

Abstract
In the present study, we aimed to decipher the mechanisms involved in the transcriptional effect of insulin on the SREBP-1c specific promoter of the human srebf-1 gene. Using luciferase reporter gene constructs in HEK-293 cells (human embryonic kidney cells), we demonstrated that the full effect of insulin requires the presence of SREs (sterol response elements) in the proximal region of the promoter. Furthermore, insulin increases the binding of SREBP-1 (sterol-regulatory-element-binding protein-1) to this promoter region in chromatin immunoprecipitation assay. We also found that the nuclear receptors LXRs (liver X receptors) strongly activate SREBP-1c gene expression and identified the LXRE (LXR-response element) involved in this effect. However, our results suggested that these LXREs do not play a major role in the response to insulin. Finally, using expression vectors and adenoviruses allowing ectopic overexpressions of the human mature forms of SREBP-1a or SREBP-1c, we demonstrated the direct role of SREBP-1 in the control of SREBP-1c gene expression in human skeletal-muscle cells. Altogether, these results strongly suggest that the SREBP-1 transcription factors are the main mediators of insulin action on SREBP-1c expression in human tissues.
Keywords: insulin, liver X receptor (LXR), promoter, sterolregulatory-element-binding protein-1c (SREBP-1c), sterol-response element, transcription
Abbreviations: ACC-2, acetyl-CoA carboxylase-2; ChIP, chromatin immunoprecipitation; ER, endoplasmic reticulum; FAS, fatty acid synthase; GSK3, glycogen synthase kinase 3; HEK-293 cells, human embryonic kidney cells; HKII, hexokinase II; Insig, insulin-induced gene; LDL, low-density lipoprotein; LXR, liver X receptor; LXRE, LXR-response element; NF-Y, nuclear factor-Y; RT–qPCR, reverse transcription–quantitative PCR; SREBP, sterol-regulatory-element-binding protein; SCAP, SREBP cleavage-activating protein; SRE, sterol-response element
INTRODUCTION
Regulation of gene expression is a major action of insulin in all its target tissues [1], as it has been highlighted in a recent report showing the changes in the mRNA levels of approx. 800 genes during a 3 h insulin infusion in the skeletal muscle of healthy individuals [2]. Several studies have evidenced important defects in the capability of insulin to regulate gene expression in peripheral tissues in Type II (non-insulin-dependent) diabetes mellitus [3,4]. Understanding how insulin exerts its transcriptional actions appears thus as a key step in the quest of the molecular causes of insulin resistance. Up to now, the transcriptional mechanisms involved in the effects of insulin on gene expression are still poorly known. Studies of the promoter regions of well-characterized insulin-regulated genes, such as FAS (fatty acid synthase) [5], ACC-2 (acetyl-CoA carboxylase-2) [6] or HKII (hexokinase II) [7], led to the conclusion that SREBP-1c (sterol-regulatory-element-binding protein-1c) is one of the key transcription factors mediating the effects of insulin [8,9].
The SREBPs are membrane-bound transcription factors of the bHLH-Zip (basic–helix–loop–helix-leucine zipper) family that have been shown to regulate gene expression of several enzymes implicated in cholesterol, lipid and glucose metabolism [9,10]. To date, three members of the SREBP family, SREBP-1a, SREBP-1c and SREBP-2, have been characterized. SREBP-1a and SREBP-1c differ in their N-terminal regions due to alternative usage of exon 1a (coding for 30 amino acids) or exon 1c (coding for six amino acids) of the srebf-1 gene under the control of specific promoters. SREBP-2 is produced from a distinct gene [11]. All SREBPs are synthesized as transcriptionally inactive precursors that are bound to the ER (endoplasmic reticulum) and nuclear envelope [11]. They are activated by proteolytic cleavage in the Golgi apparatus to produce the N-terminal mature transcription factors that migrate into the nucleus where they can bind to SREs (sterol-response elements) in the promoter region of target genes to modulate their transcription [11,12].
A large number of studies has demonstrated that SREBP-1c is tightly regulated by nutritional and hormonal status, especially at the transcriptional level, in various tissues [13,14]. Fasting decreases SREBP-1c mRNA and protein levels, whereas they are markedly increased upon feeding a high-carbohydrate diet [13,14]. Insulin itself was shown to be a potent inducer of SREBP-1c transcription in various cell models and in rodent tissues, including liver, adipose tissue and skeletal muscle [15,16]. To date, the mechanism by which insulin triggers the transcription of SREBP-1c is not fully defined. Studies of mouse and rat SREBP-1c promoters have shown that several motifs are likely to be involved for the full activation by insulin, corresponding to response elements for LXRs (liver X receptors) [LXREs (LXR-response elements)], Sp1 (stimulating protein-1), NF-Y (nuclear factor-Y) as well as for SREBPs [17–19]. The presence of SREs suggests thus that SREBP-1c gene promoter can be directly activated by nuclear SREBPs in an auto-regulatory loop [17]. In addition, the transcription of SREBP-1c can also be induced by the activation of the nuclear receptors LXRs that have been implicated in the control of lipid and cholesterol metabolism [20]. LXRs can directly promote SREBP-1c transcription through two LXRE-binding sites in the mice SREBP-1c promoter [21], and synthetic LXR agonists up-regulate SREBP-1c gene expression both in vivo in rodents [18] and in vitro in cell models, including human muscle cells [22,23]. Furthermore, investigating mouse SREBP-1c promoter in rat primary hepatocytes, Chen et al. [24] reported that LXRs might play a central role in insulin-mediated activation of SREBP-1c transcription. Their study led to the conclusion that insulin may induce SREBP-1c expression through the production of an unknown ligand that activates LXRs and consequently SREBP-1c promoter transcription, while SREBPs and NF-Y play only permissive roles [24].
