

Abstract
The AKRs (aldo-keto reductases) are a superfamily of enzymes which mainly rely on NADPH to reversibly reduce various carbonyl-containing compounds to the corresponding alcohols. A small number have been found with dual NADPH/NADH specificity, usually preferring NADPH, but none are exclusive for NADH. Crystal structures of the dual-specificity enzyme xylose reductase (AKR2B5) indicate that NAD+ is bound via a key interaction with a glutamate that is able to change conformations to accommodate the 2′-phosphate of NADP+. Sequence comparisons suggest that analogous glutamate or aspartate residues may function in other AKRs to allow NADH utilization. Based on this, nine putative enzymes with potential NADH specificity were identified and seven genes were successfully expressed and purified from Drosophila melanogaster, Escherichia coli, Schizosaccharomyces pombe, Sulfolobus solfataricus, Sinorhizobium meliloti and Thermotoga maritima. Each was assayed for co-substrate dependence with conventional AKR substrates. Three were exclusive for NADPH (AKR2E3, AKR3F2 and AKR3F3), two were dual-specific (AKR3C2 and AKR3F1) and one was specific for NADH (AKR11B2), the first such activity in an AKR. Fluorescence measurements of the seventh protein indicated that it bound both NADPH and NADH but had no activity. Mutation of the aspartate into an alanine residue or a more mobile glutamate in the NADH-specific E. coli protein converted it into an enzyme with dual specificity. These results show that the presence of this carboxylate is an indication of NADH dependence. This should allow improved prediction of co-substrate specificity and provide a basis for engineering enzymes with altered co-substrate utilization for this class of enzymes.
Keywords: aldo-keto reductase, Candida tenuis, co-substrate recognition, NADH, sequence homology, xylose reductase
Abbreviations: AKR, aldo-keto reductase; 4-NBA, 4-nitrobenzaldehyde; SDR, short-chain dehydrogenase/reductase; XR, xylose reductase; ctXR, Candida tenuis XR
INTRODUCTION
The AKRs (aldo-keto reductases) are a growing superfamily of approx. 120 enzymes, currently composed of 15 families from a wide variety of organisms [1]. These enzymes are found throughout many classes of plants, animals and prokaryotes. In almost all cases, they catalyse the reversible NADPH-dependent reduction of compounds containing ketones and/or aldehydes to the corresponding alcohols. A few of these are capable of binding NADH as well, mainly in families AKR1 (hydroxysteroid dehydrogenases) and AKR2 [the mannose reductases/XRs (xylose reductases)] and to a lesser extent in families AKR6 (aflatoxin reductases) and AKR7 (potassium channel β-subunits) [2–6]. Their conventional physiological functions vary widely and include steroid metabolism, prostaglandin synthesis, xenobiotic detoxification, metabolic assimilation of carbohydrates and participation in other secondary metabolic pathways. More divergent roles have also been found as lens crystallins [7], potassium channel regulators [8] and catalysts of lactonization [9]. For many however, a physiological function has yet to be found.
The AKRs are relatively well-studied biochemically, and representative crystal structures have also been determined which explain the ordered binding of substrates, the catalytic mechanism and the specificity for co-substrate. These aspects have been reviewed by Sanli et al. [10]. Although several dimers and tetramers have been characterized, AKRs are mostly monomeric and of approx. 35 kDa. These enzymes do not adopt a classic Rossmann fold which is often involved in dinucleotide binding but rather a (β/α)8 barrel, the most common fold known for all enzymes [11]. The nucleotide is bound across the top face with loops in β/α repeats 7 and 8 contributing to the specificity for the 2′-phosphate present in NADPH or 2′-hydroxyl present in NADH (Figure 1A). Generally, binding of the co-substrate creates an active-site pocket for the carbonyl-containing substrate to bind, reduction occurs and the alcohol product leaves followed by the consumed co-substrate.
Figure 1. The co-substrate binding by ctXR (AKR2B5) and the basis of dual co-substrate specificity.

(A) The enzyme binds the co-substrate across the top of the (β/α)8 barrel. Residue numbers for amino acids involved in binding of the 2′-phosphate are shown. (B, C) Schematic diagrams of binding of the adenosine (B) and adenosine 2′-phosphate (C) portions of NAD+ and NADP+ respectively.
Co-substrate specificity has recently become an issue in the superfamily for various reasons mainly related to potential biotechnological uses for the enzymes. In many cases, NADH is preferable due to its 10-fold lower cost and also because it is more stable than NADPH. Availability is also an issue since intracellular NADPH concentrations are also typically much lower than NADH concentrations and can be more susceptible to depletion. As a result, attempts have been made to engineer NADH utilization into the NADPH-specific enzyme 2,5-diketo-D-gluconic acid reductase A (AKR5C), which catalyses a key step in the synthesis of vitamin C [12,13]. An argument has also been made for NADH utilization in XR, yeast enzymes from family AKR2 catalysing the reduction of xylose to xylitol [14]. This activity would facilitate fermentation of the xylose present in agricultural waste products by yeast because it would enable the efficient recycling of co-substrate with the next step of a high-flux xylose assimilation pathway, an NAD+-specific xylitol dehydrogenase. Finally, the ability of AKRs to reduce carbonyls may be potentially useful in catalysing one or more steps to produce chiral compounds [15].
AKRs able to naturally utilize both NADH and NADPH efficiently as co-substrates are relatively rare. This specificity has been quantitatively demonstrated in the enzymes AKR1C9, AKR1C12, AKR1C13, AKR1C19, AKR2B1, AKR2B3 and AKR2B5 although all prefer NADPH with the exception of AKR1C19, which shows little preference [2,3,16–19]. None have been discovered that are specific for NADH, although one additional enzyme from Candida parapsilosis has been found that naturally prefers NADH to NADPH [4]. Structurally, much is generally known about the conserved manner in which NADPH binds to AKRs. Previously, structures of the dual-specificity ctXR (Candida tenuis XR)/AKR2B5 have been determined in the presence of NAD+ [20] and NADP+ [21] revealing elements in the enzyme which confer specificity for each. The primary finding was that a key glutamic acid (Glu227) was critical for recognition of the 2′- and 3′-hydroxyls of the adenosine ribose in the case of NAD+. To avoid potential electrostatic repulsion or steric collision with the 2′-phosphate, Glu227 resides on a flexible loop, which allows it to rotate out of the way to accommodate the phosphate (Figure 1). Sequence comparisons show a clearly conserved glutamate in all current members of subfamilies AKR2B, AKR2C and AKR2D, which are primarily XRs. Outside of the AKR2 family, dual-specific enzyme sequences are more difficult to align in this region but the carboxylic acid-containing residues which have potential functional homology are either in an identical position (Glu226 in AKR1C12, Glu229 in the C. parapsilosis enzyme) or within several residues (Asp224 in AKR1C9 and Asp229 in AKR1C13).
Other interactions between ctXR/AKR2B5 and the NAD+ adenosine ribose are made by the side chains from Asn276 and Arg280. The 2′-phosphate in the NADP+-bound ctXR/AKR2B5 interacts with the Lys274, Ser275 and Arg280 side chains and Asn276 main chain amide nitrogen (Figure 1C). Recognition of the 2′-phosphate in NADPH-specific enzymes such as human aldose reductase is achieved in a largely similar manner [22] but NADH fails to bind efficiently due to the lack of a residue analogous to Glu227 in ctXR. A major structural difference between the NAD(P)H- and NADPH-dependent enzymes therefore appears to be the presence of a mobile carboxylate in the former. This mobility is critical for NADPH binding, which implies that sterically blocking movement of the glutamate or aspartate away from the 2′-phosphate would result in an NADH-specific enzyme.
