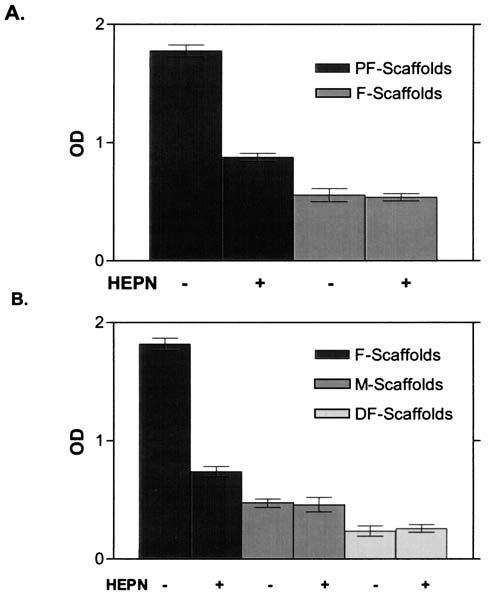
(A) PlnDI HS promote FGF-2 binding to PlnDI-collagen I fibril scaffolds. Collagen I fibril scaffolds were prepared and complexed with (PF) or without (F) PlnDI as described in Materials and Methods. Subsequently, the scaffolds were digested with heparinases as indicated, followed by incubation with FGF-2 as described in Materials and Methods. FGF-2 binding was determined by ELISA as described in Materials and Methods. The bar graphs indicate means ± SD of triplicate determinations in each case. (B) PlnDI binding to various collagen I scaffolds. Binding of biotinylated PlnDI and collagen I scaffolds was determined as described in Materials and Methods. Scaffolds were prepared with natural fibrils of collagen I (F-scaffolds), collagen I monomers (M-scaffolds), or heat-denatured fibrils of collagen I (DF-scaffolds). Subsequently, scaffolds were incubated with biotinylated PlnDI or biotinylated PlnDI predigested with heparinases I, II, and III (HEPN) as indicated. The bar graphs indicate means ± SD of triplicate determinations from a typical experiment.
To determine the effect of different collagen I forms on PlnDI binding, scaffolds were prepared with collagen I monomer, denatured collagen I fibrils, or collagen I fibrils. Significantly more biotinylated PlnDI bound to scaffolds prepared with collagen I fibrils than to scaffolds prepared with denatured collagen I fibrils or collagen I monomer (Fig. 5B) (p < 0.01). Monomer scaffolds bound significantly more biotinylated PlnDI than did DF-scaffolds (Fig. 5B) (p < 0.01). Nonetheless, scaffolds constructed of collagen I fibrils bound three to four times more PlnDI than did monomer scaffolds. Pretreatment of PlnDI with heparinases greatly reduced binding to collagen I fibril scaffolds (p < 0.01), but had no effect on binding to either collagen I monomer or denatured collagen scaffolds. Collectively, these observations demonstrated that PlnDI complexed best with scaffolds composed of collagen I fibrils and that HS was critically important in stabilizing this interaction.
FGF-2 release
In addition to considering the FGF-2-binding capacity of these scaffolds we also examined the stability of FGF-2 interaction. Scaffolds were loaded initially with 10 ng of FGF and release from scaffolds was evaluated in vitro for up to 4 weeks (Fig. 6). An early burst of release was observed with both scaffolds. Within 1 week 41 ± 2% (4.1 ± 0.2 ng) and 73 ± 4% (7.3 ± 0.4 ng) of FGF-2 were released from PF/FGF-scaffolds and F/FGF-scaffolds, respectively (Fig. 6). Nonetheless, the remaining FGF-2 associated with PF/FGF-scaffolds was more stable than F/FGF scaffolds because only 48 ± 2% of FGF-2 was released from PF/FGF scaffolds, whereas 79 ± 3% of FGF-2 was released from F/FGF-scaffolds over the 4-week time course. Thus, even after 4 weeks PF-scaffolds retained approximately 5 ng of FGF-2 whereas F-scaffolds retained approximately 2 ng. It was concluded that PF/FGF-scaffolds would provide a more stable reservoir of FGF-2 than would F/FGF-scaffolds.
FIG. 6.

FGF-2 release from collagen I fibril scaffolds. Scaffolds constructed with collagen I fibrils and complexed with (PF) or without (F) PlnDI were loaded with 10 ng of FGF-2 in 5 μL of PBS to form F/FGF-scaffolds (○) and PF/FGF-scaffolds (•). FGF-loaded scaffolds then were transferred to release medium (DMEM plus penicillin [100 U/mL] and streptomycin [100 μg/mL]), and FGF-2 release was detected at the indicated times, using a sandwich ELISA as described in Materials and Methods. The y axis refers to the percentage of 10 ng of FGF found in the medium at each indicated time (e.g., 20% = 2 ng, 80% = 8 ng, etc.). The individual points reflect means ± SD of triplicate determinations in each case.