Only few studies have been performed in tissues or cells from human origin. We have shown that hyperinsulinaemia increases SREBP-1c gene expression in vivo in skeletal muscle and in adipose tissue [3]. Moreover, insulin induces SREBP-1 protein in primary culture of human skeletal muscle [7]. However, although the organization of the SREBP-1c-proximal promoter is grossly similar in rodents and humans, differences regarding the presence of additional SRE motifs in the human gene might suggest distinct regulatory mechanisms. Therefore the aim of the present study was to investigate the regulation of the human SREBP-1c promoter in order to decipher the transcriptional mechanisms triggered by insulin in human cells. Our results indicate that insulin controls SREBP-1c transcription through SREs and that LXRs do not seems to play a major role in this effect. This suggests that insulin activation of SREBP-1c gene expression in human skeletal muscle is mainly the result of SREBP action itself.
MATERIALS AND METHODS
Materials
Human insulin was purchased from Sigma (L'Isle d'Abeau, France). The synthetic LXR agonist T0901317 was a gift from Dr D. Pruneau (Fournier-Pharma, Daix, France). Restriction enzymes and molecular biology products were from Promega (Charbonnières, France) and Roche Diagnostics (Meylan, France). Plasmids for cell transfection were prepared using Plasmid Midi kit (Qiagen, Courtaboeuf, France).
Subjects
For the study of SREBP-1 isoform mRNA expression in skeletal muscle and adipose tissue, we used total RNA preparations of tissues samples obtained from eight healthy lean volunteers who participated in a global study on insulin action on gene expression [2,3]. All participants gave their written consent after being informed of the nature, purpose and possible risks of the study. The experimental protocol (‘Clamp-Gene Study’, agreement number 2003-039/125A) was approved by the Ethical Committees of the Hospices Civils de Lyon and performed according to the French legislation (Huriet law). The description of the subjects enrolled in the study, including their anthropometric and metabolic parameters, has been presented in detail elsewhere [22].
To investigate insulin action on SREBP-1 mRNA and protein expression, the subjects were submitted to a 3 h euglycaemic hyperinsulinaemic clamp with an insulin infusion rate of 2 m-units·kg−1·min−1 as described previously in detail [3], and tissue biopsies were taken under local anaesthesia before and after the hyperinsulinaemic clamp, in a fasted condition [3].
Primary cultures of human skeletal-muscle cells
For the culture of skeletal-muscle cells (myotubes), muscle biopsies (∼200 mg wet weight) were taken under local anaesthesia from the vastus lateralis muscle of healthy subjects with the approval by the Ethical Committees. Differentiated myotubes were prepared according to the procedure previously described in detail [22].
Quantification of mRNAs
Total RNA from skeletal muscle biopsies were prepared according to a procedure based on the method of Chomczynski and Sacchi [25]. Total RNA from adipose tissue was prepared using the RNeasy kit (Qiagen). The mRNA concentrations of the target genes were determined by RT–qPCR (reverse transcription–quantitative PCR) using a Light-Cycler (Roche Diagnostics), as previously described in detail [2,13,22]. The list of the primers and real-time PCR assay conditions are available from E. L. upon request. The results were normalized using Cyclophilin mRNA concentration, measured as a reference gene in each sample using RT–qPCR.
Reporter plasmids and site-directed mutagenesis
A human genomic clone (NR1-B022) which contains NotI flanking regions corresponding to the SREBP-1c promoter was obtained from Professor Eugene R. Zabarovsky (Microbiology and Tumor Biology Center and Center for Genomics and Bioinformatics, Karolinska Institute, Stockholm, Sweden) [26] and further subcloned into the luciferase reporter gene vector pGL3-Enhancer (Promega). The longest fragment of the human SREBP-1c promoter (−1470/+90, named p1c-E) was used to generate p1c-E1 (−571/+90), p1c-E2 (−257/+90), p1c-E3 (−167/+90) and p1c-E4 (−73/+90) constructs by restriction enzyme digestion (PvuII, AatII, ApaI and PstI respectively). Mutations were created by site-directed mutagenesis using the QuikChange® kit (Stratagene, La Jolla, CA, U.S.A.). The sequences of the primers used to generate site-directed mutations are as follows (mutated bases are indicated in boldface): mLXRE1, 5′-GAGGGCCAGAGTCCGCCAGATTCCCCGGCA-3′; mLXRE2, 5′-GGCGGAAGTCCGCTAGATTCCCCAACCCC-3′; mSRE1, 5′-CCATTCAGCGCCGCGAGATAAAACTCGAGCCCCC-3′; and mSRE2, 5′-GGCCGCGCGCGCTTATCTCATGCCCGGCCCGC-3′. The sequence of each construct was verified by digestion and sequencing before use.