Engineering altered co-substrate specificity using guidance from crystal structures has been successful in changing co-substrate preference but has not been straightforward. In the case of AKR5C, a quadruple mutant was necessary to convert the NADPH-specific enzyme into one that efficiently utilized both NADPH and NADH [23]. Interestingly, none of the point mutations (F22Y/K232G/R238H/A272G) introduced a carboxylate into the phosphate-binding pocket but they disrupted phosphate-binding interactions instead. Progress has also been made in improving the NADH-dependent co-substrate specificity of ctXR/AKR2B5 by mutating residues surrounding the phosphate [24]. The best of these, a K274R/N276D double mutant, increased the NADH versus NADPH selectivity 193-fold relative to the wild-type enzyme by preventing movement of Glu227 in response to NADPH binding.
Precedents exist for varied co-substrate utilization within an enzyme superfamily. The best characterized example is probably the SDR (short-chain dehydrogenase/reductase) superfamily, which utilizes a Rossmann fold to bind the NAD(P)+ co-substrate and is unrelated to the AKRs in both sequence and structure [25]. Numerous structures of SDRs of various specificities exist in complex with co-substrate (reviewed in [26]) and have led to a well-developed understanding of how sequence dictates specificity [27]. Interestingly, a carboxylate-containing residue has been found to make interactions with the 2′- and 3′-hydroxyls of the adenosine ribose in NAD+-specific SDRs, analogous to those made by Glu227 in ctXR/AKR2B5 (Figure 1B). The co-substrate preference of SDRs has been converted from NADP+ into NAD+ in the past by changing a serine or threonine residue at this position to an aspartate residue [28]. Alternatively, mutating the carboxylate-containing residue to a serine has converted an NAD+-dependent SDR into an NADP+-dependent enzyme [29]. The present study provides evidence that similar interactions involving a carboxylate-containing residue are likely to be present in the dual-specificity and NADH-specific AKRs.
Discovery of naturally occurring NADH-dependent enzymes removes the need to engineer this specificity into those dependent on NADPH provided they accept the appropriate carbonyl substrate. Alternatively, an increased understanding of residues conferring this specificity will facilitate identification of these enzymes and provide guidance for engineering mutants when necessary. To investigate the possibility that residues analogous to Glu227 in ctXR/AKR2B5 function in other uncharacterized AKRs to allow NADH utilization, we searched sequence databases to find proteins containing a carboxylate at or near this position, suggesting dual specificity or even NADH-specific activity. Herein, we describe and test the kinetic properties of these enzymes with a particular focus on co-substrate utilization.
EXPERIMENTAL
Sequence alignments
The protein sequence of the dual-specificity ctXR [30] [NCBI (National Center for Biotechnology Information) accession number AAC25601] was used to perform a BLASTp [31] search against both SWISS-PROT and TrEMBL protein databases [32]. Nine potential AKR sequences were selected (Table 1) based on the presence, in a pairwise BLASTp sequence alignment with ctXR, of a potential NAD(H)-binding site (Figure 2B). This was mainly determined by the presence of a carboxylate within three amino acids of the position of Glu227 in ctXR and to a lesser extent by sequence divergence among the amino acids known to interact with the 2′-phosphate in NADP(H): Lys274, Ser275 and Arg280 (Figure 1C).
Table 1. Proteins selected for study.
Those proteins which were not successfully purified (designated NP) and those which did not exhibit enzymatic activity (NA) were not assigned systematic names. ORF, open reading frame.
| Name | Source organism | Gene/ORF name | NCBI protein accession no. | Molecular mass (Da) | Systematic name |
|---|---|---|---|---|---|
| at1 | A. thaliana | F19P19.12 | P93818 | 50908 | NP |
| dm1 | D. melanogaster | CG10863 | AAD38635 | 36312 | AKR2E3 |
| ec1 | E. coli | yafB | P30863 | 29436 | AKR3F2 |
| ec2 | E. coli | ydjG | P77256 | 36328 | AKR11B2 |
| sm1 | Si. meliloti | SMa1410 | 81635765 | 30893 | AKR3F3 |
| sp1 | Sch. pombe | SPAC26F1.07 | Q10494 | 36192 | NP |
| sp2 | Sch. pombe | SPAC19G12.09 | O13848 | 31569 | AKR3C2 |
| ss1 | Su. solfataricus | SSO2885 | 74556625 | 35080 | NA |
| tm1 | T. maritima | TM_1743 | Q9X265 | 31359 | AKR3F1 |
Figure 2. Aldo-keto reductase sequence alignments.

(A) Sequence alignment of the proteins studied with ctXR. Conserved catalytic residues are boxed. Residues involved in 2′-phosphate binding are light grey and the carboxylate-containing residue putatively interacting with 2′- and 3′-hydroxyls (found in pairwise sequence alignments) is shown for each protein in light grey with a star. Other residues known to interact with NADP+ in ctXR are shown in dark grey. (B) Results from pairwise sequence alignment of the region, with the carboxylate-containing residue putatively interacting with 2′- and 3′-adenosine hydroxyls (underlined). The grey area is identical with the dark grey sequence following β7 in (A).
Cloning, protein expression and purification
PCR templates were Arabidopsis thaliana cDNA [courtesy of Dr M. Bostick (Section of Molecular and Cellular Biology, University of California, Davis, CA, U.S.A.)], Drosophila melanogaster cDNA (Clontech), Schizosaccharmyces pombe cDNA [courtesy of Dr J. Nunnari (Section of Molecular and Cellular Biology, University of California, Davis, CA, U.S.A.)] or genomic DNA from Escherichia coli (strain BL21), Sulfolobus solfataricus [A.T.C.C. (American Type Culture Collection)], Sinorhizobium meliloti (A.T.C.C.) and Thermotoga maritima (A.T.C.C.) using primers sequences as listed in Table 2. In each case, the resulting fragment was inserted into the NdeI and SmaI sites of the plasmid pTYB12 (New England Biolabs) to yield the final bacterial expression vector. These were designed to produce protein with no extra residues on the C-terminus, a region known to be important in AKR substrate specificity. Sequencing of each of the plasmids was done to ensure that there were no mutations introduced by the PCR reaction. Constructs were used to transform the E. coli expression strain ER2566 and the recombinant AKRs were produced in fusion with plasmid-derived intein- and chitin-binding domains. The proteins were expressed in Luria–Bertani medium containing 100 μg/ml ampicillin and induced with 500 μM IPTG (isopropyl β-D-thiogalactoside) at 15 °C. Cells were harvested by centrifugation (8000 g for 20 min at 4 °C) 12 h after induction and lysed in Buffer A (20 mM Tris, pH 8.0, 500 mM NaCl and 0.1 mM EDTA) supplemented with 0.1% Triton X-100 using a microfluidizer (Microfluidics) at 15000 lbf/in2 (1 lbf/in2=6.9 kPa). The lysate was clarified by centrifugation and the resulting cell extracts containing AKR–intein–chitin-binding domain fusion protein were passed over a fresh approx. 15 ml column of chitin beads. This was washed with at least 20 column volumes of Buffer A supplemented with 0.1% Triton X-100 followed by 20 column volumes of Buffer A alone to remove the detergent. The AKR–intein fusion proteins were then cleaved on the chitin column by incubating at 4°C for 36 h in Buffer A supplemented with 50 mM 2-mercaptoethanol. The cleaved AKRs were eluted in Buffer A, dialysed into 20 mM Tris (pH 8.0) and further purified (when necessary) by anion-exchange chromatography on a quaternized poly(ethyleneimine) HQ column using a BioCad Sprint fast protein liquid chromatography system at pH 8.0 using a gradient of 0 to 1 M NaCl. The purified proteins (Figure 3) were then exchanged into 20 mM Hepes (pH 7.4) with 20 mM NaCl and concentrated to 1.5 mg/ml using Millipore Ultrafree spin concentrators.