To determine whether FGF-2 could be released from PF/FGF-scaffolds via enzymatic cleavage of HS chains, PF/FGF-scaffolds were incubated with and without heparinases and FGF-2 release was determined by sandwich ELISA. As summarized in Fig. 7A, the results suggest that heparinase treatment releases significantly more FGF-2 from PF/FGF-scaffolds relative to PF/FGF-scaffolds not treated (Fig. 7A) (p < 0.01). F-scaffolds bind much lower amounts of FGF-2 and release is not influenced by heparinase treatment (p > 0.05). We concluded that heparinase treatment promoted FGF-2 release, presumably because of FGF-2 binding to HS chains of PlnDI in association with collagen I scaffolds. Thus, degradation of HS chains might provide a means to modulate FGF-2 release from PF/FGF-2 scaffolds in vivo.
FIG. 7.
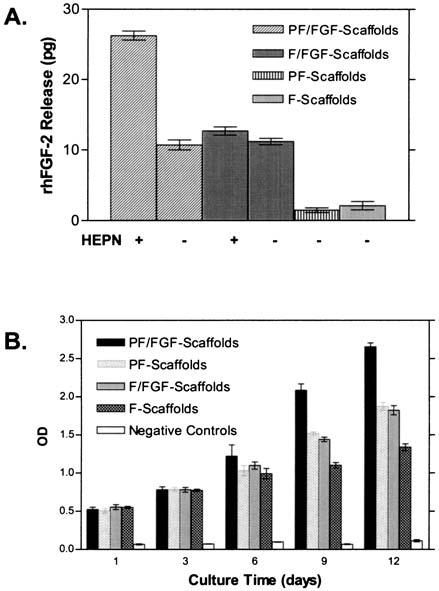
(A) Heparinase release of FGF-2 from collagen I scaffolds. Collagen I fibril scaffolds complexed without (F) or with (PF) PlnDI were constructed and nonspecific protein-binding sites were blocked with BSA as described in Materials and Methods. FGF-2 bound and released was evaluated after treatment with or without heparinases I, II, and III (HEPN) as indicated. PBS and F/FGF-scaffolds were treated with HEPN. PF-scaffolds and F-scaffolds not preincubated with FGF-2 were used as negative controls. FGF-2 released to the medium was detected in a sandwich ELISA as described in Materials and Methods. The bar graphs reflect means ± SD of triplicate determinations in a typical experiment. (B) MG63 cell proliferation in collagen I scaffolds. Scaffolds were formed with collagen I fibrils only (F) or in conjunction with PlnDI (PF) and subsequently complexed without or with FGF-2 (FGF) as described in Materials and Methods. These scaffolds then were seeded with MG63 cell suspensions (2 × 105/mL) in DMEM supplemented with 5% (v/v) FCS. Cell-seeded scaffolds were transferred to 24-well plates and incubated in DMEM containing 1% (v/v) FCS. Collagen I scaffolds without cells were used as negative control. Scaffolds were collected at the indicated times postseeding and cell proliferation was determined by BrdU incorporation assay. The results reflect means ± SD of triplicate determinations in each case.
PlnDI-containing scaffolds enhance rhFGF-2 activity
To determine the effects of rhFGF-2 bound to PlnDI associated with collagen I scaffolds on cell proliferation, the human osteoblastic cell line MG63 and hBMSCs were used. BrdU incorporation was measured as an index of cell proliferation. For MG63 cells, no significant differences were observed through 6 days of culture; however by day 9, significant differences were noted in BrdU incorporation for cells seeded on PF/FGF-scaffolds (Fig. 7B) (p < 0.01). In addition, MG63 cells seeded on PF/FGF-scaffolds displayed the highest level of BrdU incorporation compared with other kinds of scaffolds (PF-scaffolds, F/FGF-scaffolds, F-scaffolds, and negative control) (p < 0.01) on days 9 and 12 (Fig. 7B) whereas the level of BrdU incorporation by MG63 cells seeded on F-scaffolds was lower than on PF/FGF-, PF-, and F/FGF-scaffolds (p < 0.01) on days 9 and 12 (Fig. 7B). In contrast, no significant differences were detected between PF-scaffolds and F/FGF-scaffolds (p > 0.05) at all observation times (Fig. 7B). For hBMSCs, no significant differences in BrdU incorporation were detected among cells seeded on any of the scaffolds (p > 0.05) at day 1 (Fig. 8); however, by day 5, a significant difference was noted between PF/FGF-scaffolds and F/FGF-scaffolds (p < 0.01). The hBMSCs seeded on PF/FGF-scaffolds displayed the greatest BrdU incorporation whereas the BrdU incorporation of hBMSCs seeded on F-scaffolds was modest (Fig. 8). As was the case for MG63 cells, BrdU incorporation by hBMSCs seeded on PF-scaffolds and F/FGF-scaffolds was similar on days 10 and 15 (Fig. 8) (p > 0.01). Therefore, PlnDI incorporation into collagen I scaffolds not only promoted the proliferation of both MG63 cells and hBMSCs in PF-scaffolds, but also enhanced FGF-2 activity.