Transient transfections and luciferase reporter assays
HEK-293 cells (human embryonic kidney cells) were grown at 37 °C in an atmosphere of 5% CO2 in Dulbecco's modified Eagle's medium containing 25 mM glucose, 100 units/ml penicillin and 100 μg/ml streptomycin sulfate supplemented with 10% (v/v) fetal calf serum. Transfection studies were carried out with cells plated on to 12-well plates. HEK-293 cells were maintained in serum-free medium for 18–24 h before transfections that were made using Exgen 500 reagent (Euromedex, Souffelweyersheim, France) according to the manufacturer's instructions. Each culture well received 500 ng of a given SREBP-1c promoter/luciferase gene construct mixed with 1 ng of pRL-CMV vector (where CMV is cytomegalovirus; Promega). The cells were incubated, 6 h after transfection, with the indicated agents for 24 h [10−7 mol/l insulin, 1 μM T0901317 or 0.1% DMSO (vehicle)]. Firefly and Renilla luciferase activities (Dual luciferase reporter assay system; Promega) were then measured using a TD-20/20 luminometer (Turner Designs, Sunnyvale, CA, U.S.A.) as previously reported [7].
Human SREBP-1 expression plasmids
Expression vectors encoding human mature nuclear forms of SREBP-1a (named pCMV-hSREBP1a) and SREBP-1c (named pCMV-hSREBP1c) were generated by PCR amplification and ligation into the pcDNA3.1 expression vector (Invitrogen). The following primers were used: pCMV-hSREBP1a, 5′-GGCTGCGCCATGGACGAGCCAC-3′ for sense and 5′-AGCGGTCCAGCATGCCCCGGCTGTGC-3′ for antisense; pCMV-hSREBP1c, 5′-GCAGATCGCGGAGCCATGGATTGC-3′ for sense and 5′-AGCGGTCCAGCATGCCCCGGCTGTGC-3′ for antisense. The sequence of each construct was verified before being use. For SREBP-1a or SREBP-1c overexpression, 50 ng of pCMV-hSREBP1a or pCMV-hSREBP1c was co-transfected with the promoter construct plasmids. The amount of DNA was kept constant by adding pcDNA3.1 plasmid when necessary.
Generation of recombinant adenoviruses and infections
Recombinant adenoviral genomes carrying the human mature forms of SREBP-1a and SREBP-1c were generated by homologous recombination as described in [27]. Briefly, CaCl2 competent Escherichia coli BJ5183 were co-transformed with 200 ng of SwaI-linearized VmAdcDNA3 plasmid (a gift from Dr S. Rusconi, Fribourg, Switzerland) and 600 ng of linearized pCMV-hSREBP1a or pCMV-hSREBP1c. Recombinants were screened by PCR with the following set of primers: primer A, 5′-GACGGATGTGGCAAAAGTGA-3′ annealing to the leftmost part of the adenoviral genome; and primer B, 5′-ATGGGGTGGAGACTTGGAAATC-3′ annealing to the portion of the CMV promoter which is brought in by homologous recombination. Positive clones were further analysed by restriction analysis. Positive recombinants were large-scale amplified in E. coli XL-1 Blue, digested with PacI and transfected by the calcium phosphate methods in HEK-293 cells. Cytopathic effect due to virus production was observed 8–10 days after transfection. Adenoviruses were extracted by three freeze/thaw cycles and stored in PBS and 10% (v/v) glycerol at −20 °C. Viral titre of stocks was higher than 108 plaque-forming units/ml.