Table 2. Primers used for cloning and site-directed mutagenesis.
For each insert, the 5′-primer is listed on top and the 3′-primer is on the bottom. Restriction sites are underlined except for the primer pairs used for mutagenesis where the mismatched bases are underlined.
| Insert | Primer |
|---|---|
| dm1 | 5′-GGCGGCTCATATGTCCAGCAAAATTCCCAGGTAAA |
| 5′-GCGCGCGCCCGGGAAACTCGATGTTAAATGGATAGT | |
| sp2 | 5′-GGCGGCTCATATGCTTATCGCTGCTATGGGCCC |
| 5′-GCGCGCGCCCGGGTTACTCATCCATGTGCTTCATAAATGTACG | |
| sp2 D202S | 5′-CTCCCTTGGTTCGTAGTGCGCAAGGTCCTGTTG |
| 5′-CAACAGGACCTTGCGCACTACGAACCAAGGGA | |
| ec1 | 5′-GGCGGCTCATATGGCTATCCCTGCATTTGGTTTAGG |
| 5′-GCGCGCGCCCGGGTTAATCCCATTCAGGAGCCAGACC | |
| sm1 | 5′-GGCGGCTCATCATATGGGAGGACCGATGAGCGTCTATTTC |
| 5′-CGCGCGCCCGGCCCGGGCCGGTCCCAATCCGGAACCCAGGGCA | |
| tm1 | 5′-GGCGGCTCATATGTTATACAAAGAACTTGGAAGAACAGG |
| 5′-GCGCGCGCCCGGGTTAGCCAAGGCTGTCCAGTAGC | |
| tm1 E208S | 5′-GTTCTCTTCGTTTTTGATGAAAGAAGAGTTC |
| 5′-GAACTCTTCTTTCATCAAAAACGAAGAGAAC | |
| ss1 | 5′-GGCGGCTCATATGGAAAAGAAGCCATTAGGATGG |
| 5′-GCGCGCGCCCGGGTTACCTTGCGAAATAATCTATCG | |
| ss1 E208S | 5′-CTAATAGGGAGAAAGTCGGTCAGAAACGAG |
| 5′-CTCGTTTCTGACCGACTTTCTCCCTAT | |
| at1 | 5′-GGCGGCTCATATGGCGTCATCTTACGTCACC |
| 5′-GCGCGCGCCCGGGTCAAACGAAAGAAGGGTCTTTGAACC | |
| ec2 | 5′-GGCGGCTCATATGAAAAAGATACCTTTAGGCACAACG |
| 5′-GCGCGCGCCCGGGTTAACGCTCCAGGGCCTCTGCC | |
| ec2 D232A | 5′-CCGCCCGGAACGTAAGCACGAGTGATGGTGCCG |
| 5′-CACCATCACTCGTGCTTACGTTCCGGGCGG | |
| ec2 D232E | 5′-GCACCATCACTCGTGAATACGTTCCGGGCGG |
| 5′-CCGCCCGGAACGTATTCACGAGTGATGGTGC |
Figure 3. SDS/PAGE of expressed AKRs.

Lane 1, standards (molecular mass as marked); lane 2, ec2/AKR11B2; lane 3, ec1/AKR3F2; lane 4, tm1/AKR3F1; lane 5, sm1/AKR3F3; lane 6, ss1; lane 7, sp2/AKR3C2; lane 8, dm1/AKR2E3.
To determine oligomerization of each protein (and as a secondary purification step for ss1), gel filtration experiments were carried out on a Biocad Sprint perfusion chromatography system (PerSeptive Biosystems) using a Superdex 200 HR 10/30 column (Amersham Biosciences). The column was equilibrated with PBS buffer containing 137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4 and 1.4 mM KH2PO4 (pH 7.3). Purified AKRs were diluted in the same buffer (1 ml; 0.3 mg/ml) and loaded on to the column. Elution was carried out at a flow rate of 0.5 ml/min. Protein elution was detected by absorbance at 280 nm. Calibration for molecular mass determination was performed with the following protein standards (Bio-Rad): thyroglobulin (670 kDa), γ-globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa) and vitamin B12 (1.3 kDa).
Site-directed mutagenesis
An inverse PCR method was employed to generate all site-directed mutants [33]. The mutagenic oligonucleotide primers used are listed in Table 2 with the mismatched bases underlined. Plasmid DNA was sequenced in order to verify the introduction of the desired mutation and to ensure that no misincorporations of nucleotides had occurred due to DNA polymerase errors. Expression and purification of the mutagenized gene in E. coli was carried out using a method identical with that described above for the other AKRs.
Steady-state assays
All velocity measurements were done in a buffer containing 50 mM K2HPO4, 10 mM KCl and 0.5 mM EDTA (pH 7.0) and at 25 °C using a Shimadzu UV160U spectrophotometer by observing the decrease in absorbance of either NADH (ϵ340=6220 M−1·cm−1; where ϵ is molar absorption coefficient) or NADPH (ϵ340=6200 M−1·cm−1) at 340 nm in a 1 ml quartz cuvette. Measurements at 5–8 different substrate or co-substrate concentrations were taken for the substrate(s) and cosubstrate(s) that showed activity. Apparent constants were determined by varying substrates or co-substrates in the presence of saturating concentrations (>10 Km) of the other reaction component. Typically, velocities were measured for 1 min after 15 s of equilibration time. Careful attention was paid to non-enzymatic velocities and corrections were applied to measured velocities when these were significant. Km and kcat were determined by curve-fitting the Michaelis–Menten equation to the resulting values using the program DeltaGraph (SPSS Inc.). A modified Michaelis–Menten equation (eqn 1) was used to incorporate substrate inhibition observed in the cases of tm1, ec2 (wild-type and mutant), sp2 and ec1.
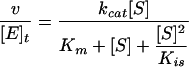 |
(1) |
Fluorescent measurements of dissociation constants
Measurements of the emission fluorescence spectra were conducted on an Aminco-Bowman Series 2 luminescence spectrometer from SIM-Aminco Spectronic Instruments and were digitized using the AB2 software V5.0. Assays were performed in a buffer containing 10 mM KCl (pH 7.1) at 25 °C in a 150 μl quartz cuvette. The emission scans were recorded between 300 and 400 nm at 60 nm/min with a continuous excitation wavelength fixed at 280 nm (bandpass 4 nm). Untransformed fluorescence data were plotted as the percentage change in fluorescence at emission λmax versus either [NADH] or [NADPH] [34]. The program DeltaGraph (SPSS Inc.) was used to fit a hyperbola to the data, which provided an estimate of the dissociation constant.
Secondary structure prediction and molecular modelling
PSIPRED V2.4 program coupled with the NCBI BLAST V.2.2.13 package was used to predict secondary structures for the seven AKR sequences described here [35,36]. The BLAST database was generated based on the whole UNIPROT sequence database of November 2005 [37].
