

Abstract
Cholesterol oxidase is a monomeric flavoenzyme that catalyses the oxidation of cholesterol to cholest-5-en-3-one followed by isomerization to cholest-4-en-3-one. The enzyme from Brevibacterium sterolicum contains the FAD cofactor covalently bound to His121. It was previously demonstrated that the H121A substitution results in a ≈100 mV decrease in the midpoint redox potential and a ≈40-fold decrease in turnover number compared to wild-type enzyme [Motteran, Pilone, Molla, Ghisla and Pollegioni (2001) Journal of Biological Chemistry 276, 18024–18030]. A detailed kinetic analysis of the H121A mutant enzyme shows that the decrease in turnover number is largely due to a corresponding decrease in the rate constant of flavin reduction, whilst the re-oxidation reaction is only marginally altered and the isomerization reaction is not affected by the substitution and precedes product dissociation. The X-ray structure of the mutant protein, determined to 1.7 Å resolution (1 Å≡0.1 nm), reveals only minor changes in the overall fold of the protein, namely: two loops have slight movements and a tryptophan residue changes conformation by a rotation of 180° about χ1 compared to the native enzyme. Comparison of the isoalloxazine ring moiety of the FAD cofactor between the structures of the native and mutant proteins shows a change from a non-planar to a planar geometry (resulting in a more tetrahedral-like geometry for N5). This change is proposed to be a major factor contributing to the observed alteration in redox potential. Since a similar distortion of the flavin has not been observed in other covalent flavoproteins, it is proposed to represent a specific mode to facilitate flavin reduction in covalent cholesterol oxidase.
Keywords: covalent flavin, flavoprotein, kinetics, protein structure, redox, structure–function relationships
Abbreviations: CO, cholesterol oxidase; BCO, cholesterol oxidase from Brevibacterium sterolicum containing FAD covalently linked to the enzyme; SCO, cholesterol oxidase from Streptomyces hygroscopicus containing FAD non-covalently linked to the enzyme; PEG, poly(ethylene glycol); rmsd, root mean square deviation; VAO, vanillyl-alcohol oxidase
INTRODUCTION
The flavoprotein CO (cholesterol oxidase, EC 1.1.3.6) is the first enzyme in the pathway of cholesterol degradation in a number of soil bacteria. It is an alcohol dehydrogenase/oxidase that catalyses the dehydrogenation of C(3)-OH of the cholestane system to yield the corresponding carbonyl product, and the subsequent isomerization of the cholest-5-en-3-one to cholest-4-en-3-one. During the reductive half-reaction, the oxidized FAD accepts a hydride from the alcohol, and in the ensuing oxidative half-reaction, the reduced flavin transfers the redox equivalents to molecular oxygen, the final acceptor.
CO has a broad range of biotechnological applications. It is widely used in analytical chemistry, in particular in clinical laboratories for the determination of serum cholesterol levels and as a probe to study membrane structure (for a review see [1]). Previously, COs have been found to exhibit larvicidal activity and are being developed as an insecticide against Coeloptera [2]. In addition, recent gene disruption studies of the CO gene in the pathogenic bacteria Rhodococcus equi have shown that the enzyme is required for infection of the host macrophage [3]. Since COs are unique to bacterium, they represent a potential target for new antibiotics.
CO exists in two forms, one where the FAD cofactor is non-covalently bound to the protein [exemplified by SCO (the Streptomyces hygroscopicus CO)] and one where the cofactor is linked to the enzyme via a histidine residue [exemplified by BCO (the Brevibacterium sterolicum CO), the enzyme used in the present study]. The non-covalent form of the enzyme is a member of the GMC (glucose/methanol/choline) oxidoreductase family [4]. The structures of the non-covalent form from R. equi (formerly classified as Brevibacterium sterolicum [5]) and Streptomyces sp. have been determined crystallographically [6–9]. The CO containing covalently bound FAD has a different primary sequence and its three-dimensional structure belongs to the subfamily of VAO (vanillyl-alcohol oxidase), a group of enzymes that contain a fold proposed to favour covalent flavinylation [10]. The covalent and non-covalent COs also differ from each other in many properties, e.g. spectral properties, the Kd for sulfite binding and the midpoint redox potentials [11]. Although the two enzymes have similar rates of flavin reduction (≈230 s−1 in the presence of 1% Thesit and 10% propan-2-ol), the turnover number at infinite oxygen and cholesterol concentrations is 2-fold higher for SCO than for BCO (202 s−1 and 105 s−1 respectively) and BCO catalysis proceeds via a Ping Pong mechanism, whereas SCO follows a sequential pathway [12]. In the last few years, extensive kinetic and mutagenesis studies on the non-covalent enzyme have been undertaken to elucidate the mechanism of substrate dehydrogenation [8,13–16].
Up to now, more than 30 flavoenzymes have been reported to contain flavin covalently linked to a histidine, cysteine or tyrosine residue of the polypeptide chain [17]. The covalent flavin–protein linkage has been proposed to associate with different functions [16]: it allows saturation of the active site with the cofactor, it plays a role in fine-tuning the redox properties and in substrate specificity, it protects the coenzyme from modification and inactivation, it modulates the electron transfer to another coenzyme, and finally it affects the protein structure (as a way to improve its stability). To elucidate the function of covalent flavin linkage in BCO, we have previously performed a functional characterization of a mutant enzyme in which the substitution of His121 (corresponding to His69 in the mature form) with an alanine prevents formation of the histidyl-FAD bond [18]. The mutant enzyme retains catalytic activity, but with a turnover rate decreased ≈40-fold. Stabilization of the flavin semiquinone and binding of sulfite are markedly decreased, and this result correlates with a lower midpoint redox potential (−204 mV compared to −101 mV for wild-type BCO). An increase in midpoint redox potential and in catalytic activity was obtained by reconstitution of BCO apoprotein with the ‘high potential’ flavin analogue 8-chloro-FAD. We thus concluded that the flavin 8α-linkage to a (N1)histidine is a pivotal factor in the modulation of the redox properties of this CO to increase its oxidative power [18].
In order to further probe the function of His121 in modulating redox activity for the covalent BCO we have pursued structural studies of the H121A mutant enzyme and performed a detailed investigation of its kinetic properties. The present study demonstrates that the covalent linkage between His121 and FAD results in a distortion of the isoalloxazine ring moiety to more closely resemble that expected for the reduced enzyme.
EXPERIMENTAL
Structure solution and refinement
Wild-type and mutant BCO were a gift from Roche Molecular Biochemicals. Crystals were grown by microseeding in hanging drops over a precipitant solution composed of 10–12% (w/v) PEG [poly(ethylene glycol)] 8000, 15% glycerol and 74 mM MnSO4 in 100 mM sodium cacodylate buffer at pH 5.2. Equal volumes of protein at 8.7 mg/ml in 10 mM Hepes (pH 7.0) and precipitant solution were mixed for each drop. The drops were streaked with a cat whisker carrying microseeds [19]. Crystallization trays were incubated at 17 °C. Rod-shaped crystals suitable for diffraction were obtained after 2 weeks; the largest crystals measured 0.5 mm×0.1 mm×0.1 mm.