FIG. 8.
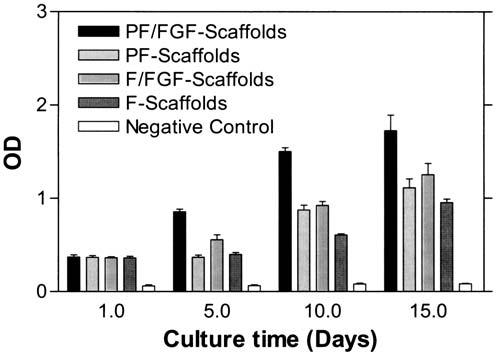
hBMSC proliferation in PlnDI-coated collagen I scaffolds. hBMSCs derived from human bone marrow were expanded as described in Materials and Methods. Collagen I fibril scaffolds were seeded with an hBMSC suspension (2 ×105/mL) in DMEM supplemented with 10% (v/v) FCS. Cell-seeded scaffolds then were cultured as described in Materials and Methods. Cell-free collagen I scaffolds served as a negative control. Scaffolds were collected on the indicated days postseeding and cell proliferation was determined by BrdU incorporation assay as described in Materials and Methods. The results reflect means ± SD of triplicate determinations in each case.
DISCUSSION
Scaffolds used for TE should facilitate cell attachment and proliferation, and ultimately promote differentiated cell functions.4,6,30,47 To improve biological activity of scaffolds, ECM component utilization in TE devices provides one approach that might better promote interactions between scaffolds and cells and potentially improve tissue morphology and regeneration. The HSPG perlecan (Pln) influences biological processes by interacting with a large number of physiologically important molecules. A key Pln function is its ability to interact with growth factors, such as FGF-2, and to regulate their activities through its GAG chains attached on domain I.19,21-23 The large size and complexity of Pln makes production and use of the intact molecule impractical; however, the ability to conveniently prepare domain I (PlnDI) with attached GAG chains coupled with the maintenance of key biological activities in this portion of Pln9,38,44,48 makes this an attractive alternative for use in TE applications.
Recombinant PlnDI expressed in and purified from transfected 293 EBNA cells displayed large size heterogeneity. Digestion with GAG lyases demonstrated that the PlnDI core protein contained both HS and chondroitin sulfate, as seen in other expression systems.38 Pln regulates FGF-2 activity in vitro19 and in vivo19,21 via HS chains on its domain I. Some studies indicate that FGF-2 interacts with HS sequences consisting of a hexasaccharide containing 2-O-sulfated iduronic acid and N-sulfated glucosamine.49 Furthermore, Pln isolated from different cell types differs significantly in its ability to interact with FGF-2, apparently because of the difference in HS sequences.50,51 We used a dot-blot assay to verify that FGF-2 could bind to the recombinant PlnDI used in these studies. FGF-2 binding to PlnDI was both robust and HS dependent.
Previous studies have demonstrated that HS and heparin interact with collagen I fibrils and promote cell attachment27 and angiogenesis.28 HS binding to collagen I relies on an N-terminal basic triple-helical domain and fibrillar configuration of collagen.26 Heat-denatured collagen fibrils fail to interact with HS and heparin.26,28 In the present experiment, biotinylated PlnDI was used to evaluate the binding to collagen I fibrils. The interaction between PlnDI and collagen fibrils was observed both in suspension and solid-phase assays and was HS dependent in both cases; however, denatured collagen I fibrils failed to interact with PlnDI in solid-phase assays (data not shown). Consistent with this observation, we found that PlnDI bound to scaffolds prepared with collagen I fibrils much better than to scaffolds prepared with denatured collagen I fibrils. Scaffolds constructed with collagen I monomers bound significantly more PlnDI than did scaffolds constructed with denatured collagen, but significantly less then scaffolds constructed with collagen I fibril preparations. Thus, it appears that the fibrillar configuration of collagen I and multiple binding sites, that is, increased avidity, for HS within collagen I fibrils both contribute to optimal PlnDI binding and retention.