Molecular modelling was performed using the automated comparative protein modelling server SWISS-MODEL [38,39]. Template(s) for each of the AKRs were selected after a BLASTp search against the entire PDB. ec1 templates are structures with PDB accession codes 1M9H, 1HW6 and 1MZR; the dm1 template is 1K8C; ec2 templates are 1PZ1, 1PYF, 1PZ0 and 1YNQ; sm1 templates are 1M9H, 1HW6 and 1VBJ; sp2 templates are 1K8C and 1YNQ; ss1 templates are 1PZ1, 1PYF, 1PZ0 and 1VP5; tm1 templates are 1K8C and 1YNQ; AKR1C9, AKR2B1 and AKR2B3 template is 1K8C; AKR1C12 and AKR1C13 templates are 2FVL, 1MRQ and 1XJB; AKR1C19 templates are 1MRQ, 1Q13, 1Q5M and 1XJB; C. parapsilosis XR templates are 1YE4 and 1YE6. Statistics for the models are summarized in Table 3. To consistently measure distances from the carboxylate-containing residue to the 2′-phosphate, NADPH was manually fitted into the models (including the NADH-specific ec2) using Swiss-PdbViewer and COOT [39,40]. Each model was first overlaid with holo NADPH-bound AKR structures: AKR2B5 (1K8C) and AKR11C1 (1YNQ). NADPH was then fitted into the active site in agreement with the conserved residues involved in the co-substrate binding [20,41].
Table 3. Modelling statistics and results for proteins in the present study and other dual-specific enzymes.
The carboxylate to phosphate distance measures the separation between the aspartate or glutamate carboxylate and phosphate oxygens that are closest to each other. 1 Å=0.1 nm.
| Procheck validation | |||||
|---|---|---|---|---|---|
| Enzyme | Modelled (%) | Residues modelled | Residues in allowed regions (%) | Residues in disallowed regions (%) | Carboxylate to phosphate distance (Å) |
| dm1/AKR2E3 | 94.9 | 3–303 out of 316 | 100.0 | 0 | 13.9 |
| ec1/AKR3F2 | 97.8 | 1–261 out of 267 | 99.6 | 0.4 | 22.5 |
| ec2/AKR11B2 | 99.7 | 1–325 out of 326 | 98.2 | 1.8 | 4.4 |
| sm1/AKR3F3 | 92.8 | 8–265 out of 277 | 97.4 | 2.6 | 18.2 |
| sp2/AKR3C2 | 66.5 | 82–112, 116–275 out of 284 | 98.8 | 1.2 | 3.4 |
| ss1 | 100.0 | 304 out of 304 | 98.6 | 1.4 | 14.5 |
| tm1/AKR3F1 | 65.7 | 34–214 out of 274 | 100.0 | 0 | 8.7 |
| AKR1C9 | 97.8 | 4–318 out of 322 | 98.6 | 1.4 | 12 |
| AKR1C12 | 100 | 322 out of 322 | 99.7 | 0.3 | 16.5 |
| AKR1C13 | 100 | 323 out of 323 | 99.7 | 0.3 | 18.7 |
| AKR1C19 | 100 | 323 out of 323 | 99.3 | 0.7 | 20.5 |
| AKR2B1 | 100 | 318 out of 318 | 100 | 0 | 5.4 |
| AKR2B3 | 99.3 | 3–318 out of 318 | 100 | 0 | 5.4 |
| ctXR | 98.2 | 6–324 out of 324 | 98.6 | 1.4 | 3.4 |
RESULTS AND DISCUSSION
Selection and expression of proteins
To investigate the possibility that analogous glutamates or aspartates function in other AKRs to allow NAD+ utilization, we initially used BLASTp to search sequence databases using the ctXR sequence as a template to find other proteins containing a carboxylate at this position. Nine potential AKRs sequences were primarily selected based on the presence of a carboxylate-containing residue within three amino acids of the position homologous with Glu227 in ctXR (Figure 2B). All of the selected sequences possessed the canonical catalytic tetrad of Tyr51, Lys80, Asp46 and His113 (numbered according to ctXR) found in the active site and conserved among the AKR superfamily, suggesting that they were catalytically active (Figure 2A) [42]. Overall sequence divergence was considerable, ranging from 19 to 45% identity when pairwise comparisons with ctXR were made (Figure 2A). The diversity is particularly large in the loop region which contains the carboxylate-containing residue (Figure 2A).
Attempts were made to express each of these nine proteins in bacteria as a fusion construct with an intein/chitin-binding domain to facilitate purification using the IMPACT system (New England Biolabs). Stable and soluble protein was obtained from seven after purification using chitin affinity chromatography. Further purification was achieved using anion exchange (for dm1 and sm1) or gel filtration (for ss1) columns. No secondary purification was found to be necessary for ec1, ec2, sp2 and tm1. All proteins were near single-band purity, as judged by a reducing SDS gel, and ran near the expected molecular mass with the exception of sm1, which ran anomalously slowly (Figure 3). Since some AKR members from families 2, 6 and 7 are known to be oligomeric, analytical gel filtration was also performed on all samples to determine quaternary structure. The proteins were all clearly monomeric with the exception of sm1, which consistently eluted in the void volume. Each was named using the first letters of the genus and species and a sequential numbering system (e.g. ec2 for the second protein from E. coli). The Arabidopsis protein, at1, consistently co-purified with a higher molecular mass protein, presumably a chaperone and was not pursued further. sp1 rapidly degraded during purification and was abandoned.
Enzymatic activity
Based on kinetic experiments measuring the oxidation of NADH or NADPH for the seven proteins, six were found to be active enzymes: ec1, sm1, dm1, sp2, tm1 and ec2 (Tables 4 and 5). ss1 exhibited no detectable reductase activity under standard conditions with the substrates DL-glyceraldehyde, 4-NBA (4-nitrobenzaldehyde), methylglyoxal and isatin. As ss1 is derived from the thermophilic organism Su. solfataricus, additional assays were carried out at 65 °C with both of the co-substrates and methylglyoxal, phenylglyoxal and 4-NBA but again yielded no activity. Although it is possible that ss1 is enzymatically active and an appropriate substrate was not found, a number of AKRs in family 6 (the K+ channel β-subunit) have been found to bind co-substrates yet exhibit no detectable catalytic activity. In order to determine the ability of this protein to bind co-substrates, tryptophan fluorescence quenching assays were done in the presence of both NADH and NADPH [34].
Table 4. Co-substrate utilization.
Kinetic or binding constants associated with different co-substrates are listed for each AKR. Saturating concentrations of designated substrate were used to measure velocities. Abbreviations: DLG, DL-glyceraldehyde; Me-Gly, methylglyoxal; NMA, no measurable activity. Errors are S.E.M. from regression analysis. ss1, Kd,NADPH=1.6 μM±0.6; ss1, Kd,NADH=5.2 μM±1.8; ss1 (E208S), Kd,NADPH=5.2 μM±1.8; ss1 (E208S), Kd,NADH=59.14 μM±7.8.
| NADPH | NADH | ||||
|---|---|---|---|---|---|
| Enzyme | Substrate | Km,NADPH (μM) | kcat,NADPH (s−1) | Km,NADH (μM) | kcat,NADH (s−1) |
| dm1/AKR2E3 | DLG | 42.6±5.6 | 25.1±1 | NMA | NMA |
| ec1/AKR3F2 | 4-NBA | 100±36 | 50.8±11.6 | NMA | NMA |
| ec2/AKR11B2 | Me-Gly | NMA | NMA | 52±2 | 5.6±0.1 |
| ec2/AKR11B2 (D233A) | Me-Gly | 110±14 | 1.95±0.05 | 83±15 | 11.4±3 |
| ec2/AKR11B2 (D232E) | Me-Gly | 135.8±17.7 | 4.1±0.27 | 28.2±4.2 | 15.7±0.64 |
| sm1/AKR3F3 | Isatin | 40.7±3.5 | 16.7±0.41 | NMA | |
| sp2/AKR3C2 | Isatin | 34±9.6 | 11.8±0.9 | 53.3±11 | 4.2±0.6 |
| sp2/AKR3C2 (D202S) | Isatin | NMA | NMA | 378.0±91.5 | 56.4±7.43 |
| ss1 | – | NMA | NMA | NMA | NMA |
| ss1 (E208S) | – | NMA | NMA | NMA | NMA |
| tm1/AKR3F1 | DLG | 5.6±0.7 | 0.75±0.01 | 26±5.8 | 3.3±0.16 |
| tm1/AKR3F1 (E208S) | Isatin | 30.1±4.5 | 17.2±0.63 | 37.8±6.3 | 11.8±0.6 |
Table 5. Carbonyl-containing substrate utilization.