Crystals used for data collection were transferred to a cryoprotectant solution containing the precipitant where the glycerol concentration was increased to 20%. Individual crystals were flash frozen as propane popsicles and stored in liquid nitrogen until they were used for diffraction experiments.
Diffraction data to 1.7 Å (1 Å=0.1 nm) resolution were obtained from a single crystal on an ADSC CCD Quantum detector at Beamline 5.0.1 at the Advanced Light Source (Lawrence Berkeley Laboratories) and processed with the HKL2000 programme suite [20]. The crystal was maintained at 100 K in a nitrogen gas cryostream during data collection. The complete dataset consisted of 182 frames of 1 degree each. The total exposure time was 4 h. Early in the data collection the region of the crystal exposed to the X-ray beam underwent a bleaching from its original yellow colour. This suggested that the enzyme was undergoing reduction in the X-ray beam, however the extent of reduction could not be monitored.
With the exception of the addition of glycerol to the precipitant solution, the crystallization conditions for the H121A mutant protein were similar to that of the wild-type enzyme, crystallized in the P21 space group [21]. Despite these similarities however, the mutant enzyme crystallized in the C2 space group, with cell dimensions a=140.6 Å, b=85.9 Å, c=78.8 Å and β=112.5°. The asymmetric unit of this lattice could hold one or two molecules of a 62 kDa protein corresponding to a Matthews coefficient of 3.5 and 1.8 or a solvent content of 65% or 30% respectively. Both are within the ranges usually observed for globular proteins.
The structure was solved by molecular replacement using chain A of the wild-type BCO as the search model [10]. The FAD, ligands and water molecules were excluded and, for those side chains modelled in two conformations, only one was chosen. The rotation function was computed using data between 15 and 4 Å resolution with the program CNS [22]. The cross rotation search gave a strong signal, 5.3 times above the second peak in the rotation function. Translation search with this orientation again resulted in one significant signal, seven times the height of the second peak. This confirmed that only one protein molecule was present in the crystal asymmetric unit, in contrast to the wild-type P21 crystals, which contained two enzyme molecules in the asymmetric unit.
The solution from molecular replacement was used as the starting model for crystallographic refinement. Initially refinement was performed using data to 2.2 Å. FAD was included in the model based on the difference electron density maps. For subsequent refinement, the data was extended to 1.6 Å resolution. Molecular replacement and earlier stages of refinement were carried out using the CNS software suite [22] and modelling was performed with the program O [23]. During the later stages of refinement, the program REFMAC from the CCP4 program suite [24] was used and rebuilding performed with XFIT from the Xtalview package [25]. Side chains in alternate conformation were added and the current model contains 44 residues with alternate conformations. The current model contains 536 amino acid residues, one FAD molecule, 696 water molecules, 2 cacodylate molecules, 3 glycerol molecules and two manganese ions. Table 1 gives the data processing and refinement statistics.
Table 1. Data processing and crystallographic refinement statistics.
| Data processing | |
| Resolution (Å) | 72.55–1.6 (1.66–1.6 Å) |
| Number of Reflections | 103989 |
| I/σ | 18.1 (5.1) |
| Completeness (%) | 96.2 (95.1) |
| R merge | 0.094 (0.549) |
| Wilson B factor | 10.965 (from Wilson) |
| Crystallographic refinement | |
| R factor (1.642–1.6Å) | 0.148 (0.180) |
| Free R factor (5% of data) | 0.167 (0.191) |
| rmsd bond length | 0.008 |
| rmsd bond angle | 1.231 |
| rmsd torsion | 5.914 |
| rmsd chiral | 0.073 |
| rmsd planarity | 0.005 |
| Average B factor (main chain) | 11.33 |
| Average B factor (side chain) | 15.49 |
| Average B factor (all atoms) | 13.76 |
| Number of non-hydrogen atoms | 5148 |
| Number of water molecules | 698 |
Kinetic measurements
Rapid kinetic measurements were performed as described previously [12,26] using a BioLogic SFM-300 stopped-flow instrument equipped with a 1-cm path length and interfaced to a J & M diode-array detector at 25 °C. All of the kinetic experiments were performed in 50 mM sodium pyrophosphate, (pH 7.5), 1% Thesit and 1% propan-2-ol. Spectra were recorded from the time of mixing until completion of the reaction in the wavelength range 190–700 nm and with a frequency of 1 spectrum/ms. Anaerobiosis of substrate (in the 0.05–0.75 mM range) and enzyme (6–12 μM) solutions was obtained in tonometers by repeated cycles of evacuation and flushing with O2-free argon. Oxygen was scrubbed from the stopped-flow apparatus by incubating with a dithionite solution for 16 h followed by rinsing with anaerobic buffer. As a further control of absence of oxygen from reaction mixtures, some of the experiments were also performed in the presence of 100 mM glucose, 6–50 nM glucose oxidase and 0.7 μM catalase [27]. For re-oxidation experiments, enzymes were reduced with a 2-fold excess of cholesterol under anaerobic conditions and then reacted with buffer solutions equilibrated with various N2/O2 mixtures [12,27].
Rate constants were determined from fitting of traces of absorbance at 446 nm versus time that were extracted from the spectra versus time data sets, as well as by global analysis of the same data sets using Specfit/32 software (Spectrum Software Associates, Chapel Hill, NC, U.S.A.). The same program was used for simulations of the entire spectral traces based on a three-step kinetic model for the reductive half-reaction (see eqn 1 below). Secondary kinetic data were analysed by least squares curve fitting procedures and graphics were generated with KaleidaGraph software (Synergy Software, Reading, PA, U.S.A.). Rates and dissociation constants were estimated as described in [28].
For isomerization experiments, the reaction was followed by monitoring the production of cholest-4-en-3-one from cholest-5-en-3-one spectrophotometrically at 240 nm (Δϵ=5.47 mM−1·cm−1 as measured in the stopped-flow apparatus). For the reaction starting from the reduced form, the enzyme (12.5 nM) was reduced with a 400-fold excess of cholesterol (5 μM) under anaerobic conditions and then reacted in the stopped-flow apparatus with different concentrations of cholest-5-en-3-one (0.025–0.5 mM concentration range); the reaction was followed at 240 nm up to 120 s. The isomerization reaction of the oxidized form was studied similarly, but in the presence of oxygen and in the absence of cholesterol. All concentrations mentioned in these experiments are final ones, i.e. those after mixing.
