

Abstract
Studies of memory impairment in humans and experimental animals have been fundamental to learning about the organization of memory and its cellular and molecular substrates. When memory impairment occurs, especially after perturbations of the nervous system, the question inevitably arises whether the impairment reflects impaired information storage or impaired accessibility. This topic has been the subject of considerable commentary and experimental work over the years. In this reappraisal, I first consider four broad areas of behavioral study from the 1970s and 1980s that led to a dominant and compelling view of memory impairment as a deficit of information storage. Second, I identify some ambiguities that arise about how the terms “storage” and “retrieval” are applied, especially when the evidence is somewhat indirect and based on a behavioral-psychological level of analysis. I then review neurobiological findings that have been largely overlooked in these discussions. The relevant studies are ones where it has been possible to monitor neurons and synapses in direct relation to behavioral memory, for example, in animals with simple nervous systems and in single cell recordings from behaving monkeys. This work provides a straightforward and illuminating perspective on the question and confirms the view that first emerged from less direct evidence.
Inhibitors of protein synthesis, bilateral hippocampal lesions, electroconvulsive shock (ECS), and a variety of other manipulations impair memory when they are introduced in proximity to initial learning. When one of these manipulations occurs before learning, a subsequent impairment in retention is termed anterograde amnesia. When the manipulation occurs after learning, impaired retention is termed retrograde amnesia. Anterograde and retrograde amnesia have been studied extensively during the past half-century, most commonly in mice, rats, monkeys, and humans. Anterograde amnesia can occur with any degree of severity, depending on the severity of the amnestic treatment. Retrograde amnesia can vary both in severity and in how far it extends into the premorbid time period. Typically, retrograde amnesia is temporally graded, such that recent memories are most affected and remote memories are spared (Squire et al. 2001; Manns et al. 2003; Frankland and Bontempi 2005). These facts are not in serious contention (for specific aspects of retrograde memory impairment that are of current interest, see Bayley et al. 2003; Clark et al. 2005; Moscovitch et al. 2005).
A fundamental question concerns the nature of memory impairment. On the one hand, memory impairment could reflect the fact that changes in the brain that represent information storage have either not occurred or have been degraded, damaged, or otherwise altered. This scenario is usually referred to as a “storage deficit.” The stored information that would ordinarily represent a specific memory is absent or partially absent. On the other hand, memory impairment could reflect a failure of memory retrieval. The changes in the brain that represent information storage are intact, but the information is not available. This scenario is usually referred to as a “retrieval deficit.” Although memory cannot be expressed, it is potentially recoverable under appropriate circumstances.
This issue—storage versus retrieval—has been the target of considerable commentary and experimental work over the years. The most active period of discussion occurred in the 1970s and early 1980s. As will be explained, penetrating experimental evidence from that period made a strong case for a storage interpretation of most memory impairments. As a result, the dominant perspective that emerged from this era was that amnesia reflects a loss of memory from storage. Nevertheless, the issue continues to be discussed, and it has been difficult to decide the matter with certainty.
There are two difficulties. First, there is some ambiguity and imprecision around the terms “storage” and “retrieval” with respect to how they are used to explain memory impairment. Second, the best-known and most-often-cited evidence is, in the end, somewhat indirect and is founded on a behavioral-psychological level of analysis.
But it is difficult to establish the nature of an amnesic condition on the basis of behavioral data alone. Questions about the nature of amnesia are ultimately biological questions, and to answer these questions, one wants to have some biological facts. One wants to know directly whether the synaptic changes that would ordinarily represent acquired information are present in the nervous system or not. In amnesia, what actually happens to the synaptic changes that carry the memory?
What follows is in three parts. First, I review the key findings that emerged from the experimental work of the 1970s and 1980s. Second, I briefly consider some ambiguities that arise about the terms “storage” and “retrieval,” which can complicate the interpretation of experimental work. Third, I review more recent neurobiological findings that have been almost entirely overlooked in discussions of storage, retrieval, and amnesia. These biological findings promise a more direct and potentially definitive account of the nature of memory impairment.
The 1970s and 1980s
During this period, one can identify four areas of work that shaped the discussion: (1) the beneficial effects of partial cues on the performance of amnesic patients; (2) recovery from retrograde amnesia in patients; (3) improved retention of experimental animals when reminder treatments were given after an amnestic treatment, most commonly ECS; and (4) improved retention of experimental animals when an inhibitor of protein synthesis was given before training and a stimulant or other drug was given some time later. This is not an exhaustive list, but it does describe four broad areas of investigation that together determined how amnesia came to be viewed.
Partial cues
A series of experiments with memory-impaired patients in the 1970s demonstrated that patients could sometimes perform normally on tests that used three-letter word stems as cues for previously presented words (Warrington and Weiskrantz 1974, 1978). For example, patients studied words such as MOTEL and BRIDE and then produced these words as frequently as did controls when given MOT and BRI as cues. This finding, that patients had apparently full access to recently studied information, led to the suggestion that amnesia is a retrieval deficit. The difficulty was that the critical importance of task instruction was not initially appreciated. Later, it was shown that only one kind of task instruction results in normal performance by amnesic patients (i.e., “complete this word stem to form the first word that comes to mind”). With conventional memory instructions (i.e., “use this cue to find the previously presented word”), normal subjects outperformed the patients (Graf et al. 1984). The fact that amnesic patients performed normally when the word stems were administered as a word-completion test came to be understood as an early demonstration of the phenomenon of priming. And priming was subsequently recognized as an example of a kind of memory that is intact in amnesia (Tulving and Schacter 1990). Accordingly, a retrieval explanation of these partial-cueing effects was set aside in favor of an explanation in terms of multiple memory systems, many of which operate normally in amnesic patients (for a history of these developments, see Squire 2004). What is impaired in amnesia is the capacity for conscious declarative memory, and an impairment is also observed when partial cues are given with instructions for explicit recall.