Saturating concentrations of each co-substrate were used to measure velocities;±values are S.E.M. associated with each parameter obtained from regression analysis. Dashes signify that a particular assay was not performed. Abbreviations: DLG, DL-glyceraldehyde; Me-Gly, methylglyoxal; NMA, no measurable activity. Trace NADPH activity associated with ec2 and DLG is discussed in the text.
| DLG | 4-NBA | Isatin | Me-Gly | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Enzyme | Co-substrate | kcat (s−1) | Km (mM) | kcat (s−1) | Km (mM) | kcat (s−1) | Km (mM) | kcat (s−1) | Km (mM) |
| dm1/AKR2E3 | NADH | NMA | NMA | NMA | NMA | NMA | NMA | – | – |
| NADPH | 40±0.8 | 9.5±0.7 | NMA | NMA | NMA | NMA | – | – | |
| ec1/AKR3F2 | NADH | – | – | NMA | NMA | – | – | – | – |
| NADPH | – | – | 74.9±40 | 1.78±0.7 | – | – | – | – | |
| ec2/AKR11B2 | NADH | 2±0.08 | 363±35 | NMA | NMA | NMA | NMA | 15.7±0.8 | 1.81±0.25 |
| NADPH | Trace | Trace | NMA | NMA | NMA | NMA | NMA | NMA | |
| ec2/AKR11B2 (D233A) | NADH | 23.6±1.75 | 1200±140 | NMA | NMA | NMA | NMA | 40.1±11 | 5.2±2 |
| NADPH | 1.5±0.1 | 218±35 | NMA | NMA | NMA | NMA | 5.1±0.64 | 5.51±1.38 | |
| ec2/AKR11B2 (D232E) | NADH | Trace | Trace | NMA | NMA | NMA | NMA | 100.1±51 | 7.8±4.6 |
| NADPH | Trace | Trace | NMA | NMA | NMA | NMA | 4.09±0.26 | 0.13±0.02 | |
| sm1/AKR3F3 | NADH | NMA | NMA | NMA | NMA | NMA | NMA | NMA | NMA |
| NADPH | 2±0.13 | 256±35 | NMA | NMA | 6.6±0.6 | 7±1.9 | NMA | NMA | |
| sp2/AKR3C2 | NADH | NMA | NMA | NMA | NMA | 1.4±0.027 | 0.017±0.0016 | – | – |
| NADPH | 2.5±0.25 | 552±99 | NMA | NMA | 33±0.63 | 0.017±0.017 | – | – | |
| sp2/AKR3C2 (D202S) | NADH | NMA | NMA | NMA | NMA | 91.7±19.9 | 0.43±0.11 | – | – |
| NADPH | Trace | Trace | Trace | Trace | Trace | Trace | – | – | |
| tm1/AKR3F1 | NADH | 4±2 | 52±20 | – | – | – | – | – | – |
| NADPH | 4.8±0.19 | 137±6.8 | – | – | – | – | – | – | |
| tm1/AKR3F1 (E208S) | NADH | 111.8±21.0 | 210.8±55.9 | – | – | 30.1±7.0 | 0.61±0.19 | – | – |
| NADPH | – | – | – | – | 17.3±0.37 | 0.047±0.005 | – | – | |
The proteins that possessed catalytic activity were assigned systematic superfamily names which are given in Table 1 according to the rules established by Jez et al. [43] where proteins within families are distinguished by having >40% amino acid identity and subfamilies have >60%. Families are designated by a number and subfamilies by a letter. Thus ctXR is a member of family 2 and the fifth member of subfamily B and is therefore designated AKR2B5.
A new subfamily, AKR3F is founded by tm1, ec1 and sm1 which have been assigned the systematic names AKR3F1, AKR3F2 and AKR3F3 respectively. Family AKR3 had previously been populated primarily by yeast AKRs so it is notable that subfamily AKR3F currently contains only prokaryotic members. Prior studies of ec1/AKR3F2 suggest the AKR3F subfamily may be responsible for metabolizing incompletely oxidized forms of glucose such as methylglyoxal or 2,5-diketo-D-gluconate [44,45]. dm1/AKR2E3 is a member of subfamily AKR2E which consists of 3-dehydroecdysone 3β-reductases from other insects [46,47], suggesting that it may play a similar role in Drosophila ecdysteroid-mediated development as well. sp2 is designated AKR3C2 and is most closely related to AKR3C, which functions as an arabinose dehydrogenase in Saccharomyces cerevisiae [48]. Finally, ec2 was assigned to family 11 (AKR11B2), which was previously composed of Bacillus AKRs of ill-defined function.
Based on kinetic experiments measuring the oxidation of NADH and NADPH for the seven proteins, three were exclusive for NADPH (ec1/AKR3F2, sm1/AKR3F3 and dm1/AKR2E3), two were dual-specific (sp2/AKR3C2 and tm1/AKR3F1) and one was specific for NADH (ec2/AKR11B2), the first time this activity has been found in an AKR (Table 4). Despite being enzymatically inactive, ss1 was dual-specific in the sense that it was able to bind both NADH and NADPH with micromolar affinities (Table 4).
The NADH-specific enzyme ec2/AKR11B2 and the role of Asp232
Unique to the AKR superfamily, ec2/AKR11B2 was found to use NADH exclusively as co-substrate when assayed with methylglyoxal as a substrate. Trace NADPH-dependent activity was found at very high concentrations of DL-glyceraldehyde but the enzyme could not be saturated with the substrate (Table 5). Based on a linear slope of a velocity versus co-substrate plot, a kcat/Km of 8.6×10−3 M−1·s−1 was estimated for DL-glyceraldehyde using saturating concentrations of NADPH. This is approx. 3 orders of magnitude worse than the NADH-dependent catalytic efficiency with DL-glyceraldehyde, a poor substrate compared with methylglyoxal. No NADPH-dependent activity was detectable using methylglyoxal as a carbonyl-containing substrate.
It is not yet clear why ec2/AKR11B2 is not dual-specific but there is one obvious possibility. Rather than binding the 2′- and 3′-ribose hydroxyls with a glutamate, ec2/AKR11B2 has an aspartate residue at this position (Figure 2). This side chain has less torsional freedom to rotate away from a negatively charged 2′-phosphate, which is likely to constrain its position and could lead to electrostatic repulsion. Less conformational flexibility in the loop on which it resides could also prevent its movement away from the 2′-phosphate. Alternatively, it could be engaged in an interaction that impedes its movement. The other known members of family 11 (AKR11A and AKR11B) are only active with NADPH [49]. The difference in co-substrate specificity between ec2/AKR11B2 and the other members of family 11 is not immediately obvious based on sequence alignments, which indicates that AKR11A has a homologous aspartate residue at position 218 and AKR11B has a glutamate residue at position 218. Structures of these proteins show that their locations are very far from the 2′-phosphate and are therefore not structurally conserved. The shortest distance between carboxylate and phosphate oxygens is 19 Å (1 Å=0.1 nm) in the case of AKR11A and 15 Å in the case of AKR11B.