RESULTS
Kinetic course of the reductive half-reaction
The kinetic mechanism of wild-type BCO with the substrate cholesterol has been studied previously by a combination of steady-state and pre-steady-state approaches [12], and was proposed to consist of a Ping Pong (binary complex) process. The steady-state kinetic behaviour of H121A BCO was studied analogously using the enzyme monitored turnover method and with cholesterol as substrate (see Table 2) [18]. The double-reciprocal (Lineweaver–Burk) plot of the turnover number as a function of substrate concentration for the H121A BCO also gave sets of parallel lines, consistent with a Ping Pong mechanism (results not shown). Enzyme monitored turnover experiments confirmed that the absence of the covalent flavin linkage in BCO induces a large decrease in the turnover number (Table 2) [18]. In the present study, the reductive half-reaction of H121A BCO was studied by reacting the oxidized form anaerobically with various concentrations of cholesterol (0.05–0.75 mM), such that pseudo-first-order conditions were maintained [conditions: 1% propan-2-ol and 1% Thesit (pH 7.5) at 25 °C]; wild-type BCO was studied under identical conditions for comparison. In both cases no spectral changes associated with formation of the encounter complex were observed. The time course of reaction, when followed at 446 nm, for wild-type BCO can be fitted with either a single or a double exponential algorithm (yielding either kobs or kobs1+kobs2 terms). The residuals are <4% and suggest the absence of major systematic errors in the evaluation; those for a 2-exponential algorithm are superior at the onset of the reaction (up to ≈20 ms, see Figure 1a). The amplitude of the first phase represents 80–85% of the total changes, in good agreement with the assumption that it reflects conversion of the oxidized enzyme (spectrum 1 in Figure 1a inset) into the E-FADred∼product complex (spectrum 2 in Figure 1a inset, see below). Data evaluation was thus done based on a two-step process. The rates of the first phase, kobs1, exhibit saturation with increasing cholesterol concentration (Figure 2a). The rate of the second phase (kobs2) is essentially substrate independent (=3.6±0.6 s−1, see Figure 2a). This last step yields a species the spectrum of which (spectrum 3 in Figure 1a inset, see below) closely resembles that of free reduced enzyme [11,12]. This is interpreted as being approximated by a three-step sequence (see Discussion section), as represented by eqn 1 [28,29] in which kobs1 reflects k2 and kobs2 corresponds to k2′:
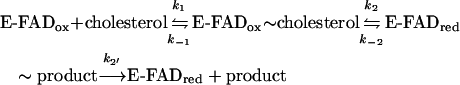 |
(1) |
Table 2. Comparison of steady-state coefficients and of reductive and oxidative half-reaction rate constants obtained for the reaction of wild-type and H121A BCO with cholesterol as substrate in stopped-flow experiments such as those of Figures 1 and 2.
Reductive half-reaction measurements were conducted in 50 mM potassium phosphate (pH 7.5), 1% propan-2-ol and 1% Thesit (some experiments also contained 100 mM glucose, 6–50 nM glucose oxidase and 0.7 μM catalase), at 25 °C. The Kd,app was obtained from the slope divided by the intercept in double-reciprocal plots of the rates of reduction versus cholesterol concentration; such a value corresponds to (k−1+k2)/k1 [it reduces to the true Kd (=k−1/k1) only when k−1≫k2]. kred is obtained from the dependence of kobs1 from [cholesterol] and k2′ corresponds to kobs2 (see Figure 2 and Eqn 1). The rate constants k1 and k−1 are estimated from the simulation of the spectral time courses using Specfit/32. The re-oxidation parameters were obtained from the double reciprocal plot reported in Figure 2(b) inset according to [12].
| Steady-state kinetics | Reductive half-reaction | Oxidative half-reaction | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Lineweaver– Burk pattern | kcat (s−1) | Km,Chol (mM) | Km,O2 (mM) | kred (∼k2) (s−1) | Kd,app (mM) | k1 (M−1·s−1) | k−1 (s−1) | k2′ (s−1) | kconv (=1/y-intercept) (s−1) | k4/k−conv (=x-intercept) (mM−1) | |
| Wild-type | Parallel* | 19.2 * | 0.21* | 0.14* | 27.2±4.5 | 0.05±0.01 | 5.7±2.1 × 105 | <0.2 | 3.6±0.6 | 170±30 | 4.8±0.8 |
| H121A | Parallel† | 0.55† | 0.47† | 0.45† | 0.31±0.06 | 0.1±0.01 | 3.0±1.2 × 103 | <0.05 | 0.04±0.01 | 75±15 | 8.2±2.0 |
Figure 1. Time courses of the anaerobic reduction of wild-type (a) and H121A (b) BCO.

Anaerobic solutions of enzymes (≈9 μM and ≈6.3 μM for wild-type and H121A BCOs respectively) and 0.7 mM cholesterol were mixed in the stopped-flow instrument, in the presence of 1% propan-2-ol/1% Thesit (pH 7.5) at 25 °C; (|) represent the data points at 446 nm. Curve (-−- −-) is the fit for a single exponential decay; curve (---) is the fit for a double exponential decay; curve (−) is the trace obtained from simulations using Specfit/32 software and based on the sequence of steps of Eqn 1, on known extinction coefficients for the oxidized and reduced enzyme forms (see insets) and on the rate constants reported in Table 2. Insets: The spectra shown are those obtained by deconvolution with Specfit/32. Spectrum 1, oxidized enzyme(s); Spectrum 2, reduced enzyme–product intermediate complex; and Spectrum 3, free reduced enzyme. The residuals are the subtraction of the experimental data points at 446 nm from the traces obtained from fit or simulation procedures.
Figure 2. Dependence of the rates of flavin reduction and re-oxidation for wild-type and mutant enzymes.

(a) Dependence of the observed rates of flavin reduction for wild-type BCO from the cholesterol concentration. The observed rate constants are obtained from fits of the absorbance change at 446 nm using a double exponential algorithm as detailed in the text (conditions as detailed in the legend of Figure 1). The data points are: (●) the values of kobs1, the rates corresponding to the transformation of species (1) into species (2) (inset of Figure 1a); (○) the values of kobs2, the rates corresponding to the transformation of species (2) into species (3) (inset of Figure 1a). Where error bars are not shown the data scatter is smaller than the size of the symbols. (b) Dependence of the rates of flavin re-oxidation for wild-type (●) and H121A BCO (○) from the oxygen concentration. The observed rate constants were obtained from fits of traces reflecting the absorbance increase at 446 nm (the time courses are essentially monophasic). Where error bars are not shown the data scatter is smaller than the size of the symbols. Inset: double reciprocal plot of the same data as reported in the main graph.
The proposed kinetic model for the reductive half-reaction is validated by the results of simulations of the spectral traces (Software Specfit/32, Figure 1a, see Table 2 for the corresponding rate constants). The spectra obtained by deconvolution analysis (see Figure 1a inset) are compatible with the species (spectrum 2) being that of the E-FADred∼product complex and the species (spectrum 3) that of free reduced enzyme. It should be stated that the second (slow) phase can be fitted equally well with a biexponential algorithm such as required for two reactions occurring in parallel, where the slow phase would belong for example to an enzyme species having substantially reduced activity. However, and in contrast to the observation, in such a case a dependence of the rate of kobs2 from [substrate] might be expected. This lends support to the scheme of eqn (1), but clearly cannot exclude other kinetic alternatives. We attribute the failure to locate such a phase in previous experiments [12,18] to the superior quality of the data obtained in the present study with a diode-array photometer and to the corresponding global fitting analysis method.