ChIP (chromatin immunoprecipitation) assay
ChIP assays were performed as previously reported but with modifications [13]. For each experimental condition, four confluent 10 cm plates of HEK-293 cells were used. Cells were cross-linked for 5 min at room temperature (21 °C) with 1% formaldehyde and the reaction was stopped by adding glycine at a final concentration of 127 mM. After 5 min, cells were scraped and washed with cold PBS containing proteinase inhibitor (1 mM PMSF). Preparation and enzymatic fragmentation of chromatin was performed using the Enzymatic Shearing kit (Active Motif, Rixensart, Belgium) according to the manufacturer's instructions. An aliquot was used to verify the fragmentation on agarose gels and to quantify the amount of DNA. For the ChIP assays, 50 μg of digested chromatin was first precleared for 1 h with Protein A–Sepharose beads (Amersham Biosciences, Orsay, France). After centrifugation, supernatants were incubated with 7 μg of anti-SREBP-1 antibody (H160) or anti-SREBP-2 (H164) or rabbit IgG (all three from Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.) or no antibody (mock condition), and incubated overnight at 4 °C under rotative agitation. The immunoprecipitated complexes were bound to Protein A–Sepharose beads during 3 h at 4 °C and washed successively in a RIPA buffer (10 mM Tris/HCl, 1 mM EDTA, 1% Triton X-100, 0.1% SDS, 140 mM NaCl, 0.1% sodium deoxycholate and 1 mM PMSF, pH 7.4), LiCl buffer (1 mM Tris/HCl, 250 mM LiCl, 0.5% Nonidet P40, 0.5% sodium deoxycholate and 1 mM EDTA, pH 7.4) and Tris–EDTA buffer (1 mM EDTA and 10 mM Tris/HCl, pH 7.4). Proteins were then eliminated using 200 μg of proteinase K (Promega) in the presence of 10% (w/v) SDS by overnight incubation at 37 °C. Chromatin DNA was extracted with phenol/chloroform followed by ethanol precipitation. Samples were dissolved in 80 μl of water. The sets of PCR primers used for the analysis of the human SREBP-1c-proximal promoter were 5′-GCTCAGGGTGCCAGCGAACCAGTG-3′ for sense and 5′-GGGTTACTAGCGGACGTCCGCC-3′ for antisense. Primer sets for the analysis of the SREBP-1a-proximal promoter were 5′-GGGACCCCTATAACTTGGATCC-3′ for sense and 5′-CTCGCGCACAATGGCAGCGGCCTCTG-3′ for antisense. Primers sets for the analysis of the FAS-proximal promoter were 5′-CGACGCTCATTGGCCTGG-3′ for sense and 5′-CTGCCGTCTCTCTGGCTC-3′ for antisense. Primers sets for the analysis of the LDL (low-density lipoprotein) receptor-proximal promoter were 5′-CACTTTCGAAGGACTGGAGTGG-3′ for sense and 5′-CCACGTCATTTACAGCATTTC-3′ for antisense. Primers sets for the analysis of a distal region of SREBP-1 gene (negative control) were 5′-CCCACTTCATCAAGGCAGACTCGC-3′ for sense and 5′-CGACCATGTGGACTGTTGCCAAGATG-3′ for antisense. PCR amplification products were analysed on ethidium bromide-stained 3%-(w/v)-agarose gels.
SREBP-1 protein analysis by Western blotting
Cells were lysed at 4 °C in 200 mM NaF, 20 mM NaH2PO4, 150 mM NaCl, 50 mM Hepes, 4 mM Na3VO4, 10 mM EDTA, 1% Triton X-100, 10% glycerol and 2 mM PMSF, and proteins were quantified by the Bradford assay (Bio-Rad). Aliquots of proteins (40 μg for cell extracts) were separated by SDS/10% PAGE and transferred to PVDF membrane. Efficiency of the transfer was confirmed by Ponceau staining. Membranes were then incubated with SREBP-1 antibody (H160; Santa Cruz Biotechnology). When indicated, Insig-1 (insulin-induced gene 1) protein was also analysed (Insig-1 antibody N-19; Santa Cruz Biotechnology). The signal was detected using a horseradish peroxidase-conjugated secondary antibody and revealed with the enhanced chemiluminescence system (Pierce, Rockford, IL, U.S.A.). After analysis, the membranes were stripped with the Re-Blot Plus solution (Chemicon International, Temecula, CA, U.S.A.) and blotted with the α-tubulin antibody (TU-02; Santa Cruz Biotechnology) to normalize the protein amount.
Statistical analysis
All results in the text and Figures are presented as means±S.E.M. Statistical significance of the results was determined using the Student's t test. The threshold for significance was set at P<0.05.
RESULTS
Effects of 3 h insulin perfusion on the expression of SREBP-1 isoforms in skeletal muscle and adipose tissue
The mRNA levels of SREBP-1a and SREBP-1c, the two isoforms produced by the srebf-1 gene, were quantified using RT–qPCR in tissues of healthy subjects, before and at the end of a 3 h hyperinsulinaemic clamp. Figure 1 shows that insulin infusion significantly increased SREBP-1c mRNA levels both in skeletal muscle and in subcutaneous abdominal adipose tissue, in agreement with a previous report [3]. In contrast with SREBP-1c, the expression of SREBP-1a was only marginally affected during the clamp, with no change in skeletal muscle and only a slight increase in adipose tissue. These results indicated therefore that insulin preferentially affects the transcriptional regulation of SREBP-1c in human tissues. In order to define its mechanisms of action, we investigated the regulation of the SREBP-1c promoter region of the human srebf-1 gene.
Figure 1. Effects of insulin infusion on the mRNA levels of SREBP-1 isoforms in skeletal muscle and adipose tissue.