In previous structural studies of the B. subtilis enzymes AKR11A and AKR11B, the 2′-phosphate of NADPH was found to be strongly stabilized by salt links from Arg282, Lys214 and Arg227 (AKR11A) and Lys214 (AKR11B) [49]. This is similar to the scheme found in the NADPH-specific human aldose reductase, which attests to the importance of these residues in 2′-phosphate recognition. These residues are lacking in ec2/AKR11B2 (an enzyme ∼40% identical with AKR11B) where Thr293, Thr228 and Arg239 are found respectively at the same positions. Furthermore, the same observation can be made with all the dual-specificity enzymes found in the present study, where the 2′-phosphate of NADPH is weakly stabilized by hydrogen-bonding but not for those enzymes which rely exclusively on NADPH consumption.
Based on these results, an aspartate residue found at a position structurally homologous with Glu227 in ctXR seems to contribute to an exclusive NADH specificity in ec2/AKR11B2. In order to test the role of this particular aspartate residue, we created a sitedirected mutant enzyme in which Asp232 was converted into an alanine (D232A) which converted ec2/AKR11B2 into a dual-specific enzyme. Based on the ratio of catalytic efficiencies, this mutation yielded an enzyme that was able to use NADPH with 13% of the catalytic efficiency of NADH (Table 4) without significantly affecting the carbonyl substrate kinetic parameters (Table 5). We ascribe this to the lack of electrostatic repulsion with the 2′-phosphate that the aspartate at this position would confer. A second site-directed mutant where the aspartate was changed into a glutamate (D232E) was also able to utilize both NADPH and NADH as co-substrates with the NADPH-dependent activity being 5% of the NADH-dependent activity based on catalytic efficiencies (Table 4). The lengthened side chain of glutamate mimics Glu227, which is responsible for dual co-substrate specificity in ctXR/AKR2B5. This implies that the carboxylate is able to move to accommodate the negative charge found on the phosphate.
Since carboxylate-containing residues that were homologous with Glu227 in ctXR from sequence alignments may not be structurally homologous, a model was constructed for ec2/AKR11B2 (and the other enzymes) as described in the Experimental section. Based on the secondary structure prediction, Asp232 is predicted to sit on a flexible region occupied by Glu227 in ctXR. Modelling studies indicate that Asp232 is 4.4 Å from the 2′-phosphate, which implicates it in hydrogen-bonding interactions with the 2′- and 3′-hydroxyls (Table 3). Interestingly, both of these mutations have very little effect on NADH utilization, suggesting that the primary role of the aspartate is in discrimination against the co-substrate 2′-phosphate.
Dual-specificity enzymes
Efficient use of both NADH and NADPH as a co-substrate is relatively rare in the AKR superfamily with only seven of the approx. 120 members having this ability. Of the seven AKRs examined, sp2/AKR3C2 and tm1/AKR3F1 were catalytically active with both co-substrates. Sequence alignments show these have respectively an aspartate (Asp202) and a glutamate (Glu208) homologous with Glu227 in ctXR (Figure 2). In order to investigate the role of these carboxylates, we made the D202S mutant in sp2/AKR3C2 and the E208S mutant in tm1/AKR3F1. The sp2/AKR3C2 D202S mutation was anticipated to improve NADPH utilization by removal of the negatively charged carboxylate adjacent to the 2′-phosphate and possibly reduce NADH utilization due to the potential loss of hydrogen-bonding with the 2′- and 3′-hydroxyls. Surprisingly, it resulted in an almost 2-fold increase in activity associated with NADH and it was unable to use NADPH as co-substrate, converting it into an exclusively NADH-specific enzyme (Table 4). Modelling of the sp2/AKR3C2 structure suggests that Asp202 should be in close proximity (3.4 Å) to the 2′-phosphate and that it resides on a flexible loop as observed for ctXR (Figure 1). Although it is difficult to reconcile the requirement for the carboxylate-containing residue with NADPH utilization, others have noted that protein carboxylates can form very strong hydrogen bonds with the protonated oxygen in phosphate and this may be the case with sp2 as well [50].
The carboxylate appeared to be relatively unimportant in tm1/AKR3F1 where the mutation of Glu208 into a serine has little effect on the selectivity constants (the catalytic efficiency of the enzyme using NADPH divided by the catalytic efficiency of the enzyme using NADH) (Table 4). The wild-type selectivity was 1.06 and the mutant selectivity was 1.83, indicating a slight preference for NADPH in both cases. Owing to poor sequence homology with known structures, the tm1/AKR3F1 model is partial and of poor quality in the active-site region, but tends to reveal that the glutamate resides at a further distance from the 2′-phosphate (8.7 Å) than was observed for Asp202 in sp2/AKR3C2.
Although no catalytic activity was observed for ss1 and no systematic name was issued as a result, fluorescence experiments indicate that it binds both NADH and NADPH with similar affinities (Table 4). The mutation of Glu208 into a serine was expected to disrupt hydrogen-bonding between the carboxylate and the 2′- and 3′-hydroxyls in NADH without introducing any impediments to NADPH binding. Fluorescence measurements were consistent with this and indicated little effect on the NADPH binding, which was decreased approx. 3-fold but showed an 11-fold loss for NADH (Table 4).
To determine whether a glutamate functions in recognition of the adenosine 2′-hydroxyl in other enzymes, modelling experiments were also done for the dual-specific AKR1C9, AKR1C12, AKR1C13, AKR1C19, AKR2B1, AKR2B3 and the C. parapsilosis XR enzymes. This revealed that the carboxylate approaches the 2′-hydroxyl in the AKR2B subfamily and C. parapsilosis enzymes and strongly indicates its function in NADH binding (Table 3). The remainder of the known members of the AKR2B, AKR2C and AKR2D subfamilies (primarily XRs) have a clearly conserved glutamate/aspartate in a conserved sequence context (results not shown), implying that they are also capable of using NADH, a compound with an intracellular concentration typically approx. 100-fold greater than NADPH. The physiological importance of this may be associated with the fact that these function in xylose assimilation, which can be a high flux catabolic pathway. Such processes are frequently mediated by NADH-dependent enzymes. Importantly, the NAD+ product of the reaction can be recycled by the next step in the pathway: an NAD+-dependent xylitol dehydrogenase oxidizing the xylitol to yield xylulose. Most other AKRs mediate anabolic pathways (which often use NADPH-dependent enzymes) with relatively low flux. Other members of the AKR2 family, subfamilies AKR2A (sorbitol-6-phosphate reductase and mannose-6-phosphate reductase) and AKR2E (3-dehydroecdysone 3β-reductases) lack a carboxylate and do not appear to function as XRs. Modelling does not support the carboxylate functioning in NADH binding in the AKR1C subfamily. These results, however may be due to poor sequence alignments in the region coupled with conformational flexibility in the loop. Upon examination of the AKR1C models, a non-homologous carboxylate is found to be close (<5 Å) to the 2′-phosphate in the cases of AKR1C12 (Glu276), AKR1C13 (Glu276) and AKR1C19 (Glu276) and is a possible candidate for mediating NADH binding in these enzymes.
NADPH-specific enzymes
dm1/AKR2E3, ec1/AKR3F2 and sm1/AKR3F3 are NADPH-specific despite having a carboxylate-containing residue from the pairwise sequence alignment (Figure 2B). Further investigation into the location of these carboxylates was done via modelling, and the distances separating the closest oxygens between the carboxylate and phosphate ranged from 13.9 to 22.5 Å. This suggests that their locations do not correspond well to the sequence alignments in the region (Table 3). Sequence homology is therefore not apparently reflected in structural homology in these cases. Interestingly in these three models, the aspartate or glutamate residue homologous with Glu227 in ctXR is predicted to reside in a helical region, as observed for the Asp288 in the structure of the NADPH-specific AKR11A. It therefore seems that AKRs able to utilize NADH as co-substrate appear to have the carboxylate located on a coil region instead of a helix.