In the case of the H121A BCO mutant, a fit of the experimental traces at 446 nm for the reductive half-reaction based on a biexponential term is required only at high cholesterol concentration (>0.5 mM) where the separation between the rates is relatively large. The maximal rate of flavin reduction estimated at high [cholesterol] is ≤0.5 s−1, i.e. significantly lower than that observed for the wild-type enzyme. As with wild-type BCO (see Experimental section for details) the spectral courses at different cholesterol concentrations were simulated using Specfit/32 (global analysis) based on the set-up of eqn (1). The trace shown in Figure 1(b) was obtained using the rate constants reported in Table 2; it does not show relevant trends in the residuals, indicating a reasonable correspondence of model and experimental data. The simulation results also show that the decrease in the rate of enzyme reduction (k2) for the H121A mutant enzyme compared to wild-type BCO is accompanied by a decrease in rate constants for substrate binding/dissociation (k1 and k−1) and in the rate constant for the second phase of the reductive half-reaction (k2′).
Kinetic course of the oxidative half-reaction
The re-oxidation reaction of BCO was studied in the stopped-flow spectrophotometer by following the appearance, in the spectrum, of the oxidized enzyme when the uncomplexed reduced enzyme was treated with varying oxygen concentrations in buffer solutions containing 1% Thesit and 1% propan-2-ol. With up to 21% oxygen saturation, the time courses of flavin re-oxidation are similar for wild-type and H121A BCO, whereas at higher oxygen saturation the reaction is slower for the mutant. For both BCO forms, the rate of re-oxidation shows saturation with increasing oxygen concentration (Figure 2b), in agreement with the inferred presence of an intermediate [12]:
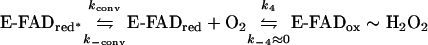 |
(2) |
The y-axis-intercept of the corresponding double reciprocal plot (inset of Figure 2b) yields 1/kconv and the x-axis-intercept corresponds to −k4/k−conv. These values are only slightly modified by the H121A substitution (Table 2).
The isomerisation reaction
The Δ5–6→Δ4–5 isomerization reaction that is represented by eqn 3:
 |
(3) |
can be followed directly by monitoring the absorbance changes at 240 nm that accompany the formation of cholest-4-en-3-one, the final product. With SCO this reaction is catalysed by both the oxidized and the reduced forms, although at a 2000-fold lower rate for the latter [12,28]. Firstly, and in order to assess whether reduced BCO catalyses the isomerization reaction, wild-type and H121A enzymes (2 μM) were reduced anaerobically by the addition of a 25-fold excess of cholesterol. The spectral course of the isomerization reaction was then followed in a diode array spectrophotometer for up to 30 min upon addition of 50 μM cholest-5-en-3-one. The extent of absorbance increase at 240 nm was similar for both BCO forms and the spectra collected during the reaction course confirm that the flavin remains in the reduced state during the entire process for both BCO forms.
The kinetics of the isomerization reaction was then studied using the stopped-flow instrument by mixing the oxidized or the reduced forms of wild-type and H121A BCOs with increasing concentrations of cholest-5-en-3-one under the conditions used for the reductive half-reaction (see above). The rates extracted from the 240 nm absorbance versus time traces are plotted in Figure 3 as a function of [cholest-5-en-3-one] (in the 0.025–0.5 mM range). The results summarized in Table 3 show that the rate of the isomerization reaction is not affected by the presence of the H121-flavin covalent bond and by the state of oxidation of the flavin itself. Comparison of the rate constants for the isomerization reaction (Table 3) with those for the dehydrogenation of cholesterol (Table 2), show that for H121A BCO the Δ5–6→Δ4–5 rearrangement is not rate-limiting, whereas it is close to the rate of flavin reduction for the wild-type BCO at saturating substrate concentration [12,28].
Figure 3. Dependence of the observed rate of isomerization of cholest-5-en-3-one into cholest-5-en-4-one from the substrate concentration for the oxidized (●) and reduced (○) forms of H121A BCO.
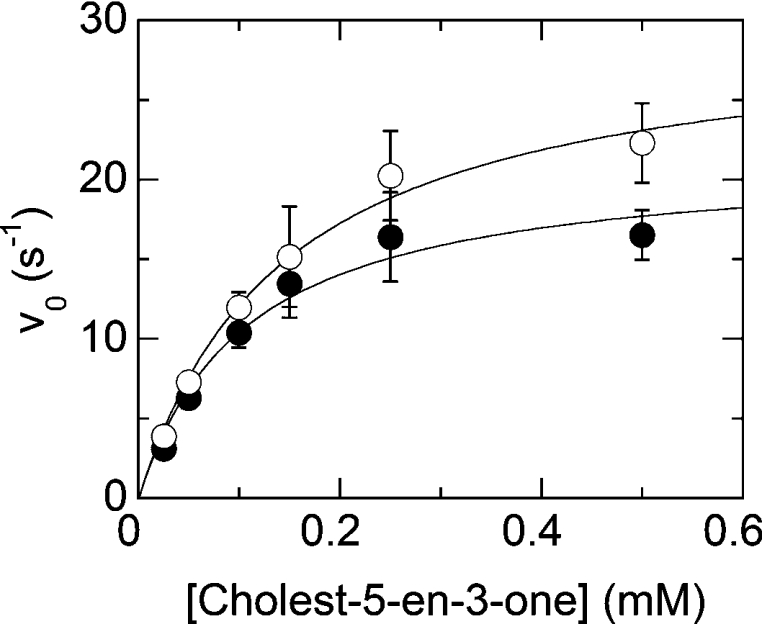
The data points (vo) are the rates of the absorbance changes at 240 nm that reflect isomerization as obtained in stopped-flow experiments (see text for details). Where error bars are not shown the data scatter is smaller than the size of the symbols.
Table 3. Comparison of steady-state parameters obtained for the isomerization reaction of wild-type and H121A BCO with cholest-5-en-3-one as substrate in stopped-flow experiments.
Measurements were in 50 mM potassium phosphate buffer (pH 7.5), 1% propan-2-ol, 1% Thesit and 100 mM glucose (with the addition of 6 nM glucose oxidase and 0.7 μM catalase for the measurements starting from the reduced enzyme form), at 25 °C.
| kcat (s−1) | Km,chol-5-en-3-one (mM) | ||
|---|---|---|---|
| Wild-type | Oxidized | 32.3±3.5 | 0.14±0.04 |
| Reduced | 23.3±2.5 | 0.17±0.01 | |
| H121A | Oxidized | 21.5±1.9 | 0.11±0.03 |
| Reduced | 29.8±1.8 | 0.14±0.02 |
Crystallography
In order to ascertain what structural effects the removal of the covalent linkage has on the enzyme, we undertook a crystallographic study of the mutant protein. Surprisingly, under identical crystallization conditions, the mutant enzyme crystallized in a different space group from that of the wild-type enzyme, suggesting some possible significant changes on the surface of the protein that affect the packing of the molecules in the crystal. Analysis of the structure revealed that the overall fold of the mutant form is identical to the native enzyme. The mutant protein crystallizes with one molecule in the asymmetric unit of the C2 cell whereas the native BCO crystallizes with two molecules in the asymmetric unit of the P21 cell. Comparison of the packing for the native and mutant structures shows that the non-crystallographic dimer of the native structure becomes a crystallographic dimer in the mutant structure. Identical crystal contacts, utilizing two manganese atoms and an arsenate molecule, are maintained in both crystal forms. A superposition of secondary structure elements between the native and the mutant protein resulted in an rmsd (root mean square deviation) of 0.4 Å. The region of the structure that lies on the side of the dimethylbenzene ring of the FAD makes the most significant movement (in particular the loops consisting of residues 116–128) and appears to open somewhat relative to the wild-type enzyme structure. This is assigned to the absence of the covalent linkage between His121 and the FAD molecule. Other significant movements from the native position occur in loops 74–90 and 422–431 (Figures 4A and 4B).