A related finding from the same era that appeared initially to suggest a retrieval account of amnesia was that the performance of amnesic patients on tests of factual knowledge sometimes improved dramatically when prompts were provided. For example, the noted patient H.M., who became amnesic in 1953, was markedly impaired at identifying the faces of persons who became famous in the 1950s and 1960s. Yet he improved to near normal levels when given semantic or phonetic prompts for the names (Marslen-Wilson and Teuber 1975). In this case, controls scored almost perfectly with the prompts, so that it was not known how much better controls might have performed if the test had allowed higher scores.
One sometimes hears the perspective that, if amnesia reflects a deficit in storage, prompts and other cues should not be particularly effective for recovering memories. And, if prompting can eliminate or greatly attenuate the amnesia, then amnesia might be better viewed as a retrieval deficit. This view neglects the fact that amnesia seldom presents as an absolute absence of memory ability. Even a patient as severely impaired as H.M. has some capacity for new learning, and H.M. has acquired significant knowledge of people who became famous after the onset of his amnesia (O'Kane et al. 2004). The point is, so long as a little information can be stored, no matter how little, it should always be the case that easier memory tests, more effective cuing, and repeated probing should uncover more information than can be uncovered with difficult tests that are given without prompting. As George Talland wrote about patients with Korsakoff syndrome: “Like other men and women they too are more likely to succeed in recognition than in unaided recall, but in all tests of memory their capacity and reliability are abnormally small” (Talland 1965).
A study of the moderately impaired, diencephalic amnesic patient N.A. illustrates just this point (Squire and Slater 1977; Squire et al. 1989a). N.A. and eight controls first provided as many facts as they could about 25 news events that had occurred between 1970 and 1975 (N.A. became amnesic in 1960). Controls produced an average of 45 facts, but N.A. produced only 10. Each participant was then read on average 109 additional facts about the same 25 news events. N.A. now endorsed 30 of the additional facts, but the controls did much better and endorsed 63 of the additional facts. (In related tests that included incorrect statements about news events, N.A. and his controls had a similar false-alarm rate.) These results provide no basis for supposing that amnesia reflects a retrieval deficit that can be reversed by prompting procedures. Procedures that improve the performance of amnesic patients also improve the performance of normal subjects.
Subsequently, in more formal tests, it was demonstrated that the benefit to amnesic patients of recognition memory procedures over free recall procedures is exactly proportional to the advantage that recognition procedures have over recall procedures for healthy individuals (Haist et al. 1992). Thus, recall and recognition scores are tightly linked manifestations of declarative memory. In the case of studies involving cues and prompts, providing amnesic patients with additional information appears to improve memory performance about as much as would be expected, given the benefit that these procedures afford to persons with intact memory abilities. There are no reports of a procedure that can fully reinstate the memory performance of amnesic patients to normal levels. The straightforward interpretation is that in amnesia an abnormally small amount of information is in storage, and that what is stored benefits from cues and prompts in the same way that normal human memory benefits from these procedures.
Recovery from retrograde amnesia
In their classic study of traumatic amnesia following closed head injury, Russell and Nathan (1946) reported that memory loss for events from before the injury occurred with high frequency (839 of the 873 cases that had some degree of post-traumatic amnesia). Retrograde amnesia was often very long initially, but the amnesia then shrank until it reached its final duration. Although the final duration of retrograde amnesia was typically brief, there was almost always a limit to the recovery. For example, permanent retrograde amnesia was observed in 77% of 200 cases, ranging from 1–30 min. It was noted that the distant memories returned first and the most recent memories returned last.
Subsequent to these classic clinical observations, the phenomenon of shrinking retrograde amnesia has been documented with formal tests in a number of settings. In one study, psychiatric patients receiving bilateral electroconvulsive therapy (ECT) for depressive illness initially exhibited temporally graded retrograde amnesia for public events that covered 1–3 yr. The amnesia had largely subsided by 1–2 wk after the completion of treatment (Squire et al. 1975). Recovery from retrograde amnesia for personal, autobiographical memories was also documented after ECT (Squire et al. 1981) and also following an episode of transient global amnesia (TGA) (Kritchevsky and Squire 1989). In both instances, retrograde amnesia initially covered many years, but significant recovery occurred during the subsequent weeks and months. Importantly, as in the case of amnesia from closed head injury, recovery was not complete. Following both ECT and TGA, some permanent retrograde amnesia remained for the immediately preceding hours and days.
The facts of shrinking retrograde amnesia indicate directly that many of the memories that were initially lost were not erased but were temporarily inaccessible. One could take this recovery as indication that the amnesia can be fully explained as a retrieval deficit, but an emphasis on retrieval does not tell a complete story. Note that memories were affected in relation to their age at the time of treatment. Specifically, when the disruptive condition (head trauma, ECT, or TGA) occurred long after learning, memory was not affected. When the disruption occurred at an intermediate time, memory was reversibly impaired. When the disruption occurred shortly after learning, memory was permanently lost. Thus, retrograde amnesia revealed differences among memories of different ages that must be related to their neural organization and to their status in storage at different times after learning.
These regularities were part of the inspiration for modern formulations of the historic concept of memory consolidation (McClelland et al. 1995; Squire and Alvarez 1995; McGaugh 2000). According to these formulations, medial temporal lobe structures are needed for the formation and maintenance of memory during a lengthy period of reorganization. After reorganization is complete, these structures are not needed. This process has been illuminated by recent studies with immediate early genes, which demonstrate greater activation of widespread cortical areas after remote, compared with recent, memory tests (Frankland et al. 2004; Maviel et al. 2004). These findings are consistent with an increasingly important role for neocortex as time passes after learning, and they suggest that memories are not fully stored and consolidated into neocortex at times after learning when retrograde amnesia can still occur.
The facts of shrinking retrograde amnesia show that if a transient disruption of medial temporal lobe function occurs before the process of reorganization and consolidation is complete, then consolidation can sometimes continue after the disruption has subsided. And as the disruption subsides, recovery from amnesia can occur. From a biological perspective, the terms “storage” and “retrieval” are not particularly apt for describing recovery from retrograde amnesia. If those terms are to be used, one would better describe the deficit as a transient loss of the mechanism that supports both storage and retrieval. When it becomes possible to assess directly the neural substrates of memory storage during this period of transient loss, one should expect to find that essential aspects of the memory store itself are absent or incomplete and that the memory is therefore not retrievable.