It was of interest to determine whether PlnDI-collagen I fibril complexes provide improved systems for growth factor binding, retention, and delivery. To test this, we used FGF-2 as a prototypical HS-binding growth factor. The results revealed that significantly more FGF-2 bound to complexes of PlnDI and collagen I fibrils than to collagen I fibrils alone. The markedly reduced FGF-2 binding observed with heparinase-digested PlnDI-collagen I fibril complexes demonstrates that HS is critical for this interaction. Whereas studies examining the HS structural features required for FGF-2 binding are available,52,53 no studies are available for HS binding to collagen I fibrils. Thus, it is not clear if there are distinct binding sites on HS chains for FGF-2 and collagen I fibrils. We observed low, but significant, binding between FGF-2 and collagen I fibrils when compared with negative controls, an interaction that is not reduced by pretreatment with heparinases. The mechanism involved in the interaction between FGF-2 and collagen I fibrils is not clear.
Growth factors and their carriers are needed for engineered tissue and design of TE scaffolds.54 TE scaffolds can be designed to mimic natural ECM in order to regulate local concentrations of growth factors at application sites and to protect them from proteolysis.5,47,54 In our experiment, PF-scaffolds immobilized significantly more FGF-2 than did F-scaffolds. This suggests that PF-scaffolds can be efficient carriers not only for FGF-2 within scaffolds, but also potentially for other heparin-binding growth factors, for example, VEGF, hepatocyte growth factor (HGF), platelet-derived growth factor (PDGF), and heparin-binding epidermal growth factor (HB-EGF).55 In addition, FGF-2 binding to PF-scaffolds was greatly reduced after pretreatment with heparinase. These results indicate that the HS chains of PlnDI in PF-scaffolds are involved in the immobilization of FGF-2.
Controlled release of growth factors from scaffolds has been explored in tissue engineering to improve angiogenesis of implants, cell proliferation, and tissue regeneration.8,37,47,54 For these purposes, isolated GAGs have been immobilized covalently to collagen I scaffolds to sustain FGF-2 release.8,37,47 In the present study, GAG-bearing PlnDI formed complexes with collagen I fibrils by a self-assembly process. This allowed us to avoid the use of chemical reagents to provide a more natural, and potentially more biocompatible, complex. Our results show that PF-scaffolds sustain FGF-2 release more effectively than do F- or H-scaffolds. Prolonged growth factor release may be especially beneficial for application in tissue engineering, such as that of cartilage and bone, which require weeks to months for new tissue formation.
Both extracellular heparanases and proteases potentially could promote FGF release from these scaffolds.2,3,18,54 Heparanase releases FGF-2 from Pln and modulates the bioavailability of FGF-2.16 Thus, it is reasoned that FGF-2 release from PF/FGF-scaffolds would be promoted by endogenous heparanases after scaffolds are implanted in vivo, a process that would be compatible with host wound-healing processes. To test this hypothesis, PF/FGF-scaffolds were treated with heparinases in vitro. This resulted in much reduced FGF-2 retention by these scaffolds. Therefore, it is proposed that FGF-2 release from PF/FGF-scaffolds would be promoted by heparanases expressed by cells penetrating or surrounding these scaffolds in vivo.
FGFs produce their mitogenic and angiogenic effects on target cells by signaling through cell surface tyrosine kinase receptors. In addition, it is now generally recognized that HSPGs play an important role in signaling by FGF family members apparently via interactions with both FGFs and FGF receptors.20-22 Some studies have demonstrated that HS or heparin can enhance the proliferation of certain cells, including endothelial cells37 and osteoblasts.56 MG63 cells are an osteosarcoma cell line that has been used extensively as a model of osteoblast function in the testing of biomaterials.44,57 In addition, hBMSCs derived from bone marrow are attractive cell sources for engineered tissue constructs, such as bone and cartilage, and have broad therapeutic potential.42,43 Therefore, both MG63 cells and hBMSCs were employed to test the biological function of collagen scaffolds coated with PlnDI with or without FGF. PF/FGF-scaffolds enhanced MG63 cell proliferation relative to other scaffolds tested on days 9 and 12 (Fig. 7B). In addition, hBMSC proliferation in PF/FGF-scaffolds was enhanced by days 5, 10, and 15 (Fig. 8). MG63 cell proliferation in PF-scaffolds was comparable to proliferation in F/FGF-scaffolds on days 9 and 12 (p > 0.01; Fig. 7B). Similarly, hBMSC proliferation in PF-scaffolds was not significantly different from the proliferation observed in F/FGF-scaffolds by days 5, 10, and 15 (p > 0.01; Fig. 8). These observations suggest that PlnDI incorporated into PF-scaffolds promotes the activity of heparin-binding growth factors in both an osteoblastic cell line and primary cultures of bone marrow-derived cells. The increase in activity most likely reflects the ability of PlnDI to bind, concentrate, and retain the growth factor at the material surface. When heparin-binding growth factors are produced by cells in the scaffold, PlnDI also might serve to enhance autocrine/paracrine responses by concentrating these molecules at the sites of production.