Figure 4. Comparison of the structures of wild-type and H121A BCO.

(a) Stereo figure showing a superposition of the trace of wild-type BCO and the H121A mutant enzyme. The three loops that exhibit the largest change between the two structures are coloured in magenta (for the mutant BCO) and cyan (for the wild-type BCO). The remaining structural elements are coloured in light (mutant enzyme) and dark (wild-type BCO) grey. The FAD molecules are coloured in the same way as the loops of the corresponding structure. The superposition was carried out using the SSM algorithm [46] incorporated into COOT [47]. (b). Stereo view of key residues in the loop regions that have altered positions in the native and mutant enzyme structure. The colouring is as detailed above. The side-chains for His121, Ala121 (mutant BCO), Trp80 and Arg429 for both structures are included. (c) Side view of the superposed isoalloxazine moiety for the native enzyme (cyan) and the H121A mutant BCO (magenta). The atoms in the pyrimidine ring were used for the superposition.
Both the mutant protein crystals and the native crystals rapidly bleach upon exposure to X-rays most likely due to formation of the reduced flavin. Given that both forms of the enzyme undergo bleaching, a direct comparison of the structures is appropriate and relevant as they both reflect the reduced enzyme form. The electron density map at position 121 of the mutant enzyme structure clearly shows the absence of the histidine side-chain and the subsequent loss of a covalent linkage between the protein and C8 of the cofactor (Figure 5A). Furthermore, the electron density features for the FAD cofactor are very clearly delineated (Figure 5B). A comparison of the isoalloxazine ring system of the FAD cofactor for the native and mutant protein structures reveals significant differences (Figure 4C). In the native enzyme the isoalloxazine ring adopts a butterfly twist about the N5 and N10 atoms: in particular the dimethylbenzene ring is twisted significantly away from planarity. This twist is a consequence of the covalent linkage between the 8-methyl group of the dimethylbenzene ring and the ND1 atom of His121; the electron density for the H121A mutant structure clearly reveals a planar isoalloxazine ring system (Figure 4C).
Figure 5. The 2 Fo−Fc electron density map for the H121A mutant protein.

The map, contoured at 1.5 σ, shows density features for (a) the FAD cofactor and residue 121 and (b) a side view of the isoalloxazine ring moiety.
Other significant movements in the structure of the mutant enzyme are apparent in three loop regions situated near the dimethylbenzene ring system of the cofactor (77–86, 120–127 and 423–430) (Figure 4B). The loop containing residues 120–127 includes the mutated histidine residue. The largest movement in this loop is the position of a glycine residue directly following position 121, thus redirecting the main chain for the mutant protein structure. This deviation from the native structure repositions the carbonyl oxygen atom of Ala121 to make a hydrogen bond to O3 of the ribityl chain of FAD. In the native structure this main chain oxygen atom is rotated away from the ribityl chain and makes a hydrogen bond with a water molecule which in turn hydrogen bonds to the main chain of Ser81. Thus the bound water in the native structure is displaced by the repositioned main chain, and the carbonyl oxygen atom of Ala121 in the mutant protein structure rotates to hydrogen bond directly with the ribityl chain of the cofactor (Figure 6). This conformational change in the main chain also results in a repositioning of Cα of Gly122 nearer to the C8M carbon of the dimethylbenzene ring of the cofactor (4.6 Å relative to 6.2 Å for the wild-type enzyme). Interestingly, Arg117 of this region becomes sensitive to trypsin cleavage in the unfolding intermediate obtained at 2 M urea [30].
Figure 6. Interactions in the region of residues 121 and 122.

Stereo view showing the intramolecular interactions in the region of 121–122 for (a) the native enzyme and (b) the mutant BCO. Hydrogen bonds are depicted as green broken lines.
A second loop (residues 77–86) behind that of 120–127 also undergoes a conformational shift, placing the main chain region at the tip of the loop approximately 2 Å closer to the dimethylbenzene ring of the cofactor. In addition, the side chain of Trp80 undergoes a rotation about χ2 of 180° compared to the native structure (Figure 4B). Thus although the indole ring is still positioned perpendicular to the C8M atom of the cofactor, the opposite side of the ring now faces the cofactor compared to the native enzyme. This rotation of the side chain fills up the volume occupied by the histidine residue in the native structure. The side chain of Trp80 is sandwiched between the methyl groups of the flavin cofactor and the side chain of Arg429. The reorientation of Trp80 is correlated with a reorientation of Arg429 such that the guanidinium group π stacks over the aromatic indole ring system. The alterations of the tertiary structure induced by the removal of the flavin-His121 covalent link are reflected by the spectral properties. Thus the tryptophan fluorescence emission maximum is shifted from 331 to 335 nm and increased in intensity in the mutant enzyme. The latter exhibits differences in the near-UV CD spectra such as a higher ellipticity at 285 nm and a lower one at 253 nm [30].
DISCUSSION
In an attempt to understand the factors that govern the redox chemistry of CO we undertook the present detailed kinetic and structural study of a mutant of BCO where the histidine residue responsible for the covalent linkage between the enzyme and the cofactor was replaced with an alanine (H121A). Analysis of the structure revealed that the overall fold of the H121A mutant enzyme is identical to that of the native BCO: a superposition of secondary structure elements between the native and the mutant BCO resulted in an rmsd of 0.4 Å. The most significant movements are observed in the region close to the dimethylbenzene ring of the FAD, in particular the loops consisting of residues 74–90, 116–128 and 422–431.