Reminder treatments
During the 1970s, a major point of discussion concerned the basis of amnesia in experimental animals (Miller and Springer 1973; Gold and King 1974). The most common paradigm for studying amnesia in that period involved rats trained in an inhibitory avoidance task and then made amnesic by ECS. Thus rats were punished by footshock for entering a dark compartment or for drinking water. They were then given ECS, and amnesia was later evident as a tendency to enter the dark compartment or to drink water more readily than were animals that were also punished but not given ECS. No finding attracted more interest than reports that certain “reminder” treatments given after ECS (e.g., a re-exposure to the training apparatus or a “noncontingent” footshock given in a different apparatus) could substantially improve the retention performance of an otherwise amnesic animal.
As Miller and Springer (1973) wrote: “… these recovery of memory studies indicate that at least some instances of experimental amnesia are cases of retrieval failure …” “Experimental amnesia is viewed as a failure to retrieve information from long-term memory rather than a failure of the information to be consolidated in long-term memory.” Yet attenuation of amnesia by reminder treatments does not contradict a storage view of amnesia (Cherkin 1972; Gold et al. 1973; Haycock et al. 1973). An amnestic treatment need not have an all-or-none effect, and a reminder treatment might summate with a weak memory to raise it above threshold for behavioral expression. Indeed, only rats with partial amnesia benefited from the reminder treatment.
The key experiment from this period involved rats that were only weakly trained, as the result of having received very mild footshock, and that did not receive ECS (Fig. 1). When these rats were given a noncontingent footshock in a novel apparatus, their retention scores improved. These animals were not amnesic. As the result of having received only a mild footshock, they exhibited weak memory that was comparable to the weak memory of rats given strong training plus ECS. Yet these weakly trained animals, similar to the animals with ECS-produced amnesia, improved their performance scores following the noncontingent footshock. As Gold et al. (1973) wrote: “… reminder treatments do not release a [well-stored] memory from an ECS-produced retrieval block. Instead, noncontingent ‘reminder’ treatments provide a learning experience that adds to the performance of animals which would otherwise have poor retention…. Such treatments are effective whether animals have partial retrograde amnesia for the training, or are simply poorly trained”.
Figure 1.

Rats were given inhibitory avoidance training and tested for retention 24 h (T1) and 48 h (T2) after training. Long latencies to respond at retention signify good memory of the training, and short latencies signify poor memory. Rats given strong footshock (strong FS) remembered well at both retention tests, and rats given electroconvulsive shock immediately after strong footshock (strong FS-ECS) were amnesic. A non-contingent footshock given in a different apparatus 1 h after T1 (FSECS+NCFS) markedly improved retention at T2. Importantly, other animals performed about as poorly as the amnesic animals at T1 and T2 by virtue of receiving only a weak footshock at training (weak FS). These animals also markedly improved their retention score at T2 when a non-contingent footshock was given after T1 (weak FS-NCFS). (Adapted from Gold et al. 1973).
While there were many reports of recovery from, or attenuation of, experimental amnesia during this period, none of them provided compelling support for a retrieval view of amnesia. If amnesia does reflect a block of retrieval, then reports of recovery should include instances where memory recovered to full (normal) strength and then decayed at a normal rate across time. Yet one does not find such reports. Taken together, the literature on reminding effects can be understood as effects that summate with a weak memory to improve performance.
Inhibitors of protein synthesis followed by drug treatment
Another important focus of discussion in the 1970s followed on the discovery that inhibitors of brain protein synthesis impaired retention of long-term memory when the inhibitors were introduced during or shortly after training. The literature from this period has been thoroughly reviewed (Davis and Squire 1984). The discussion concerned whether impaired long-term retention in this circumstance should be viewed as a failure to form long-term memory or as a failure of memory retrieval. This issue arose because some studies found that stimulants, hormones, or other drugs that modulate catecholamine metabolism could attenuate the memory impairment when the drugs were given after training (for review, see Quartermain 1976).
The issue was considerably clarified by a study of the effects of amphetamine (Barondes and Cohen 1968). This study built on the basic phenomenon that mice given a protein synthesis inhibitor (cycloheximide) before training exhibited good memory up to 3 h after training but poor memory at 6 h and 7 d after training. (Cycloheximide inhibits ∼95% of brain protein synthesis for 1–2 h.) Thus, in the presence of cycloheximide, a short-term process that is independent of protein synthesis can support retention for at least 3 h after training. The key finding was that the amnesia ordinarily observed at 7 d after training could be largely prevented by giving amphetamine before the short-term memory process had faded (i.e., at 3 h after training but not at 6 h or 7 d). In addition, re-establishment of protein synthesis inhibition at 2.5 h after training blocked this attenuating effect of amphetamine. It was also found that the attenuating effects of caffeine and nicotine were blocked by extending the duration of protein synthesis inhibition (Flood et al. 1978). Together, these findings indicate that if short-term memory is available, along with a capacity for protein synthesis, then drugs such as amphetamine can establish long-term memory, perhaps by creating a state of arousal. Accordingly, when drugs given shortly after training are found to attenuate the amnesia associated with protein synthesis inhibition, the most likely explanation is that the drugs promote the formation of long-term memory by the usual protein synthesis-dependent mechanism. There is no need to suppose that memories are formed in the presence of inhibitors but cannot be retrieved.
Other studies found that drugs could attenuate the amnesia associated with protein synthesis inhibition, even when the drugs were given long after training, just before the retention test (Quartermain 1976). Because short-term memory is not available after such long intervals, such a finding might seem at first glance to support a retrieval view of amnesia. However, an alternative interpretation is that the drugs facilitated performance during the retention test, independently of the amnesic effects of protein synthesis inhibition. A study of the effects of cycloheximide and amphetamine on memory in mice illustrates this point (Squire 1979). In this study, mice given cycloheximide before object discrimination training were markedly impaired on a retention test 24 h later. Giving amphetamine 1 h before the retention test improved the performance scores of mice given cycloheximide but improved the scores of control mice even more. In addition, mice were given a reversal test 24 h after training (i.e., the object that was originally incorrect was now correct) (Fig. 2). Here, as expected, control mice performed poorly on the reversal test because of their memory for the competing habit. Further, mice given cycloheximide before training were amnesic and did not remember the competing habit. As a result, these mice performed better on reversal testing than did control mice.