Conclusions
The presence of a carboxylate-containing amino acid structurally homologous with Glu227 in ctXR appears to be an indication of NADH dependence among the AKRs. Studies described here used only sequence homologies but were still able to identify four new enzymes with this unusual ability out of seven surveyed. Unexpectedly, one which relies exclusively on NADH was found among these. This novel specificity for the AKR superfamily was shown to be dependent on an aspartate at a position poised to interact with the 2′- and 3′-adenosine hydroxyls of NADH. Previously, only a few of the approx. 120 AKRs were able to bind efficiently or utilize NADH and all were dual-specific. It appears that the success rate is substantially less than the 93–97% observed for prediction of co-substrate specificity in SDRs [27], primarily because sequence homology fails to predict structural homology in the more divergent AKRs. These new data should allow improved prediction of co-substrate specificity in AKRs and provide a basis for improved engineering of enzymes with altered co-substrate utilization.
Acknowledgments
Thanks are due to Professor Trevor Penning (Department of Pharmacology, University of Pennsylvania Medical Center, PA, U.S.A.) for discussions and maintenance of the AKR superfamily nomenclature. This work was supported by NIH (National Institutes of Health) grant GM66135 to D.K.W. and the Keck Foundation.
References
- 1.Jez J. M., Penning T. M. The aldo-keto reductase (AKR) superfamily: an update. Chem. Biol. Int. 2001;130–132:499–525. doi: 10.1016/s0009-2797(00)00295-7. [DOI] [PubMed] [Google Scholar]
- 2.Ratnam K., Ma H., Penning T. M. The arginine 276 anchor for NADP(H) dictates fluorescence fluorescence kinetic transients in 3α-hydroxysteroid dehydrogenase, a representative aldo-keto reductase. Biochemistry. 1999;38:7856–7864. doi: 10.1021/bi982838t. [DOI] [PubMed] [Google Scholar]
- 3.Neuhauser W., Haltrich D., Kulbe K. D., Nidetzky B. NAD(P)H-dependent aldose reductase from the xylose-assimilating yeast Candida tenuis. Isolation, characterization and biochemical properties of the enzyme. Biochem. J. 1997;326:683–692. doi: 10.1042/bj3260683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee J. K., Koo B. S., Kim S. Y. Cloning and characterization of the xyl1 gene, encoding an NADH-preferring xylose reductase from Candida parapsilosis, and its functional expression in Candida tropicalis. Appl. Environ. Microbiol. 2003;69:6179–6188. doi: 10.1128/AEM.69.10.6179-6188.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ellis E. M., Hayes J. D. Substrate specificity of an aflatoxin-metabolizing aldehyde reductase. Biochem. J. 1995;312:535–541. doi: 10.1042/bj3120535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu S.-Q., Jin H., Zacarias A., Srivastava S., Bhatnagar A. Binding of pyridine nucleotides to the β-subunit of the voltage-sensitive K+ channel. J. Biol. Chem. 2001;276:11812–11820. doi: 10.1074/jbc.M008259200. [DOI] [PubMed] [Google Scholar]
- 7.Fujii Y., Watanabe K., Hayashi H., Urade Y., Kuramitsu S., Kagamiyama H., Hayaishi O. Purification and characterization of rho-crystallin from Japanese common bullfrog lens. J. Biol. Chem. 1990;265:9914–9923. [PubMed] [Google Scholar]
- 8.Xu J., Li M. Auxiliary subunits of shaker-type potassium channels. Trends Cardiovasc. Med. 1998;8:229–234. doi: 10.1016/s1050-1738(98)00011-5. [DOI] [PubMed] [Google Scholar]
- 9.Yokochi N., Yoshikane Y., Trongpanich Y., Ohnishi K., Yagi T. Molecular cloning, expression, and properties of an unusual aldo-keto reductase family enzyme, pyridoxal 4-dehydrogenase, that catalyzes irreversible oxidation of pyridoxal. J. Biol. Chem. 2004;279:37377–37384. doi: 10.1074/jbc.M405344200. [DOI] [PubMed] [Google Scholar]
- 10.Sanli G., Dudley J. I., Blaber M. Structural biology of the aldo-keto reductase family of enzymes: catalysis and cofactor binding. Cell Biochem. Biophys. 2003;38:79–101. doi: 10.1385/CBB:38:1:79. [DOI] [PubMed] [Google Scholar]
- 11.Branden C. I. The TIM barrel – the most frequently occurring folding motif in proteins. Curr. Opin. Struct. Biol. 1991;1:978–983. [Google Scholar]
- 12.Khurana S., Powers D. B., Anderson S., Blaber M. Crystal structure of 2,5-diketo-D-gluconic acid reductase A complexed with NADPH at 2.1 Å resolution. Proc. Natl. Acad. Sci. U.S.A. 1998;95:6768–6773. doi: 10.1073/pnas.95.12.6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanli G., Banta S., Anderson S., Blaber M. Structural alteration of cofactor specificity in Corynebacterium 2,5-diketo-D-gluconic acid reductase. Protein Sci. 2004;13:504–512. doi: 10.1110/ps.03450704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hahn-Hagerdal B., Wahlbom C. F., Gardonyi M., van Zyl W. H., Otero R. R. C., Jonsson L. J. Metabolic engineering of Saccharomyces cerevisiae for xylose utilization. Adv. Biochem. Eng. Biotechnol. 2001;73:53–84. doi: 10.1007/3-540-45300-8_4. [DOI] [PubMed] [Google Scholar]
- 15.Kratzer R., Leitgeb S., Wilson D. K., Nidetzky B. Probing the substrate binding site of Candida tenuis xylose reductase (AKR2B5) with site-directed mutagenesis. Biochem. J. 2006;393:51–58. doi: 10.1042/BJ20050831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ikeda S., Okuda-Ashitaka E., Masu Y., Suzuki T., Wantanabe K., Nakao M., Shingu K., Ito S. Cloning and characterization of two novel aldo-keto reductases (AKR1C12 and AKR1C13) from mouse stomach. FEBS Lett. 1999;459:433–437. doi: 10.1016/s0014-5793(99)01243-0. [DOI] [PubMed] [Google Scholar]
- 17.Verduyn C., Van Kleef R., Frank J., Schreuder H., van Dijken J. P., Scheffers W. A. Properties of the NAD(P)H-dependent xylose reductase from the xylose-fermenting yeast Pichia stipitis. Biochem. J. 1985;226:669–677. doi: 10.1042/bj2260669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Verduyn C., van-Dijken J. P., Scheffers W. A. Multiple forms of xylose reductase in Pachysolen tannophilus CBS4044. FEMS Microbiol. Lett. 1985;30:313–317. [Google Scholar]
- 19.Ishikura S., Horie K., Sanai M., Matsumoto K., Hara A. Enzymatic properties of a member (AKR1C19) of the aldo-keto reductase family. Biol. Pharm. Bull. 2005;28:1075–1078. doi: 10.1248/bpb.28.1075. [DOI] [PubMed] [Google Scholar]
- 20.Kavanagh K. L., Klimacek M., Nidetzky B., Wilson D. K. Structure of xylose reductase bound to NAD+ and the basis for single and dual co-substrate specificity in family 2 aldo-keto reductases. Biochem. J. 2003;373:319–326. doi: 10.1042/BJ20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kavanagh K. L., Klimacek M., Nidetzky B., Wilson D. K. The structure of apo and holo forms of xylose reductase, a dimeric aldo-keto reductase from Candida tenuis. Biochemistry. 2002;41:8785–8795. doi: 10.1021/bi025786n. [DOI] [PubMed] [Google Scholar]