As deduced from previous kinetic measurements, H121A BCO works by a binary complex mechanism, similar to that reported for native enzyme [12], however a large decrease in the turnover number is observed for the mutant enzyme [18]. In the present study we demonstrate that the decrease in activity is mainly due to a ≥80-fold decrease in the rate of flavin reduction/cholesterol dehydrogenation, k2 in eqn (1). The oxidative half-reaction of wild-type BCO is quite complex since the saturation behaviour of the observed rate constants for re-oxidation indicates the presence of a definite intermediate preceding re-formation of the oxidized form (eqn 2). However, the reactivity of the reduced enzyme with oxygen is only slightly altered by the replacement of His121 with an alanine (2.5 fold change in the observed parameters, see Table 2). Km and Kd,app for cholesterol are also slightly altered by removal of the flavin-His121 covalent bond. A significant decrease in the magnitude of the rate of substrate binding to the oxidized form of the enzyme is evident for the H121A mutant compared to wild-type enzyme. Interestingly, there are no changes in the position of active-site residues that can affect substrate binding. Based on the high average temperature factors, the loop connecting α7 and β14 (residues 423–430) in wild-type BCO was proposed as a possible entrance for cholesterol into the active site [10]. Figure 4(B) shows a movement of this loop in the H121A protein (note the reorientation of Arg429) compared to the wild-type enzyme and thus could be responsible for the slower substrate binding/product dissociation observed in this mutant BCO. In line with the absence of main changes in the active site among wild-type and H121A forms, the presence of the flavin covalent bond appears not to affect the rates of isomerization and the corresponding Km values. In contrast to the case of non-covalent COs (e.g. SCO) [12], in BCO the isomerization reaction is catalysed with a similar efficiency by the reduced and oxidized forms (Table 3). It is also faster than the second phase observed in the reductive half-reaction experiments (k2′ in eqn 1) (Table 2). This second phase could be due to processes involved in release of product(s), however its exact attribution is uncertain in view of the complexity of the system (see above), and is beyond the scope of the present study. These results indicate that the chemical step of the Δ5–6→Δ4–5 rearrangement (kisom in eqn 4) is not altered by the geometry of the flavin cofactor (see below) and occurs prior to release of the final product(s) (k3 in eqn 4):
 |
(4) |
Since the catalytic mechanism proposed for BCO [12] is of the Ping Pong type this requires k3 to be faster than kcat, i.e. that release of the final product cholest-4-en-3-one from the reduced enzyme via k3 is not rate-limiting. Consequently the oxidative half-reaction starts from the free reduced enzyme (k4 in eqn 4 and eqn 2). Based on this we conclude that the main effect of the flavin covalent attachment to His121 is on the redox potential of the flavin itself that in turn affects the rates of substrate dehydrogenation.
A crystallographic analysis of the mutant protein was carried out to a significantly high resolution to visualize structural details that may be associated with the differences in redox properties of the enzyme. As the mutant protein crystal was exposed to X-rays a rapid bleaching was observed. The identical phenomenon was observed for the native crystals when cryoprotected with glycerol. This photobleaching has been reported for ribonucleotide reductases, where the rate of reduction was highly dependent on the concentration of glycerol, an effective electron hole-trapping agent [31,32]. We have also observed a bleaching in a glycerol concentration dependent fashion for the non-covalent form of CO (P. Lario and A. Vrielink, unpublished work). As this bleaching most likely reflects the presence of the reduced enzyme, a comparison can be made between the native and H121A structures in the reduced enzyme form: a significant difference is observed in the planarity of the isoalloxazine ring system of the cofactor (Figure 4C).
A number of covalently linked flavoenzymes have been structurally characterized recently [10,33–36]. The factors that modulate redox activity and, particularly, the role that the covalent modification may play in the redox activity of the cofactor are a focus of study for this sub-class of enzymes. Of the covalent flavoenzymes where the reduction potentials are known [1,37–40], BCO has the lowest Em. Comparisons of the redox potentials in the presence and absence of the covalent linkage have only been carried out for VAO and BCO [18,37]. In the case of VAO the midpoint redox potential is +55 mV for the native enzyme and −65 mV for a mutant enzyme where the covalent linkage has been removed (H422A), this corresponding to a decrease of around 110 mV [37]. Structural analysis of this mutant enzyme shows no changes to the conformation of the flavin ring system. In the case of BCO, the difference of the Em between covalently and noncovalently bound FAD is of the order of 100 mV (−101 mV for wild-type BCO compared with −204 mV for the H121A mutant enzyme). The differences in Em corresponding to approx. 100 mV between the two enzymes are very similar and could therefore be attributed to the covalent linkage. It should be recalled that the substitution at position 8α with a histidine residue induces a change in the flavin Em of ≈50 mV (−209 and −154 mV for FAD and 8-imidazolylriboflavin respectively) [41,42]. This effect is, however, pH dependent and reflects the state of ionization of the imidazole substituent. Since the latter can be influenced by the protein environment, it is difficult to differentiate between effects originating with the 8α-substitution and effects brought about by the protein environment, indeed these effects will be interconnected, at least to some extent. A quantitative interpretation of the 100 mV difference in midpoint redox potential in terms of reaction rates (changes in free energy) is not straight-forward. To our knowledge within flavoproteins, the most comprehensive treatment of the dependence of the rate(s) of flavin reduction from the redox potential of the flavin cofactor itself is from Yorita et al. [43]. Therein it is shown that with the enzyme lactate oxidase there is a linear dependence on Em of the flavin coenzyme, although upper limits/plateaus occur that probably correspond to rate control by diffusion. The slopes of the dependence(s) varies markedly depending on the substrate (e.g. Δk/100 mV≈1000 for L-lactate and ≈15 for L-mandelate) [43]. No molecular explanation was put forward to rationalize this observation in molecular terms. However, it appears reasonable to assume that electronic effects will not necessarily be translated quantitatively into effects on rates. The extent of the effect might depend strongly on the geometry of the transition state(s) and on the mode(s) of orbital reorganization during the redox process(es). In conclusion the effect estimated in the present work appears to be intermediate to those reported by Yorita et al. [43] and might be interpreted assuming that the redox potential is an important contributor, albeit not the sole contributor, to the modulation of the rate of substrate dehydrogenation.
It is the sum of many effects exerted by the microenvironment around the FAD cofactor that modulates its redox potential. VAO has a number of interactions between the isoalloxazine ring system and the protein: a hydrogen bond between the O2 of FAD and the guanidinium moiety of Arg504 and an interaction between N5 of the FAD and the carboxylate of Asp170. Mutation of Asp170 to a serine residue significantly decreases the midpoint redox potential of the enzyme [44]. In contrast, for BCO there are relatively few contacts between the isoalloxazine ring system and the protein. Only the pyrimidine N3, O2 and O4 make hydrogen bond contacts to main chain oxygen and nitrogen atoms; N5 has no hydrogen bond partner. This suggests that modulation of the redox potential is substantially different between VAO and BCO. In the latter, the finding of an Em for the noncovalent flavin corresponding to that of free FAD suggests that the effect of the protein is a minor one. On the other hand, for BCO, the covalent linkage between His121 and the flavin could play a role in modulating the geometry of the isoalloxazine ring and in fixation of the flavin such that the chemistry involving the cofactor becomes more facile, i.e. in decreasing the activation energy needed for reduction of the flavin. Theoretical studies by Miller and Walsh [45] have shown that flavin reduction potentials are dependent on the conformation of the isoalloxazine ring; a planar conformation is energetically preferred for the oxidized species and a bent conformation is preferred for the reduced state. Our structural studies support these theoretical findings. The protein therefore acts to fine-tune the environment around the isoalloxazine moiety in order to optimize the orientation required for orbital overlap during hydride transfer (as made evident by the observed decrease in the rate of flavin reduction following the removal of the flavin covalent link). In the case of BCO, His121 facilitates maintaining the isoalloxazine ring system in a bent conformation more favorable for the reduced enzyme. The absence of this covalent linkage in the mutant structure, and the rearrangement of the protein environment around the dimethylbenzene ring of FAD (movement of Trp80 and the loops around the dimethylbenzene portion), results in the cofactor adopting a less favourable planar conformation in the reduced state. A difference in the flavin geometry for the native and mutant enzymes is not seen for other covalently linked flavoenzymes: it is probably due to the specific FAD-apoprotein interactions that finely modulate the redox properties in different flavoenzymes.