Figure 2.

Mice were trained for 20 trials on an object discrimination task and were then retested 24 h later by giving 20 trials of reversal training (the object that was previously incorrect was now correct). During initial training, normal animals achieved a score of 7.6 trials correct. Reversal performance was easier for mice given cycloheximide before training than for control mice, because the mice given cycloheximide were amnesic for the original task. Mice given cycloheximide before training and amphetamine before the reversal test performed even better. If memory of the original discrimination had recovered, it would have interfered with reversal performance. Thus, amphetamine did not recover memory but facilitated reversal performance independently of the effects of cycloheximide. The drugs indicated above the bars were given 30 min before training. The drugs indicated below the bars were given 1 h before retest. SAL indicates saline; CXM, cycloheximide; dAMP, d-amphetamine. Brackets show SEM. (Adapted from Squire 1979).
The key group were mice given cycloheximide before training and amphetamine before the reversal test. If amphetamine acts to improve the retrieval of previously acquired information, then memory of the original discrimination should be revived and should interfere with performance during reversal testing. Yet, the finding was that amphetamine facilitated reversal performance. Indeed, mice given cycloheximide before training and then amphetamine before reversal testing performed significantly better than did any other group. These results show that amphetamine did not improve retrieval. Amphetamine simply facilitated performance, both on tests that assessed retention of original learning and on tests of the ability to reverse what had been learned. Accordingly, findings that drugs given just before a retention test can sometimes attenuate the amnesic effects of protein synthesis inhibition are best understood as facilitatory effects, which are superimposed on amnesic effects. In this case, the behavior observed on a retention test is a joint product of effects that can act independently to either attenuate or facilitate performance.
Ambiguities
From the beginning, discussions about storage and retrieval have been beset by certain ambiguities and fuzziness about the terms (Squire 1980). There are a number of difficulties. It is one thing to construct behavioral studies to determine whether information persists in storage but cannot be retrieved, or whether what has been learned has in fact been abolished. It is another thing to know exactly what has happened in the nervous system to the ensemble of synaptic changes that together represent stored information. Does a storage view of amnesia require that the synaptic changes that originally represented acquired information are no longer present in the nervous system? Can a storage view also accommodate a situation where synaptic changes remain but they no longer belong to a coherent working ensemble, because important connections between neuronal elements have been lost? Does a retrieval view mean that all the synaptic changes that originally represented acquired information are present, and that successful retrieval is only a matter of encountering the appropriate cues? Can a retrieval view also accommodate a situation where some of the synaptic changes have been lost (and the stored information is degraded), but retrieval can still succeed, although only when extra cues are given and more time is allowed? How does one view a circumstance where so much noise (e.g., new synaptic weight changes) has been added to the system that the original representation is no longer viable? The information-carrying pattern of acquired synaptic weights is now embedded within a network of meaningless code. Does failure to remember in this instance reflect a storage failure or a retrieval failure?
The point is that one can imagine scenarios where there will be some uncertainty about how to apply the terms “storage” and “retrieval.” Nevertheless, it can only help to learn more about what actually happens in the nervous system when memory impairment occurs. History shows that as biological information about mechanism becomes available, explanation becomes more concrete and terminology becomes less critical. With additional biological information, perhaps the matter could be resolved. The next section considers recent neurobiological findings that have not previously been part of discussions about storage and retrieval. This work provides a fresh and illuminating perspective on our question.
Neurobiological evidence
The instructive case of forgetting
There has been long debate in psychological science about what actually happens during forgetting. Does forgetting involve the true loss of what was originally stored (like the melting of an ice cube or the loss of branches from a tree)? Or does forgetting reflect mainly a loss of accessibility such that the information is there but for the moment is not available. This matter has been difficult to settle on the basis of behavioral experiments alone. One wants to know some biological facts, for example, whether the cellular and synaptic modifications that record memory do or do not weaken or disappear with time.
Concrete evidence to answer this question requires obtaining direct access to the neurons and synapses involved in behavioral memory. Not surprisingly, these requirements have been difficult to satisfy except in animals with relatively simple nervous systems. But in these more favorable conditions, pertinent information has been obtained. The marine mollusk Aplysia californica exhibits long-term sensitization of its gill- and siphon-withdrawal reflex (Pinsker et al. 1973). Long-term sensitization is induced by 4 d of spaced training, is retained at a high level for 1 wk, and then weakens but is still present after 3 wk. The neuronal circuit for the withdrawal reflex is known, and long-term sensitization involves synaptic changes within the circuit, including enhanced efficacy at the synapses made by sensory neurons onto identified interneurons and motorneurons. To determine what occurs during behavioral forgetting, the morphology of sensory neuron synapses within the reflex circuit was examined in animals at different times after training and compared with the time course of behavioral memory (Bailey and Chen 1989).
A number of structural changes were identified at the pre-synaptic varicosities (boutons) of the sensory neurons, some of which were transient and some of which paralleled the time course of behavioral forgetting. Initially, 1–2 d after the completion of long-term training, there was an increase in the length of active zones and an increase in the vesicle complement at the active zones (focal regions of membrane specialization at boutons that mediate transmitter release). These changes regressed and were absent after 1 wk. In contrast, there were also increases in the number of varicosities per sensory neuron and in the number of active zones per varicosity. The increase in the number of varicosities and in the number of active zones persisted unchanged for at least 1 wk, and the increase in active zone number was only partly reversed after 3 wk. These morphological changes describe sensory neurons within the reflex circuit that grew more extended processes as the result of training and made more synaptic connections on interneurons and on the motorneurons that execute the reflex. As time passed, these newly formed branches gradually regressed in parallel with behavioral forgetting, and the number of synaptic contacts made by sensory neurons returned toward baseline. Thus, the reflex circuit itself changed, becoming stronger when sensitization occurred and weaker with forgetting. In Aplysia, forgetting is a matter of loss of information from storage. Because original learning was strong, a substantial record of the learning remained, even after 3 wk. Yet, forgetting occurred continuously, gradually weakening what had been learned.