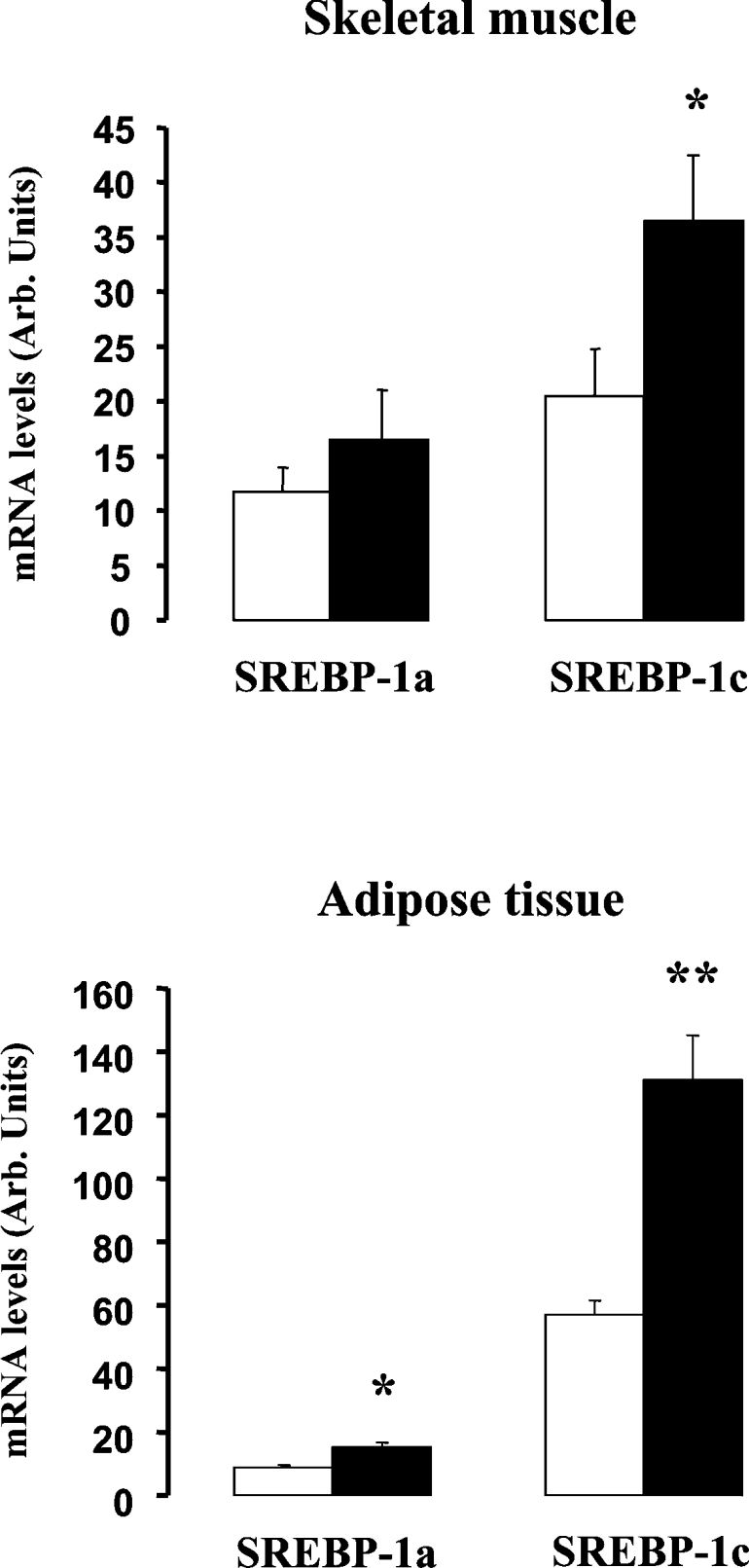
The mRNA levels of SREBP-1a and SREBP-1c were determined by quantitative real-time PCR in skeletal-muscle (upper panel) and adipose tissue (lower panel) biopsies of healthy subjects before (open bars) and at the end (black bars) of a 3 h hyperinsulinaemic euglycaemic clamp. Results are presented as means±S.E.M. *P<0.05 after 3 h of insulin infusion versus before 3 h of insulin infusion; **P<0.01 after 3 h of insulin infusion versus before 3 h of insulin infusion.
Functional characterization of the human SREBP-1c promoter and effect of insulin
It was also noticeable in Figure 1 that SREBP-1c expression was lower in skeletal muscle than in adipose tissue. The observed acute response during hyperinsulinaemic clamp (Figure 1), together with in vivo data in rodents during nutritional transitions [13], indicate that SREBP-1c expression is tightly regulated in muscle, even if this tissue possesses only limited lipogenic capacities. This might suggest additional roles for SREBP-1c in skeletal muscle other than the control of lipogenesis-related genes [3]. Up to now, the regulation of SREBP-1 has been mostly studied in cells and tissues characterized by high rate of lipogenesis, such as hepatocytes or adipocytes. In the present study, to investigate the regulation of the human SREBP-1c promoter, we decided to utilize a human cell line with similar expression patterns of the SREBP-1 variants and comparable response to insulin with that of skeletal muscle (i.e. similar mRNA levels of SREBP-1a and SREBP-1c, less than 2-fold induction of SREBP-1c by insulin, and no induction of SREBP-1a). Among different cell lines, HEK-293 cells showed very similar SREBP-1 expression and regulation to what was observed in muscle (Figure 2, inset). This cell line is also easily transfectable and widely used for promoter studies and hormone action investigations.