ACKNOWLEDGMENTS
We greatly appreciate the many helpful discussions with members of the Carson and Farach-Carson laboratories. We thank Ms. JoAnne Julian and Ms. Anissa Brown for helpful technical suggestions and critical reading of this manuscript. In addition, we are grateful to the late Dr. Rupert Timpl for generously providing the PlnDI expression construct. We thank Ms. Sharron Kingston and Mrs. Margie Barrett for excellent secretarial and graphics assistance, respectively. These studies were supported by NIH NRSA grant F32-AG20078-03 (to R.R.G.) and by NIH grant R01 DE13542 (to D.D.C. and M.C.F.-C.).
REFERENCES
- 1.Badylak SF. The extracellular matrix as a scaffold for tissue reconstruction. Semin. Cell Dev. Biol. 2002;13:377. doi: 10.1016/s1084952102000940. [DOI] [PubMed] [Google Scholar]
- 2.Kresse H, Schonherr E. Proteoglycans of the extracellular matrix and growth control. J. Cell. Physiol. 2001;189:266. doi: 10.1002/jcp.10030. [DOI] [PubMed] [Google Scholar]
- 3.Rabenstein DL. Heparin and heparan sulfate: structure and function. Nat. Prod. Rep. 2002;19:312. doi: 10.1039/b100916h. [DOI] [PubMed] [Google Scholar]
- 4.Shin H, Jo S, Mikos AG. Biomimetic materials for tissue engineering. Biomaterials. 2003;24:4353. doi: 10.1016/s0142-9612(03)00339-9. [DOI] [PubMed] [Google Scholar]
- 5.Bottaro DP, Liebmann-Vinson A, Heidaran MA. Molecular signaling in bioengineered tissue microenvironments. Ann. N.Y. Acad. Sci. 2002;961:143. doi: 10.1111/j.1749-6632.2002.tb03068.x. [DOI] [PubMed] [Google Scholar]
- 6.LeBaron RG, Athanasiou KA. Extracellular matrix cell adhesion peptides: Functional applications in orthopedic materials. Tissue Eng. 2000;6:85. doi: 10.1089/107632700320720. [DOI] [PubMed] [Google Scholar]
- 7.Lee CR, Grodzinsky AJ, Spector M. Modulation of the contractile and biosynthetic activity of chondrocytes seeded in collagen-glycosaminoglycan matrices. Tissue Eng. 2003;9:27. doi: 10.1089/107632703762687500. [DOI] [PubMed] [Google Scholar]
- 8.Pieper JS, Hafmans T, van Wachem PB, van Luyn MJ, Brouwer LA, Veerkamp JH, van Kuppevelt TH. Loading of collagen-heparan sulfate matrices with bFGF promotes angiogenesis and tissue generation in rats. J. Biomed. Mater. Res. 2002;62:185. doi: 10.1002/jbm.10267. [DOI] [PubMed] [Google Scholar]
- 9.French MM, Smith SE, Akanbi K, Sanford T, Hecht J, Farach-Carson MC, Carson DD. Expression of the heparan sulfate proteoglycan, perlecan, during mouse embryogenesis and perlecan chondrogenic activity in vitro. J. Cell Biol. 1999;145:1103. doi: 10.1083/jcb.145.5.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aviezer D, Hecht D, Safran M, Eisinger M, David G, Yayon A. Perlecan, basal lamina proteoglycan, promotes basic fibroblast growth factor-receptor binding, mitogenesis, and angiogenesis. Cell. 1994;79:1005. doi: 10.1016/0092-8674(94)90031-0. [DOI] [PubMed] [Google Scholar]
- 11.Iozzo RV. Matrix proteoglycans: From molecular design to cellular function. Annu. Rev. Biochem. 1998;67:609. doi: 10.1146/annurev.biochem.67.1.609. [DOI] [PubMed] [Google Scholar]
- 12.Kosir MA, Quinn CC, Wang W, Tromp G. Matrix glycosaminoglycans in the growth phase of fibroblasts: More of the story in wound healing. J. Surg. Res. 2000;92:45. doi: 10.1006/jsre.2000.5840. [DOI] [PubMed] [Google Scholar]
- 13.Gustafsson E, Aszodi A, Ortega N, Hunziker EB, Denker HW, Werb Z, Fassler R. Role of collagen type II and perlecan in skeletal development. Ann. N.Y. Acad. Sci. 2003;995:140. doi: 10.1111/j.1749-6632.2003.tb03217.x. [DOI] [PubMed] [Google Scholar]
- 14.Noonan DM, Fulle A, Valente P, Cai S, Horigan E, Sasaki M, Yamada Y, Hassell JR. The complete sequence of perlecan, a basement membrane heparan sulfate proteoglycan, reveals extensive similarity with laminin A chain, low density lipoprotein-receptor, and the neural cell adhesion molecule. J. Biol. Chem. 1991;266:22939. [PubMed] [Google Scholar]