Storage and retrieval: Neurobiological findings from invertebrates
The preceding section shows that in Aplysia, direct and concrete biological evidence is available about the basis of natural forgetting. Can studies of this simple nervous system also illuminate the nature of memory impairment? Does impaired memory reflect a loss of information from storage or a failure of accessibility? There is work that bears on this question. In dissociated cell cultures, synaptic connections between the sensory and motor neuron undergo long-term (24-h) facilitation in response to serotonin, a modulatory neurotransmitter normally released by sensitizing stimuli during behavioral testing (Montarolo et al. 1986). This facilitation is a model of long-term behavioral sensitization in the intact animal. In culture, as well as in the semi-intact animal, these functional changes are blocked by inhibitors of transcription or translation (actinomycin D or anisomycin), when the inhibitors are applied during the acquisition phase (Montarolo et al. 1986; Castellucci et al. 1989). These findings parallel the behavioral findings with inhibitors in vertebrates and show that the results in vertebrates do not depend on complex circuitry but arise from intrinsic characteristics of single nerve cells and their synaptic connections. Thus, in Aplysia, the inhibitors block the physiological change that supports the cellular analog of long-term behavioral memory, and this impairment is an impairment in information storage.
Importantly, transcription and translation are also required for the structural changes that accompany long-term facilitation (Bailey et al. 1992). Ordinarily, applications of serotonin to sensory neuron–motor neuron synapses enhance the excitatory, post-synaptic potential (EPSP) and also increase the number of synapses made by sensory neurons onto motor neurons. In the presence of inhibitors of protein (anisomycin) or RNA (actinomycin D) synthesis, this increase in synaptic number is blocked, along with the increase in synaptic strength (Fig. 3). These are the same drugs that block the formation of long-term behavioral memory in mammals (Davis and Squire 1984). The work in Aplysia provides strong and direct evidence that translational and transcriptional inhibitors block memory because they prevent the synaptic modifications that would ordinarily carry the memory. The biological substrate of information storage does not develop. In Aplysia, questions about the nature of memory impairment can be brought to biology and posed directly. Here, the answers are straightforward.
Figure 3.
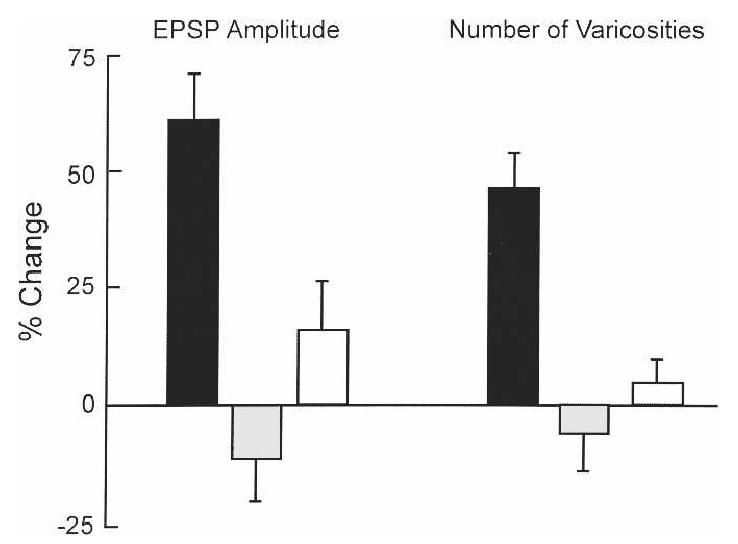
The physiological and structural changes associated with long-term behavioral modification in Aplysia were established in cultures of sensory neurons and the gill motor neuron L7. These neurons are components of the reflex circuit for long-term sensitization. Applications of serotonin enhanced the physiological efficacy of the sensorimotor synapse 24 h after treatment and increased the number of sensory neuron varicosities making contact with neuron L7 (black bars). Varicosities are structural features on neurites that identify synaptic contacts. Addition of inhibitors of protein synthesis (gray bars) or RNA synthesis (white bars) 1 h before application of serotonin blocked both the physiological and the structural changes. For the physiological change, the bars show the change in the amplitude of the excitatory post-synaptic potential (EPSPs) in neuron L7 24 h after serotonin. Brackets show SEM. (Adapted with permission from Cell Press © 1992, Bailey et al. 1992.)
Storage and retrieval: Neurobiological findings from mammals
Is there biological evidence in mammals that is as relevant as the data from invertebrates? The following passage describes an early view of the matter.
“If the hippocampus is unable to participate in establishing long-term memory, then representations that had been established in short-term memory might literally be lost, or they might achieve an unorganized state. In the case of TGA (amnesia associated with ECT) the events that occur during the period of anterograde amnesia are not subsequently remembered after recovery from amnesia. New learning again becomes possible, but events from the amnesic episode do not return to memory. One would therefore think that in the absence of the hippocampus, representations in memory must achieve some abnormal fate. Memories do not become retrievable at a later time” (Squire et al. 1989b).
Recent work in monkeys raises the possibility of approaching the matter more directly. Here, the objective was to study long-term, medial temporal lobe-dependent memory in neocortex by recording from neurons that are part of learned ensembles (Higuchi and Miyashita 1996). In this situation, one can ask what happens to these neurons when the monkeys become amnesic as the result of medial temporal lobe damage. Two monkeys were first given transections of the anterior commissure to disconnect the anterior temporal cortex in one hemisphere from the other. Then the monkeys were trained to learn 12 different pairs of colored patterns. The 24 patterns were arbitrarily arranged into 12 pairs, and the monkeys gradually learned the patterns in each pair, that is 1 and 1′, 2 and 2′, … 12 and 12′. Specifically, the task involved presenting one of the 24 patterns as a cue for 1 sec (e.g., 2 or 11′), and then 4 sec later presenting its associate (2′ or 11) together with one of the other patterns. Monkeys could obtain a reward by touching the correct paired associate.