Acknowledgments
We thank Artem Lyubimov (Molecular, Cell and Developmental of Biology, University of California, Santa Cruz, CA, U.S.A.) and Ibrahim Moustafa (Department of Chemistry and Biochemistry, University of California, Santa Cruz, CA, U.S.A.) for numerous useful discussions. In addition we thank Artem Lyubimov for technical assistance. This work is supported by NIH (National Institutes of Health) Grant GM63262 (to A.V.), by FAR 2003 and 2004, and Fondazione Cariplo 2004 (to L.P.), and by a grant from the Deutsche Forschungsgemeinschaft to S.G. (Gh 2/13-2).
References
- 1.MacLachlan J., Wotherspoon A. T. L., Ansell R. O., Brooks C. J. W. Cholesterol oxidase: sources, physical properties and analytical applications. J. Steroid Biochem. Mol. Biol. 2000;72:169–195. doi: 10.1016/s0960-0760(00)00044-3. [DOI] [PubMed] [Google Scholar]
- 2.Corbin D. R., Greenplate J. T., Purcell J. P. The identification and development of proteins for control of insects in genetically modified crops. HortScience. 1998;33:614–617. [Google Scholar]
- 3.Navas J., Gonzalez-Zorn B., Ladron N., Garrido P., Vazquez-Boland J. A. Identification and mutagenesis by allelic exchange of choE, encoding a cholesterol oxidase from the intracellular pathogen Rhodococcus equi. J. Bacteriol. 2001;183:4796–4805. doi: 10.1128/JB.183.16.4796-4805.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J., Vrielink A., Brick P., Blow D. M. Crystal structure of cholesterol oxidase complexed with a steroid substrate: Implications for flavin adenine dinucleotide dependent alcohol oxidases. Biochemistry. 1993;32:11507–11515. [PubMed] [Google Scholar]
- 5.Ladron N., Fernandez M., Aguero J., Gonzalez Zorn B., Vazquez-Boland J. A., Navas J. Rapid identification of Rhodococcus equi by a PCR assay targeting the choE gene. J. Clin. Microbiol. 2003;41:3241–3245. doi: 10.1128/JCM.41.7.3241-3245.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vrielink A., Lloyd L. F., Blow D. M. Crystal structure of cholesterol oxidase from Brevibacterium sterolicum refined at 1.8Å resolution. J. Mol. Biol. 1991;219:533–554. doi: 10.1016/0022-2836(91)90192-9. [DOI] [PubMed] [Google Scholar]
- 7.Yue Q. K., Kass I. J., Sampson N. S., Vrielink A. Crystal structure determination of cholesterol oxidase from Streptomyces and structural characterization of key active site mutants. Biochemistry. 1999;38:4277–4286. doi: 10.1021/bi982497j. [DOI] [PubMed] [Google Scholar]
- 8.Lario P. I., Sampson N., Vrielink A. Sub-atomic resolution crystal structure of cholesterol oxidase: what atomic resolution crystallography reveals about enzyme mechanism and the role of the FAD cofactor in redox activity. J. Mol. Biol. 2003;326:1635–1650. doi: 10.1016/s0022-2836(03)00054-8. [DOI] [PubMed] [Google Scholar]
- 9.Coulombe R., Yue K. Q., Ghisla S., Vrielink A. Oxygen access to the active site of cholesterol oxidase through a narrow channel is gated by an Arg-Glu pair. J. Biol. Chem. 2001;276:30435–30441. doi: 10.1074/jbc.M104103200. [DOI] [PubMed] [Google Scholar]
- 10.Fraaije M. W., van Berkel W. J. H., Benen J. A. E., Visser J., Mattevi A. A novel oxidoreductase family sharing a conserved FAD-binding domain. Trends Biochem. Sci. 1998;23:206–207. doi: 10.1016/s0968-0004(98)01210-9. [DOI] [PubMed] [Google Scholar]
- 11.Gadda G., Wels G., Pollegioni L., Zucchelli S., Ambrosius D., Pilone M. S., Ghisla S. Characterization of cholesterol oxidase from Streptomyces hygroscopicus and Brevibacterium sterolicum. Eur. J. Biochem. 1997;250:369–376. doi: 10.1111/j.1432-1033.1997.0369a.x. [DOI] [PubMed] [Google Scholar]
- 12.Pollegioni L., Wels G., Pilone M. S., Ghisla S. Kinetic mechanisms of cholesterol oxidase from Streptomyces hygroscopicus and Brevibacterium sterolicum, Eur. J. Biochem. 1999;263:1–13. doi: 10.1046/j.1432-1327.1999.00586.x. [DOI] [PubMed] [Google Scholar]
- 13.Kass I. J., Sampson N. S. Evaluation of the role of His447 in the reaction catalyzed by cholesterol oxidase. Biochemistry. 1998;37:17990–18000. doi: 10.1021/bi982115+. [DOI] [PubMed] [Google Scholar]
- 14.Kass I. J., Sampson N. S. The isomerization catalyzed by Brevibacterium sterolicum cholesterol oxidase proceeds stereospecifically with one base. Biochem. Biophys. Res. Commun. 1995;206:688–693. doi: 10.1006/bbrc.1995.1097. [DOI] [PubMed] [Google Scholar]
- 15.Yin Y., Sampson N. S., Vrielink A., Lario P. I. The presence of a hydrogen bond between asparagine 485 and the pi system of FAD modulates the redox potential in the reaction catalyzed by cholesterol oxidase. Biochemistry. 2001;40:13779–13787. doi: 10.1021/bi010843i. [DOI] [PubMed] [Google Scholar]
- 16.Kass I. J., Sampson N. S. The importance of Glu361 position in the reaction catalyzed by cholesterol oxidase. Bioorg. Med. Chem. Lett. 1998;8:2663–2668. doi: 10.1016/s0960-894x(98)00478-8. [DOI] [PubMed] [Google Scholar]
- 17.Mewies M., McIntire W. S., Scrutton N. S. Covalent attachment of flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN) to enzymes: The current state of affairs. Protein Sci. 1998;7:7–20. doi: 10.1002/pro.5560070102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Motteran L., Pilone M. S., Molla G., Ghisla S., Pollegioni L. Cholesterol oxidase from Brevibacterium sterolicum – The relationship between covalent flavinylation and redox properties. J. Biol. Chem. 2001;276:18024–18030. doi: 10.1074/jbc.M010953200. [DOI] [PubMed] [Google Scholar]
- 19.Stura E., Wilson I. A. Analytical and production seeding techniques, Methods: A companion to methods in enzymology 1. 1990:38–49. [Google Scholar]
- 20.Otwinowski Z., Minor W. Methods in Enzymology. In: Carter C. W. J., Sweet R. M., editors. Boston: Academic Press; 1997. pp. 307–325. [Google Scholar]
- 21.Croteau N., Vrielink A. Crystallization and preliminary X-ray analysis of cholesterol oxidase from Brevibacterium sterolicum containing covalently bound FAD. J. Struct. Biol. 1996;116:317–319. doi: 10.1006/jsbi.1996.0047. [DOI] [PubMed] [Google Scholar]