- 15.Murdoch AD, Dodge GR, Cohen I, Tuan RS, Iozzo RV. Primary structure of the human heparan sulfate proteoglycan from basement membrane (HSPG2/perlecan): A chimeric molecule with multiple domains homologous to the low density lipoprotein receptor, laminin, neural cell adhesion molecules, and epidermal growth factor. J. Biol. Chem. 1992;267:8544. [PubMed] [Google Scholar]
- 16.Whitelock JM, Murdoch AD, Iozzo RV, Underwood PA. The degradation of human endothelial cell-derived perlecan and release of bound basic fibroblast growth factor by stromelysin, collagenase, plasmin, and heparanases. J. Biol. Chem. 1996;271:10079. doi: 10.1074/jbc.271.17.10079. [DOI] [PubMed] [Google Scholar]
- 17.Saksela O, Moscatelli D, Sommer A, Rifkin DB. Endothelial cell-derived heparan sulfate binds basic fibroblast growth factor and protects it from proteolytic degradation. J. Cell Biol. 1988;107:743. doi: 10.1083/jcb.107.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vlodavsky I, Miao HQ, Medalion B, Danagher P, Ron D. Involvement of heparan sulfate and related molecules in sequestration and growth promoting activity of fibroblast growth factor. Cancer Metastasis Rev. 1996;15:177. doi: 10.1007/BF00437470. [DOI] [PubMed] [Google Scholar]
- 19.Aviezer D, Iozzo RV, Noonan DM, Yayon A. Suppression of autocrine and paracrine functions of basic fibroblast growth factor by stable expression of perlecan antisense cDNA. Mol. Cell. Biol. 1997;17:1938. doi: 10.1128/mcb.17.4.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Loo BM, Salmivirta M. Heparin/heparan sulfate domains in binding and signaling of fibroblast growth factor 8b. J. Biol. Chem. 2002;277:32616. doi: 10.1074/jbc.M204961200. [DOI] [PubMed] [Google Scholar]
- 21.Sharma B, Handler M, Eichstetter I, Whitelock JM, Nugent MA, Iozzo RV. Antisense targeting of perlecan blocks tumor growth and angiogenesis in vivo. J. Clin. Invest. 1998;102:1599. doi: 10.1172/JCI3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vlodavsky I, Miao HQ, Atzmon R, Levi E, Zimmermann J, Bar-Shavit R, Peretz T, Ben-Sasson SA. Control of cell proliferation by heparan sulfate and heparin-binding growth factors. Thromb. Haemost. 1995;74:534. [PubMed] [Google Scholar]
- 23.Allen BL, Filla MS, Rapraeger AC. Role of heparan sulfate as a tissue-specific regulator of FGF-4 and FGF receptor recognition. J. Cell Biol. 2001;155:845. doi: 10.1083/jcb.200106075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kadler KE, Holmes DF, Trotter JA, Chapman JA. Collagen fibril formation. Biochem. J. 1996;316:1. doi: 10.1042/bj3160001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyahara M, Hayashi K, Berger J, Tanzawa K, Njieha FK, Trelstad RL, Prockop DJ. Formation of collagen fibrils by enzymic cleavage of precursors of type I collagen in vitro. J. Biol. Chem. 1984;259:9891. [PubMed] [Google Scholar]
- 26.Koda JE, Bernfield M. Heparan sulfate proteoglycans from mouse mammary epithelial cells: Basal extracellular proteoglycan binds specifically to native type I collagen fibrils. J. Biol. Chem. 1984;259:11763. [PubMed] [Google Scholar]
- 27.Koda JE, Rapraeger A, Bernfield M. Heparan sulfate proteoglycans from mouse mammary epithelial cells: Cell surface proteoglycan as a receptor for interstitial collagens. J. Biol. Chem. 1985;260:8157. [PubMed] [Google Scholar]