After training, recordings from neurons in anterior inferotemporal cortex indicated that neurons had acquired the associations (Fig. 4A). That is, neurons that fired to one pattern in a pair tended also to fire to that pattern's associate, and overall, the two patterns in each pair elicited correlated neuronal responses. Unilateral lesions of perirhinal and entorhinal cortex were then introduced, and recordings of inferotemporal neurons were made on the same side as the lesion. (Because of the sectioned commissures, the intact side of the brain could support behavior, and the effect of the lesion on neuronal responses could be explored on the lesioned side, independently of behavioral performance.) Critically, the lesion did not affect the spontaneous discharge rate of the neurons or their maximum discharge rate, and the neurons continued to respond selectively to the visual stimuli. However, the correlated responsiveness of the neurons that was present before the lesion was lost after the lesion, and neurons no longer exhibited a tendency to fire to the two members of each pair (Fig. 4B,C). Similar results were obtained when the monkeys learned 12 new pairs of colored patterns after the lesion (Higuchi and Miyashita 1996). That is, neurons in inferotemporal cortex did not develop correlated responses to the two members of each stimulus pair.
Figure 4.

Neuronal activity in neocortex and long-term declarative memory. Monkeys learned 12 pairs of visual patterns (1 and 1′, 2 and 2′, and so on). (A) The gray bars show the response of a neuron in inferotemporal cortex to each of the 12 cue patterns, and the black bars show the firing rate to each pattern's learned associate. This neuron fired to both patterns 5 and 5′. Because the pairings were arbitrary, this result shows that the neuron has “learned” the pairings. (B) After lesions of perirhinal and entorhinal cortex, neurons still exhibited selective responding to the patterns (this neuron fired to patterns 7 and 9), but the neurons no longer coded pair associations. (C) The correlation coefficient shows the relationship between a neuron's response to each pattern and its paired associate. Neurons were recorded either before the lesion (white bar, n = 92), after the lesion using the same patterns (gray bar, n = 72), or after the lesion with a new set of patterns (black bar, n = 75). Brackets show SEM. (A) (Adapted with permission from Nature © 1991, Sakai and Miyashita 1991; (B,C) adapted with permission from National Academy of Sciences, U.S.A. © 1996, Higuchi and Miyashita 1996).
In related studies (Messinger et al. 2001, 2005), neurons in inferotemporal cortex were recorded while the paired stimuli were being learned. Changes in neuronal responsiveness to paired stimuli proceeded with a time course that paralleled behavioral learning. Because similar responses to paired visual stimuli within a single neuron have not been found in large numbers outside of the temporal lobe, it seems reasonable to suppose that these pair-coding neuronal responses originate in, and reside in, the inferotemporal cortex. Accordingly, the pair-coding neurons sampled from the inferotemporal cortex are likely to be part of long-term memory representations. These studies show directly that, when medial temporal lobe structures are damaged, the physiological signature of long-term synaptic modification in the adjacent inferotemporal cortex is abolished. The medial temporal lobe cortex is needed to maintain recently acquired representations in memory, and these representations are lost when the medial temporal lobe is damaged. Further, the representations are not acquired at all if the training occurs after the damage.
This work provides the best evidence to date in mammals regarding the fundamental nature of memory impairment. Visual paired-associate memory in monkeys is known to depend on medial temporal lobe cortex (Murray et al. 1993). If one accepts the likelihood that the pair-coding neuronal responses are in fact a part of long-term representations in declarative memory, then the loss of paired associate memory after medial temporal lobe damage reflects the literal loss of information from storage, and the failure to acquire new paired-associate memory reflects a failure to place new information into storage.
Conclusions
Beginning in the 1970s, many behavioral studies addressed the fundamental question as to whether impaired memory typically reflects impaired information storage or impaired accessibility. The findings of these experiments were supportive of the view that amnesia reflects a storage deficit. Yet the experiments were necessarily indirect, and their interpretation was not unanimous. In addition, what one really wanted to know—in memory impairment, what actually happens to the synaptic changes that record memory—could not be addressed. Beginning in the late 1980s, it became possible to study neurons and synapses in direct relation to behavioral memory, initially in animals with simple nervous systems and later in single neuron recordings from behaving monkeys. These studies have provided direct, concrete evidence for impairment of information storage. The findings, of course, do not mean that every instance of memory failure is a failure of storage. Indeed, many phenomena of normal human memory appear to reflect a transient loss of accessibility, not a loss of stored information. In this group, one could include retrieval-induced forgetting (Anderson et al. 1994; MacLeod and Macrae 2001), state-dependent effects, and other cases where context has a significant influence on the ability to remember. In addition, the non-neurological condition of functional amnesia reflects a loss of accessibility, inasmuch as patients sometimes fully recover (Kritchevsky et al. 2004). Further, some instances of what have been termed “reconsolidation” effects also appear to reflect transient loss of access (Lattal and Abel 2004; Power et al. 2006). But in circumstances where perturbations of the nervous system close to the time of learning cause anterograde or retrograde memory impairment (e.g., ECS, hippocampal lesions, inhibitors of protein and RNA synthesis), the evidence is in and it all tells the same story.
Acknowledgments
I thank Craig Bailey, Robert Clark, Jeffrey Gold, Hilde Lechner, and John Wixted for their helpful comments. Work was supported by the Medical Research Service of the Department of Veterans Affairs, NIMH grant 24600, and the Metropolitan Life Foundation.