Figure 2. Activation of human SREBP-1c promoter by insulin.

HEK-293 cells were transfected with a luciferase reporter gene driven by different constructs of the human SREBP-1c gene promoter. Cells were incubated for 24 h with (black bars) or without insulin (10−7 mol/l) (open bars). Relative luciferase activity was calculated using a dual luciferase assay as indicated in the Materials and methods section. Results are expressed by reference to the basal luciferase activity of p1c-E and presented as the means±S.E.M. for at least three independent transfection experiments. *P<0.05 in the presence of insulin versus basal conditions. Inset: effect of insulin (10−7 mol/l) on the mRNA levels of SREBP-1a, SREBP-1c and SREBP-2 in HEK-293 cells. The mRNA levels were determined by quantitative real-time PCR. Results are presented as means±S.E.M. (n=4). *P<0.05 compared with untreated cells.
Figure 2 shows the activity of various fragments of the 5′-flanking region of the human SREBP-1c promoter in HEK-293 cells. For the presentation of the results, the luciferase activity of the different constructs, measured in the absence or in the presence of insulin, was expressed by reference to p1c-E, the longest promoter fragment (−1470/+90). In the absence of insulin, the deletion of the 5′-end dramatically reduced luciferase activity when the sequence between −71 and −257 bp was deleted (Figure 2), indicating that this region contains elements that are essential for the basal activity of the SREBP-1c promoter. Insulin treatment for 24 h (10−7 M) significantly increased (1.8±0.2-fold, P=0.01) luciferase activity of the p1c-E construct (Figure 2). This induction was similar to what was observed for the stimulation of SREBP-1c mRNA by insulin in HEK-293 cells (Figure 2, inset). This stimulation by insulin was also observed with constructs p1c-E1 (1.6±0.1-fold, P<0.05) and p1c-E2 (1.6±0.2-fold, P<0.05), whereas further deletion of the 5′-end (p1c-E3) completely abolished insulin effect (Figure 2). These results indicated thus that the region spanning −257 to −167 bp contains cis-acting sequences responsible for the response of the human SREBP-1c promoter to insulin.
Effects of mutations of LXREs on SREBP-1c promoter activity
Two neighbouring LXREs (located at −311/−296 bp and at −260/−245 bp) were present in the −571/−257 bp region that appeared to be essential for the basal promoter activity. Their implication in the basal activity and/or in the response to insulin was investigated by site-directed mutagenesis (Figure 3). Mutation of the distal (p1c-E1m1), the proximal (p1c-E1m2) or both (p1c-E1m1/2) LXREs significantly reduced the basal activity (∼4-fold) of SREBP-1c promoter. This inhibitory effect was similar to the effect of the −257 deletion observed with the p1c-E2 construct (Figure 2). Treatment of the cells with T0901317 (1 μM; 24 h), a synthetic activator of the LXRs, produced a more than 3-fold increase in the basal activity of SREBP-1c promoter (Figure 3A). Mutations of the two LXREs totally abolished the effect of T0901317, demonstrating the role of these motifs in the LXR response. The individual mutations of the LXREs also reduced the effect of the LXR agonist, but this was more pronounced when the proximal LXRE (–260/−245) was modified (Figure 3A). These results indicate that the two LXR-binding sites are functional and play an important role in the basal activity of the human SREBP-1c promoter.
Figure 3. Mutations of the LXREs in the SREBP-1c promoter reduce its response to T0901317 but not to insulin.

The luciferase reporter gene p1c-E1 and LXRE mutated constructs were obtained as described in the Materials and methods section. After transfection, HEK-293 cells were incubated for 24 h with or without T0901317 (1 μM) (A) and with or without insulin (10−7 mol/l) (B). Results are expressed by reference to the basal activity of p1c-E1 and are presented as the means±S.E.M. (n=3). *P<0.05 in the presence of T0901317 or insulin versus basal conditions.
The same mutated constructs were then used to investigate the role of the LXREs in the effect of insulin. Figure 3(B) clearly shows that the mutations of the LXREs did not modify the stimulatory effect of insulin on SREBP-1c promoter activity. Despite major reduction of the basal activity, insulin significantly increased luciferase activity with all the mutated constructs to a similar extent to what was obtained with the wild-type construct (1.5±0.1-fold with p1c-E1; 1.5±0.2-fold with p1c-E1m1; 1.5±0.1-fold with p1c-E1m2; and 1.5±0.1-fold with p1c-E1m1/2). These results demonstrated therefore that the functional LXREs are not involved in the stimulatory effect of insulin on the human SREBP-1c promoter.