- 22.Wilson D. K., Bohren K. M., Gabbay K. H., Quiocho F. A. An unlikely sugar substrate site in the 1.65 Å structure of the human aldose reductase holoenzyme implicated in diabetic complications. Science. 1992;257:81–84. doi: 10.1126/science.1621098. [DOI] [PubMed] [Google Scholar]
- 23.Banta S., Swanson B. A., Wu S., Jarnagin A., Anderson S. Optimizing an artificial metabolic pathway: engineering the cofactor specificity of Corynebacterium 2,5-diketo-D-gluconic acid reductase for use in vitamin C biosynthesis. Biochemistry. 2002;41:6226–6236. doi: 10.1021/bi015987b. [DOI] [PubMed] [Google Scholar]
- 24.Petschacher B., Leitgeb S., Kavanagh K. L., Wilson D. K., Nidetzky B. The coenzyme specificity of Candida tenuis xylose reductase (AKR2B5) explored by site-directed mutagenesis and X-ray crystallography. Biochem. J. 2005;385:75–83. doi: 10.1042/BJ20040363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jornvall H., Hoog J. O., Persson B. SDR and MDR: completed genome sequences show these protein families to be large, of old origin, and of complex nature. FEBS Lett. 1999;445:261–264. doi: 10.1016/s0014-5793(99)00130-1. [DOI] [PubMed] [Google Scholar]
- 26.Oppermann U., Filling C., Hult M., Shafqat N., Wu X., Lindh M., Shafqat J., Nordling E., Kallberg Y., Persson B., Jornvall H. Short-chain dehydrogenases/reductases (SDR): the 2002 update. Chem. Biol. Interact. 2003;143–144:247–253. doi: 10.1016/s0009-2797(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 27.Persson B., Kallberg Y., Oppermann U., Jornvall H. Coenzyme-based functional assignments of short-chain dehydrogenases/reductases (SDRs) Chem. Biol. Interact. 2003;143–144:271–278. doi: 10.1016/s0009-2797(02)00223-5. [DOI] [PubMed] [Google Scholar]
- 28.Nakanishi M., Matsuura K., Kaibe H., Tanaka N., Nonaka T., Mitsui Y., Hara A. Switch of coenzyme specificity of mouse lung carbonyl reductase by substitution of threonine 38 with aspartic acid. J. Biol. Chem. 1997;272:2218–2222. doi: 10.1074/jbc.272.4.2218. [DOI] [PubMed] [Google Scholar]
- 29.Cho H., Oliveira M. A., Tai H. H. Critical residues for the coenzyme specificity of NAD+-dependent 15-hydroxyprostaglandin dehydrogenase. Arch. Biochem. Biophys. 2003;419:139–146. doi: 10.1016/j.abb.2003.09.019. [DOI] [PubMed] [Google Scholar]
- 30.Haecker B., Habenicht A., Kiess M., Mattes R. Xylose utilisation: cloning and characterisation of the xylose reductase from Candida tenuis. Biol. Chem. 1999;380:1395–1403. doi: 10.1515/BC.1999.179. [DOI] [PubMed] [Google Scholar]
- 31.Altschul S. F., Madden T. L., Schaffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boeckmann B., Bairoch A., Apweiler R., Blatter M. C., Estreicher A., Gasteiger E., Martin M. J., Michoud K., O'Donovan C., Phan I., et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hemsley A., Arnheim N., Toney M. D., Cortopassi G., Galas D. J. A simple method for site-directed mutagenesis using the polymerase chain reaction. Nucleic Acids Res. 1989;17:6545–6551. doi: 10.1093/nar/17.16.6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jez J. M., Schlegel B. P., Penning T. M. Characterization of the substrate binding site in rat liver 3alpha-hydroxysteroid/dihydrodiol dehydrogenase. The roles of tryptophans in ligand binding and protein fluorescence. J. Biol. Chem. 1996;271:30190–30198. doi: 10.1074/jbc.271.47.30190. [DOI] [PubMed] [Google Scholar]
- 35.Jones D. T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 36.Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 37.Bairoch A., Apweiler R., Wu C. H., Barker W. C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R., Magrane M., et al. The Universal Protein Resource (UniProt) Nucleic Acids Res. 2005;33:D154–D159. doi: 10.1093/nar/gki070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schwede T., Kopp J., Guex N., Peitsch M. C. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Guex N., Peitsch M. C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 40.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 41.Marquardt T., Kostrewa D., Balakrishnan R., Gasperina A., Kambach C., Podjarny A., Winkler F. K., Balendiran G. K., Li X. D. High-resolution crystal structure of AKR11C1 from Bacillus halodurans: an NADPH-dependent 4-hydroxy-2,3-trans-nonenal reductase. J. Mol. Biol. 2005;354:304–316. doi: 10.1016/j.jmb.2005.09.067. [DOI] [PubMed] [Google Scholar]
- 42.Kratzer R., Kavanagh K. L., Wilson D. K., Nidetzky B. Studies of the enzymic mechanism of Candida tenuis xylose reductase (AKR 2B5): X-ray structure and catalytic reaction profile for the H113A mutant. Biochemistry. 2004;43:4944–4954. doi: 10.1021/bi035833r. [DOI] [PubMed] [Google Scholar]
- 43.Jez J. M., Flynn T. G., Penning T. M. New York: Plenum Press; 1996. A Nomenclature System for the Aldoketo Reductase Superfamily, Vol. 6. [Google Scholar]
- 44.Ko J., Kim I., Yoo S., Min B., Kim K., Park C. Conversion of methylglyoxal to acetol by Escherichia coli aldo-keto reductases. J. Bacteriol. 2005;187:5782–5789. doi: 10.1128/JB.187.16.5782-5789.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yum D.-Y., Lee B.-Y., Pan J. G. Identification of the yqhE and yafB genes encoding two 2,5-diketo-D-gluconate reductases in Escherichia coli. Appl. Environ. Microbiol. 1999;65:3341–3346. doi: 10.1128/aem.65.8.3341-3346.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen J. H., Turner P. C., Rees H. H. Molecular cloning and characterization of hemolymph 3-dehydroecdysone 3β-reductase from the cotton leafworm, Spodoptera littoralis. A new member of the third superfamily of oxidoreductases. J. Biol. Chem. 1999;274:10551–10556. doi: 10.1074/jbc.274.15.10551. [DOI] [PubMed] [Google Scholar]
- 47.Lundstrom A., Kang D., Liu G., Fernandez C., Warren J. T., Gilbert L. I., Steiner H. A protein from the cabbage looper, Trichoplusia ni, regulated by a bacterial infection is homologous to 3-dehydroecdysone 3-β-reductase. Insect Biochem. Mol. Biol. 2002;32:829–837. doi: 10.1016/s0965-1748(01)00145-x. [DOI] [PubMed] [Google Scholar]
- 48.Kim S. T., Huh W. K., Lee B. H., Kang S. O. D-arabinose dehydrogenase and its gene from Saccharomyces cerevisiae. Biochem. Biophys. Acta. 1998;1429:29–39. doi: 10.1016/s0167-4838(98)00217-9. [DOI] [PubMed] [Google Scholar]
- 49.Ehrensberger A., Wilson D. K. Expression, crystallization and activities of the two family 11 aldo-keto reductases from Bacillus subtilis. Acta Crystallogr. Sect. D Biol. Crystallogr. 2003;59:375–377. doi: 10.1107/s0907444902072001. [DOI] [PubMed] [Google Scholar]
- 50.Luecke H., Quiocho F. A. High specificity of a phosphate transport protein determined by hydrogen bonds. Nature. 1990;347:402–406. doi: 10.1038/347402a0. [DOI] [PubMed] [Google Scholar]