References
- Anderson MC, Bjork RA, Bjork EL. Remembering can cause forgetting: Retrieval dynamics in long-term memory. J. Exp. Psychol. Learn. Mem. Cogn. 1994;20:1063–1087. doi: 10.1037//0278-7393.20.5.1063. [DOI] [PubMed] [Google Scholar]
- Bailey CH, Chen M. Time course of structural changes at identified sensory neuron synapses during long-term sensitization in Aplysia. J. Neurosci. 1989;9:1774–1780. doi: 10.1523/JNEUROSCI.09-05-01774.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey CH, Montarolo P, Chen M, Kandel ER, Schacher S. Inhibitors of protein and RNA synthesis block structural changes that accompany long-term heterosynaptic plasticity in Aplysia. Neuron. 1992;9:749–758. doi: 10.1016/0896-6273(92)90037-e. [DOI] [PubMed] [Google Scholar]
- Barondes SH, Cohen HD. Arousal and the conversion of “short-term” to “long-term” memory. Proc. Natl. Acad. Sci. 1968;61:923–929. doi: 10.1073/pnas.61.3.923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayley PJ, Hopkins RO, Squire LR. Successful recollection of remote autobiographical memories by amnesic patients with medial temporal lobe lesions. Neuron. 2003;37:135–144. doi: 10.1016/s0896-6273(03)00156-9. [DOI] [PubMed] [Google Scholar]
- Castellucci VF, Blumenfeld H, Goelet P, Kandel ER. Inhibitor of protein synthesis blocks long-term behavioral sensitization in the isolated gill-withdrawal reflex of Aplysia. J. Neurobiol. 1989;20:1–9. doi: 10.1002/neu.480200102. [DOI] [PubMed] [Google Scholar]
- Cherkin A. Retrograde amnesia in the chick: Resistance to the reminder effect. Physiol. Behav. 1972;8:949–955. [Google Scholar]
- Clark RE, Broadbent NJ, Squire LR. The hippocampus and remote spatial memory in rats. Hippocampus. 2005;15:260–272. doi: 10.1002/hipo.20056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis HP, Squire LR. Protein synthesis and memory: A review. Psychol. Bull. 1984;96:518–559. [PubMed] [Google Scholar]
- Flood JF, Bennett EL, Orme AE, Rosenzweig MR, Jarvik ME. Memory: Modification of anisomycin-induced amnesia by stimulants and depressants. Science. 1978;199:324–326. doi: 10.1126/science.619461. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Bontempi B. The organization of recent and remote memories. Nat. Rev. Neurosci. 2005;6:119–130. doi: 10.1038/nrn1607. [DOI] [PubMed] [Google Scholar]
- Frankland PW, Bontempi B, Talton LE, Kaczmarek L, Silva AJ. The involvement of the anterior cingulate cortex in remote contextual fear memory. Science. 2004;304:881–883. doi: 10.1126/science.1094804. [DOI] [PubMed] [Google Scholar]
- Gold PE, King RA. Retrograde amnesia: Storage failure versus retrieval failure. Psychol. Rev. 1974;81:465–469. doi: 10.1037/h0036949. [DOI] [PubMed] [Google Scholar]
- Gold PE, Haycock JW, Macri J, McGaugh JL. Retrograde amnesia and the “reminder effect”: An alternative interpretation. Science. 1973;180:1199–1201. doi: 10.1126/science.180.4091.1199. [DOI] [PubMed] [Google Scholar]
- Graf P, Squire LR, Mandler G. The information that amnesic patients do not forget. J. Exp. Psychol. Learn. Mem. Cogn. 1984;10:164–178. doi: 10.1037//0278-7393.10.1.164. [DOI] [PubMed] [Google Scholar]
- Haist F, Shimamura AP, Squire LR. On the relationship between recall and recognition memory. J. Exp. Psychol. Learn. Mem. Cogn. 1992;18:691–702. doi: 10.1037//0278-7393.18.4.691. [DOI] [PubMed] [Google Scholar]
- Haycock JW, Gold PE, Macri J, McGaugh JL. Noncontingent footshock attenuation of retrograde amnesia: A generalization effect. Physiol. Behav. 1973;11:99–102. doi: 10.1016/0031-9384(73)90129-7. [DOI] [PubMed] [Google Scholar]
- Higuchi S, Miyashita Y. Formation of mnemonic neuronal responses to visual paired associates in inferotemporal cortex is impaired by perirhinal and entorhinal lesions. Proc. Natl. Acad. Sci. 1996;93:739–743. doi: 10.1073/pnas.93.2.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kritchevsky M, Squire LR. Transient global amnesia: Evidence for extensive, temporally-graded retrograde amnesia. Neurology. 1989;39:213–218. doi: 10.1212/wnl.39.2.213. [DOI] [PubMed] [Google Scholar]
- Kritchevsky M, Chang J, Squire LR. Functional amnesia: Clinical description and neuropsychological profile of 10 cases. Learn. Mem. 2004;11:213–226. doi: 10.1101/lm.71404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattal KM, Abel T. Behavioral impairments caused by injections of the protein synthesis inhibitor anisomycin after contextual retrieval reverse with time. Proc. Natl. Acad. Sci. 2004;101:4667–4672. doi: 10.1073/pnas.0306546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod MD, Macrae CN. Gone but not forgotten: The transient nature of retrieval-induced forgetting. Psychol. Sci. 2001;12:148–152. doi: 10.1111/1467-9280.00325. [DOI] [PubMed] [Google Scholar]
- Manns R, Hopkins RO, Squire LR. Semantic memory and the human hippocampus. Neuron. 2003;37:127–133. doi: 10.1016/s0896-6273(03)00146-6. [DOI] [PubMed] [Google Scholar]
- Marslen-Wilson WD, Teuber HL. Memory for remote events in anterograde amnesia: Recognition of public figures from news photographs. Neuropsychologia. 1975;13:353–364. doi: 10.1016/0028-3932(75)90013-5. [DOI] [PubMed] [Google Scholar]
- Maviel T, Durkin TP, Menzaghi F, Bontempi B. Sites of neocortical reorganization critical for remote spatial memory. Science. 2004;305:96–99. doi: 10.1126/science.1098180. [DOI] [PubMed] [Google Scholar]
- McClelland JL, McNaughton BL, O'Reilly RC. Why there are complementary learning systems in the hippocampus and neocortex: Insights from the successes and failures of connectionist models of learning and memory. Psychol. Rev. 1995;102:419–457. doi: 10.1037/0033-295X.102.3.419. [DOI] [PubMed] [Google Scholar]