Effect of mutations in the SREs on SREBP-1c promoter activity
Data from Figure 2 suggested that insulin response relies on cis-acting elements that are located within the −257/−73 region of the SREBP-1c promoter. Two putative SREs are located in this region, at −228/−218 bp and at −127/−117 bp from the transcription start. The sequences of theses motifs (TTCACCCCGC and CTCACCCCAT for the distal and the proximal motifs respectively) are close to the consensus sequence ATCACCCCAC (number M00221 in Transfac) for SREs. Figure 4 shows the effect of mutations of these two potential SREBP binding sites. The mutations of the SREs (alone or in combination) had only minor effect on the basal activity of the p1c-E2 construct, but they completely suppressed the stimulatory effect of insulin (Figure 4). These results strongly suggested that the SREs motifs are directly involved in the action of insulin on the human SREBP-1c gene promoter.
Figure 4. Mutations of the SREs in the proximal SREBP-1c promoter suppress transcriptional stimulation by insulin.

The luciferase reporter gene p1c-E2 and SRE mutated constructs were obtained as described in the Materials and methods section. After transfection, HEK-293 cells were incubated for 24 h with or without insulin (10−7 mol/l). Results are expressed by reference to the basal activity of p1c-E2 and are presented as the means±S.E.M. (n=3). *P<0.05 in the presence of insulin versus basal conditions.
SREBPs proteins bind the SREs on the SREBP-1c promoter
As SREs are binding sites for SREBPs, we further aimed to demonstrate that SREBP-1 could actually bind to and activate the SREBP-1c promoter. ChIP assay was used to demonstrate the interaction between SREBP-1 and the promoter region of SREBP-1c in HEK-293 cells. Chromatin was extract from untreated and insulin-treated cells (10−7 M for 6 h), and the H160 SREBP-1 polyclonal antibody was used to immunoprecipitate the fragmented chromatin before PCR amplifications with srebf-1 gene-specific primers. As identified motifs on the human SREBP-1c promoter are also potential targets for SREBP-2 and because SREBP-2 is expressed in the HEK-293 cells (Figure 2, inset), we also made ChIP experiments using anti-SREBP2 antibody. In addition, control experiments were done using the well-characterized LDL receptor and FAS promoters. The quantitative aspect of the PCR amplifications was verified using serial dilution of the input, as shown in the bottom panel of Figure 5.
Figure 5. ChIP assay of SREBP-1 protein association with the SREBP-1c promoter.

HEK-293 cells were incubated for 6 h in control medium or in medium supplemented with insulin (10−7 mol/l). After cross-linking chromatin DNA to the interacting proteins, specific immunoprecipitations with anti-SREBP-1 or anti-SREBP-2 or non-specific (IgG) antibodies were performed as indicated in the Materials and methods section. The PCR products were generated by the amplifications of the SREBP-1c promoter, the SREBP-1a promoter, the FAS promoter, the LDL receptor (LDLr) promoter and the SREBP-1 exons 4/5 (negative control). Amplification products were resolved in 3%-agarose gels stained with ethidium bromide. The Figure is representative of three independent experiments. Abbreviations: Mock, no antibody condition; C, untreated cells; I, cells treated with insulin. The lower part of the Figure shows the quantitative aspect of the PCR amplification using serial dilutions of the input.
Using a set of primers that are specific to the SREBP-1c promoter region, we found that endogenous SREBP-1 proteins interact with the promoter under the control conditions and that treatment with insulin increased this association (Figure 5). The observed increase in the PCR signal upon insulin treatment (as estimated from the input dilution test) indicated that insulin enhances the binding of SREBP-1 on the SREBP-1c promoter. The use of control PCR primers that are specific from exons 4/5 of srebf-1 gene (at ∼5 kb from the SREBP-1c promoter) indicated that the amplifications obtained for the SREBP-1c promoter were not due to contaminating genomic DNA in the immunoprecipitate. Furthermore, there was no amplification for the SREBP-1a promoter region, demonstrating that SREBP-1 binding to the srebf-1 gene is restricted to the SREBP-1c promoter, in the absence and in the presence of insulin. Using anti-SREBP-2 antibody, a clear binding to SREBP-1c promoter was also shown. However, insulin did not increase SREBP-2 binding to SREBP-1c promoter, suggesting that the insulin regulation of SREBP-1c expression involved SREBP-1 and not SREBP-2.
Ectopic expression of SREBP-1a and SREBP-1c increased SREBP-1c promoter activity
To support further the implication of SREBP-1 transcription factors in regulating the SREBP-1c gene expression, we constructed expression plasmids for the mature active forms of the human SREBP-1a and SREBP-1c proteins. Figure 6 clearly shows that the expression of both SREBP-1a and SREBP-1c in HEK-293 cells produced a strong activation of the p1c-E2 promoter construct. Mutation of the distal SRE site (p1c-E2m1) reduced the effect of the SREBP-1 proteins, but the activations remained significant (8.3±0.3-fold with SREBP-1a, P<0.01, and 3.8±0.2-fold with SREBP-1c, P<0.01). Mutation of the proximal SRE site alone (p1c-E2m2) or in combination with the other SRE mutations (p1c-E2m1/2) completely abolished SREBP-1 proteins effects on SREBP-1c promoter (Figure 6). Expression of the recombinant SREBP-1a and SREBP-1c active proteins also increased the promoter activity of the p1c-E1 construct (3.6±0.4- and 3.2±0.5-fold respectively) and this effect was not affected by mutations of the LXREs (results not shown). Altogether, these results confirmed the role of the SREs in the response of the human SREBP-1c promoter to SREBP-1 proteins.