- 28.Sweeney SM, Guy CA, Fields GB, San Antonio JD. Defining the domains of type I collagen involved in heparin-binding and endothelial tube formation. Proc. Natl. Acad. Sci. U.S.A. 1998;95:7275. doi: 10.1073/pnas.95.13.7275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.San Antonio JD, Lander AD, Karnovsky MJ, Slayter HS. Mapping the heparin-binding sites on type I collagen monomers and fibrils. J. Cell Biol. 1994;125:1179. doi: 10.1083/jcb.125.5.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mikos AG, McIntire LV, Anderson JM, Babensee JE. Host response to tissue engineered devices. Adv. Drug Deliv. Rev. 1998;33:111. doi: 10.1016/s0169-409x(98)00023-4. [DOI] [PubMed] [Google Scholar]
- 31.Pieper JS, Hafmans T, Veerkamp JH, van Kuppevelt TH. Development of tailor-made collagen-glycosaminoglycan matrices: EDC/NHS crosslinking, and ultrastructural aspects. Biomaterials. 2000;21:581. doi: 10.1016/s0142-9612(99)00222-7. [DOI] [PubMed] [Google Scholar]
- 32.Pieper JS, van Wachem PB, van Luyn MJA, Brouwer LA, Hafmans T, Veerkamp JH, van Kuppevelt TH. Attachment of glycosaminoglycans to collagenous matrices modulates the tissue response in rats. Biomaterials. 2000;21:1689. doi: 10.1016/s0142-9612(00)00052-1. [DOI] [PubMed] [Google Scholar]
- 33.Pieper JS, Oosterhof A, Dijkstra PJ, Veerkamp JH, van Kuppevelt TH. Preparation and characterization of porous crosslinked collagenous matrices containing bioavailable chondroitin sulphate. Biomaterials. 1999;20:847. doi: 10.1016/s0142-9612(98)00240-3. [DOI] [PubMed] [Google Scholar]
- 34.van Susante JLC, Pieper J, Buma P, van Kuppevelt TH, van Beuningen H, van Der Kraan PM, Veerkamp JH, van den Berg WB, Veth RPH. Linkage of chondroitin-sulfate to type I collagen scaffolds stimulates the bioactivity of seeded chondrocytes in vitro. Biomaterials. 2001;22:2359. doi: 10.1016/s0142-9612(00)00423-3. [DOI] [PubMed] [Google Scholar]
- 35.Kang HW, Tabata Y, Ikada Y. Fabrication of porous gelatin scaffolds for tissue engineering. Biomaterials. 1999;20:1339. doi: 10.1016/s0142-9612(99)00036-8. [DOI] [PubMed] [Google Scholar]
- 36.Ibusuki S, Fujii Y, Iwamoto Y, Matsuda T. Tissue-engineered cartilage using an injectable and in situ gelable thermoresponsive gelatin: Fabrication and in vitro performance. Tissue Eng. 2003;9:371. doi: 10.1089/107632703764664846. [DOI] [PubMed] [Google Scholar]
- 37.Wissink MJ, Beernink R, Scharenborg NM, Poot AA, Engbers GH, Beugeling T, van Aken WG, Feijen J. Endothelial cell seeding of (heparinized) collagen matrices: Effects of bFGF pre-loading on proliferation (after low density seeding) and pro-coagulant factors. J. Control. Release. 2000;67:141. doi: 10.1016/s0168-3659(00)00202-9. [DOI] [PubMed] [Google Scholar]
- 38.Costell M, Mann K, Yamada Y, Timpl R. Characterization of recombinant perlecan domain I and its substitution by glycosaminoglycans and oligosaccharides. Eur. J. Biochem. 1997;243:115. doi: 10.1111/j.1432-1033.1997.t01-1-00115.x. [DOI] [PubMed] [Google Scholar]
- 39.Carson DD, Julian J, Jacobs AL. Uterine stromal cell chondroitin sulfate proteoglycans bind to collagen type I and inhibit embryo outgrowth in vitro. Dev. Biol. 1992;149:307. doi: 10.1016/0012-1606(92)90286-p. [DOI] [PubMed] [Google Scholar]
- 40.Ohan MP, Weadock KS, Dunn MG. Synergistic effects of glucose and ultraviolet irradiation on the physical properties of collagen. J. Biomed. Mater. Res. 2002;60:384. doi: 10.1002/jbm.10111. [DOI] [PubMed] [Google Scholar]
- 41.Mizuno S, Glowacki J. Three-dimensional composite of demineralized bone powder and collagen for in vitro analysis of chondroinduction of human dermal fibroblasts. Biomaterials. 1996;17:1819. doi: 10.1016/0142-9612(96)00041-5. [DOI] [PubMed] [Google Scholar]
- 42.Mauney JR, Kaplan DL, Volloch V. Matrix-mediated retention of osteogenic differentiation potential by human adult bone marrow stromal cells during ex vivo expansion. Biomaterials. 2004;25:3233. doi: 10.1016/j.biomaterials.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 43.Gronthos S, Zannettino AC, Hay SJ, Shi S, Graves SE, Kortesidis A, Simmons PJ. Molecular and cellular characterisation of highly purified stromal stem cells derived from human bone marrow. J. Cell Sci. 2003;116:1827. doi: 10.1242/jcs.00369. [DOI] [PubMed] [Google Scholar]