- McGaugh JL. Memory: A century of consolidation. Science. 2000;287:248–251. doi: 10.1126/science.287.5451.248. [DOI] [PubMed] [Google Scholar]
- Messinger A, Squire LR, Zola SM, Albright TD. Neuronal representations of stimulus associations develop in the temporal lobe during learning. Proc. Natl. Acad. Sci. 2001;98:12239–12244. doi: 10.1073/pnas.211431098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messinger A, Squire LR, Zola SM, Albright TD. Neural correlates of knowledge: Stable representation of stimulus associates across variations in behavioral performance. Neuron. 2005;48:358–371. doi: 10.1016/j.neuron.2005.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RR, Springer AD. Amnesia, consolidation, and retrieval. Psychol. Rev. 1973;80:69–79. doi: 10.1037/h0033897. [DOI] [PubMed] [Google Scholar]
- Montarolo P, Goelet P, Castellucci VF, Morgan J, Kandel ER, Schacher S. A critical period for macromolecuar synthesis in long-term heterosynaptic facilitation in Aplysia. Science. 1986;234:1249–1254. doi: 10.1126/science.3775383. [DOI] [PubMed] [Google Scholar]
- Moscovitch M, Rosenbaum RS, Gilboa A, Addis DR, Westmacott R, Grady C, McAndrews MP, Levine B, Black S, Winocur G, et al. Functional neuroanatomy of remote episodic, semantic and spatial memory: A unified account based on multiple trace theory. J. Anat. 2005;207:35–66. doi: 10.1111/j.1469-7580.2005.00421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray EA, Gaffan D, Mishkin M. Neural substrates of visual stimulus-stimulus association in rhesus monkeys. J. Neurosci. 1993;13:4549–4561. doi: 10.1523/JNEUROSCI.13-10-04549.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Kane G, Kensinger EH, Corkin S. Evidence for semantic learning in profound amnesia: An investigation with the patient H.M. Hippocampus. 2004;14:417–425. doi: 10.1002/hipo.20005. [DOI] [PubMed] [Google Scholar]
- Pinsker H, Hening W, Carew TJ, Kandel ER. Long-term sensitization of a defensive withdrawal reflex in Aplysia. Science. 1973;182:1039–1042. doi: 10.1126/science.182.4116.1039. [DOI] [PubMed] [Google Scholar]
- Power AE, Berlau DJ, McGaugh JL, Steward O. Anisomycin infused into the hippocampus fails to block “reconsolidation” but impairs extinction: The role of re-exposure duration. Learn. Mem. 2006;13:27–34. doi: 10.1101/lm.91206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quartermain D. The influence of drugs on learning and memory. In: Rosenzweig MR, Bennett EL, editors. Neural mechanisms of learning and memory. MIT Press; Cambridge, MA: 1976. pp. 508–518. [Google Scholar]
- Russell WR, Nathan PW. Traumatic amnesia. Brain. 1946;69:280–300. doi: 10.1093/brain/69.4.280. [DOI] [PubMed] [Google Scholar]
- Sakai K, Miyashita Y. Neural organization for the long-term memory of paired associates. Nature. 1991;354:152–155. doi: 10.1038/354152a0. [DOI] [PubMed] [Google Scholar]
- Squire LR. Cerebral protein synthesis inhibition and discrimination training: Effects of d-amphetamine. Brain Res. 1979;177:401–406. doi: 10.1016/0006-8993(79)90794-7. [DOI] [PubMed] [Google Scholar]
- Squire LR. Specifying the defect in human amnesia: Storage, retrieval, and semantics. Neuropsychologia. 1980;18:368–372. doi: 10.1016/0028-3932(80)90134-7. [DOI] [PubMed] [Google Scholar]
- Squire LR. Memory systems of the brain: A brief history and current perspective. Neurobiol. Learn. Mem. 2004;82:171–177. doi: 10.1016/j.nlm.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Squire LR, Alvarez P. Retrograde amnesia and memory consolidation: A neurobiological perspective. Curr. Opin. Neurobiol. 1995;5:169–177. doi: 10.1016/0959-4388(95)80023-9. [DOI] [PubMed] [Google Scholar]
- Squire LR, Slater PC. Remote memory in chronic anterograde amnesia. Behav. Biol. 1977;20:398–403. doi: 10.1016/s0091-6773(77)90953-1. [DOI] [PubMed] [Google Scholar]
- Squire LR, Slater PC, Chace PM. Retrograde amnesia: Temporal gradient in very long-term memory following electroconvulsive therapy. Science. 1975;187:77–79. doi: 10.1126/science.1109228. [DOI] [PubMed] [Google Scholar]
- Squire LR, Slater PC, Miller PL. Retrograde amnesia and bilateral electroconvulsive therapy: Long-term follow-up. Arch. Gen. Psychiatry. 1981;38:89–95. doi: 10.1001/archpsyc.1981.01780260091010. [DOI] [PubMed] [Google Scholar]
- Squire LR, Amaral DG, Zola-Morgan S, Kritchevsky M, Press G. Description of brain injury in the amnesic patient N.A. based on magnetic resonance imaging. Exp. Neurol. 1989a;105:23–35. doi: 10.1016/0014-4886(89)90168-4. [DOI] [PubMed] [Google Scholar]
- Squire LR, Shimamura AP, Amaral DG. Memory and the hippocampus. In: Byrne J, Berry W, editors. Neural models of plasticity. Academic Press; New York: 1989b. pp. 208–239. [Google Scholar]
- Squire LR, Clark RE, Knowlton BJ. Retrograde amnesia. Hippocampus. 2001;11:50–55. doi: 10.1002/1098-1063(2001)11:1<50::AID-HIPO1019>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Talland GA. Deranged memory. Academic Press; New York: 1965. [Google Scholar]
- Tulving E, Schacter DL. Priming and human memory systems. Science. 1990;247:301–306. doi: 10.1126/science.2296719. [DOI] [PubMed] [Google Scholar]
- Warrington EK, Weiskrantz L. The effects of prior learning on subsequent retention in amnesic patients. Neuropsychologia. 1974;12:419–428. doi: 10.1016/0028-3932(74)90072-4. [DOI] [PubMed] [Google Scholar]
- Warrington EK, Weiskrantz L. Further analysis of the prior learning effect in amnesic patients. Neuropsychologia. 1978;16:169–177. doi: 10.1016/0028-3932(78)90104-5. [DOI] [PubMed] [Google Scholar]