- 22.Brünger A. T., Adams P. D., Clore G. M., DeLano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J.-S., Kuszewski J., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., Warrem G. L. Crystallography and NMR System: A new software suite for macromolecular structure determination. Acta Crystallogr. 1998;D54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 23.Jones T. A., Zou J.-Y., Cowan S. W., Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. 1991;A47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 24.Murshudov G. N., Vagin A. A., Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystall. D Biol. Crystallography. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 25.McRee D. E. XtalView/Xfit – A versatile program for manipulating atomic coordinates and electron density. J. Struct. Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- 26.Boselli A., Sacchi S., Job V., Pilone M. S., Pollegioni L. Role of Tyr238 in the active site of Rhodotorula gracilis D-amino acid oxidase. A site-directed mutagenesis study. Eur. J. Biochem. 2002;269:4762–4771. doi: 10.1046/j.1432-1033.2002.t01-1-03173.x. [DOI] [PubMed] [Google Scholar]
- 27.Pollegioni L., Langkau B., Tischer W., Ghisla S., Pilone M. S. Kinetic mechanism of D-amino acid oxidases from Rhodotorula gracilis and Trigonopsis variabilis. J. Biol. Chem. 1993;268:13850–13857. [PubMed] [Google Scholar]
- 28.Strickland S., Palmer G., Massey V. Determination of dissociation constants and specific rate constants of enzyme-substrate (or protein-ligand) interactions from rapid reaction kinetic data. J. Biol. Chem. 1975;250:4048–4052. [PubMed] [Google Scholar]
- 29.Fierke C. A., Hammes G. G. Transient kinetic approaches to enzyme mechanisms. Methods Enzymol. 1995;249:3–37. doi: 10.1016/0076-6879(95)49029-9. [DOI] [PubMed] [Google Scholar]
- 30.Caldinelli L., Iametti S., Barbiroli A., Bonomi F., Fessas D., Molla G., Pilone M. S., Pollegioni L. Dissecting the structural determinants of the stability of cholesterol oxidase containing covalently bound flavin. J. Biol. Chem. 2005;280:22572–22581. doi: 10.1074/jbc.M500549200. [DOI] [PubMed] [Google Scholar]
- 31.Davydov R., Kuprin S., Gräslund A., Ehrenberg A. Electron paramagnetic resonance study of the mixed-valent diiron center in Escherichia coli ribonucleotide reductase produced by reduction of radical-free protein R2 at 77 K. J. Am. Chem. Soc. 1994;116:11120–11128. [Google Scholar]
- 32.Eriksson M., Jordan A., Eklund H. Structure of Salmonella typhimurium nrdF ribonucleotide reductase in its oxidized and reduced forms. Biochemistry. 1998;37:13359–13369. doi: 10.1021/bi981380s. [DOI] [PubMed] [Google Scholar]
- 33.Mattevi A., Fraaije M. W., Mozzarelli A., Olivi L., Coda A., van Berkel W. J. H. Crystal structures and inhibitor binding in the octameric flavoenzyme vanillyl-alcohol oxidase: the shape of the active-site cavity controls substrate specificity. Structure. 1997;5:907–920. doi: 10.1016/s0969-2126(97)00245-1. [DOI] [PubMed] [Google Scholar]
- 34.Cunane L. M., Chen Z. W., Shamala N., Mathews F. S., Cronin C. N., McIntire W. S. Structures of the flavocytochrome p-cresol methylhydroxylase and its enzyme-substrate complex: gated substrate entry and proton relays support the proposed catalytic mechanism. J. Mol. Biol. 2000;295:357–374. doi: 10.1006/jmbi.1999.3290. [DOI] [PubMed] [Google Scholar]
- 35.Trickey P., Basran J., Lian L. Y., Chen Z., Barton J. D., Sutcliffe M. J., Scrutton N. S., Mathews F. S. Structural and biochemical characterization of recombinant wild type and a C30A mutant of trimethylamine dehydrogenase from methylophilus methylotrophus (sp. W(3)A(1)) Biochemistry. 2000;39:7678–7688. doi: 10.1021/bi9927181. [DOI] [PubMed] [Google Scholar]
- 36.Trickey P., Wagner M. A., Jorns M. S., Mathews F. S. Monomeric sarcosine oxidase: structure of a covalently flavinylated amine oxidizing enzyme. Structure. 1999;7:331–345. doi: 10.1016/s0969-2126(99)80043-4. [DOI] [PubMed] [Google Scholar]
- 37.Fraaije M. W., van den Heuvel R. H. H., van Berkel W. J. H., Mattevi A. Covalent flavinylation is essential for efficient redox catalysis in vanillyl-alcohol oxidase. J. Biol. Chem. 1999;274:35514–35520. doi: 10.1074/jbc.274.50.35514. [DOI] [PubMed] [Google Scholar]
- 38.Gutman M., Bonomi F., Pagani S., Cerletti P., Kroneck P. Modulation of the flavin redox potential as mode of regulation of succinate dehydrogenase activity. Biochim. Biophys. Acta. 1980;591:400–408. doi: 10.1016/0005-2728(80)90171-1. [DOI] [PubMed] [Google Scholar]
- 39.Barber M. J., Pollock V., Spence J. T. Microcoulometric analysis of trimethylamine dehydrogenase. Biochem. J. 1988;256:657–659. doi: 10.1042/bj2560657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gomez-Moreno C., Choy M., Edmondson D. E. Purification and properties of the bacterial flavoprotein: thiamin dehydrogenase. J. Biol. Chem. 1979;254:7630–7635. [PubMed] [Google Scholar]
- 41.Williamson F., Edmondson D. E. Effect of pH on oxidation-reduction potentials of 8 α-N-imidazole-substituted-flavins. Biochemistry. 1985;24:7790–7797. doi: 10.1021/bi00347a043. [DOI] [PubMed] [Google Scholar]
- 42.Edmondson D. E., Newton-Vinson P. The covalent FAD of monoamine oxidase: structural and functional role and mechanism of the flavinylation reaction. Antioxid. Redox Signal. 2001;3:789–806. doi: 10.1089/15230860152664984. [DOI] [PubMed] [Google Scholar]
- 43.Yorita K., Misaki H., Palfey B. A., Massey V. On the interpretation of quantitative structure-function activity relationship data for lactate oxidase. Proc. Natl. Acad. Sci. U.S.A. 2000;97:2480–2485. doi: 10.1073/pnas.040559797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van den Heuvel R. H. H., Fraaije M. W., Mattevi A., van Berkel W. J. H. Asp-170 is crucial for the redox properties of vanillyl-alcohol oxidase. J. Biol. Chem. 2000;275:14799–14808. doi: 10.1074/jbc.275.20.14799. [DOI] [PubMed] [Google Scholar]
- 45.Walsh J. D., Miller A. F. Flavin reduction potential tuning by substitution and bending. J. Mol. Struct. 2003;623:185–195. [Google Scholar]
- 46.Krissinel E., Henrick K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2003;60:2256–2268. doi: 10.1107/S0907444904026460. [DOI] [PubMed] [Google Scholar]
- 47.Emsley P., Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]