- 44.Fini M, Giavaresi G, Aldini NN, Torricelli P, Botter R, Beruto D, Giardino R. A bone substitute composed of polymethylmethacrylate and α-tricalcium phosphate: Results in terms of osteoblast function and bone tissue formation. Biomaterials. 2002;23:4523. doi: 10.1016/s0142-9612(02)00196-5. [DOI] [PubMed] [Google Scholar]
- 45.Bilbe G, Roberts E, Birch M, Evans DB. PCR phenotyping of cytokines, growth factors and their receptors and bone matrix proteins in human osteoblast-like cell lines. Bone. 1996;19:437. doi: 10.1016/s8756-3282(96)00254-2. [DOI] [PubMed] [Google Scholar]
- 46.Schoof H, Apel J, Heschel I, Rau G. Control of pore structure and size in freeze-dried collagen sponges. J. Biomed. Mater. Res. 2001;58:352. doi: 10.1002/jbm.1028. [DOI] [PubMed] [Google Scholar]
- 47.Ellis DL, Yannas IV. Recent advances in tissue synthesis in vivo by use of collagen-glycosaminoglycan copolymers. Biomaterials. 1996;17:291. doi: 10.1016/0142-9612(96)85567-0. [DOI] [PubMed] [Google Scholar]
- 48.French MM, Gomes RR, Jr., Timpl R, Hook M, Czymmek K, Farach-Carson MC, Carson DD. Chondrogenic activity of the heparan sulfate proteoglycan perlecan maps to the N-terminal domain I. J. Bone Miner. Res. 2002;17:48. doi: 10.1359/jbmr.2002.17.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guimond S, Maccarana M, Olwin BB, Lindahl U, Rapraeger AC. Activating and inhibitory heparin sequences for FGF-2 (basic FGF). Distinct requirements for FGF-1, FGF-2, and FGF-4. J. Biol. Chem. 1993;268:23906. [PubMed] [Google Scholar]
- 50.Knox S, Melrose J, Whitelock J. Electrophoretic, biosensor, and bioactivity analyses of perlecans of different cellular origins. Proteomics. 2001;1:1534. doi: 10.1002/1615-9861(200111)1:12<1534::aid-prot1534>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 51.Knox S, Merry C, Stringer S, Melrose J, Whitelock J. Not all perlecans are created equal: Interactions with fibroblast growth factor (FGF) 2 and FGF receptors. J. Biol. Chem. 2002;277:14657. doi: 10.1074/jbc.M111826200. [DOI] [PubMed] [Google Scholar]
- 52.Ashikari-Hada S, Habuchi H, Itoh N, Reddi AH, Kimata K. Characterization of growth factor-binding structures in heparin/heparan sulfate using an octasaccharide library. J. Biol. Chem. 2004;279:12346. doi: 10.1074/jbc.M313523200. [DOI] [PubMed] [Google Scholar]
- 53.Raman R, Venkataraman G, Ernst S, Sasisekharan V, Sasisekharan R. Structural specificity of heparin binding in the fibroblast growth factor family of proteins. Proc. Natl. Acad. Sci. U.S.A. 2003;100:2357. doi: 10.1073/pnas.0437842100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tabata Y. The importance of drug delivery systems in tissue engineering. Pharm. Sci. Technol. Today. 2000;3:80. doi: 10.1016/s1461-5347(00)00242-x. [DOI] [PubMed] [Google Scholar]
- 55.Norrby K. Mast cells and angiogenesis. APMIS. 2002;110:355. doi: 10.1034/j.1600-0463.2002.100501.x. [DOI] [PubMed] [Google Scholar]
- 56.Blanquaert F, Barritault D, Caruelle JP. Effects of heparan-like polymers associated with growth factors on osteoblast proliferation and phenotype expression. J. Biomed. Mater. Res. 1999;44:63. doi: 10.1002/(sici)1097-4636(199901)44:1<63::aid-jbm7>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 57.Di Palma F, Douet M, Boachon C, Guignandon A, Peyroche S, Forest B, Alexandre C, Chamson A, Rattner A. Physiological strains induce differentiation in human osteoblasts cultured on orthopaedic biomaterial. Biomaterials. 2003;24:3139. doi: 10.1016/s0142-9612(03)00152-2. [DOI] [PubMed] [Google Scholar]