Figure 6. Effects of human SREBP-1a or SREBP-1c overexpression on the SREBP-1c promoter.

Each reporter gene construct was co-transfected with an empty expression vector pcDNA3.1 (control condition) or an expression vector for mature form of the human SREBP-1a (pCMV-hSREBP1a) or the human SREBP-1c (pCMV-hSREBP1c) in HEK-293 cells. Luciferase activity was measured 24 h after transfection. Results are expressed by reference to the basal activity of p1c-E2 and are presented as the means±S.E.M. (n=3). *P<0.05 in the presence of SREBP-1a or SREBP-1c versus control conditions.
Overexpression of SREBP-1 proteins in primary culture of human muscle cells
To ascertain the role of SREBP-1 proteins in the regulation of SREBP-1c promoter in a muscle context, we produced adenoviruses expressing mature forms of SREBP-1a or -1c and overexpressed these proteins in human differentiated muscle cells (myotubes) prepared from muscle biopsies of healthy individuals. As shown in Figure 7(A), increasing amounts of both adenoviruses led, as expected, to a proportional accumulation of nuclear SREBP-1 and, more importantly, to a dose-dependent increase in the SREBP-1 precursor protein (125 kDa). The induction of Insig-1 protein, another SREBP-1 target gene [28], was studied as a control. These experiments demonstrated therefore that the addition of mature nuclear forms of SREBP-1 protein (either 1a or 1c) induces the expression of srebf-1 gene in human muscle cells, as evidenced by the production of SREBP-1 precursor protein. The lack of specific SREBP-1a and SREBP-1c antibody did not allow us to determine which isoform was induced. To do so, we monitored the changes in SREBP-1a and SREBP-1c mRNAs upon adenoviral infections using RT–qPCR. Due to sequence identities between the endogenous gene and the adenoviruses, SREBP-1c transcripts were measured when SREBP-1a adenovirus was used and, conversely, SREBP-1a mRNA was determined when SREBP-1c mature protein was overexpressed. Figure 7(B) shows a clear induction of SREBP-1c mRNA and no change in SREBP-1a transcript when overexpressing the mature active forms of SREBP-1. Together with the ChIP experiment (Figure 5), these results strongly suggest that the regulation of srebf-1 gene by SREBP-1 proteins is restricted to the SREBP-1c isoform.
Figure 7. Effects of adenoviral expression of human SREBP-1a or SREBP-1c in differentiated skeletal-muscle cells.

Human skeletal-muscle cells were infected with increasing concentrations of adenovirus expressing SREBP1a or SREBP1c (Ad-SREBP1a or Ad-SREBP1c) as indicated in the Materials and methods section. Total cell lysates were prepared, 24 h after infection, and analysed for the presence of precursor and mature forms of SREBP-1 and for INSIG-1 using specific antibodies (A). α-Tubulin is shown as a loading control. In parallel, total RNA was prepared from muscle cells overexpressing the SREBP-1 isoforms (24 h after adenofection) and analysed for the expression of SREBP-1a and SREBP-1c transcripts using RT–qPCR (B). Due to sequence identities between the endogenous gene and the adenoviruses, SREBP-1c transcripts were measured when SREBP-1a adenovirus was used and, conversely, SREBP-1a mRNA was determined when SREBP-1c mature protein was overexpressed. Results are presented as means±S.E.M. (n=3). *P<0.05.
DISCUSSION
In the present study, we attempted to decipher the mechanisms involved in the transcriptional effect of insulin on the SREBP-1c-specific promoter of the human srebf-1 gene. We demonstrate that the full effect of insulin requires the presence of SREs in the proximal promoter and, using ChIP assay, that insulin increases the binding of SREBP-1 to this promoter region. Furthermore, we found that the nuclear receptor LXRs, which strongly activate SREBP-1c gene expression, do not play a major role in the effect of insulin, in contrast with what was recently observed with the rodent srebf-1 gene [19,24]. Our results suggest therefore that the transcription factors SREBP-1 are the main mediators of insulin action on SREBP-1c expression in human tissues.
Using primary hepatocytes, it was demonstrated that insulin activates the mouse SREBP-1c promoter primarily by increasing the activity of LXRs, the nuclear SREBP-1 playing only a permissive role [24]. Such a finding was further confirmed with the rat SREBP-1c promoter, although this report also demonstrated the contribution of LXR-independent mechanisms involving SREBP-1 [19]. In agreement with these and other previous studies [18,21], we observed in the present study that activation of LXRs with a selective agonist robustly increases SREBP-1c transcription in human cell models. We also identified the LXRE motifs that mediate LXR action in the human promoter region. The mutation of these sites suppressed the effect of the LXR agonist, but did not influence the response to insulin. We therefore concluded that the LXRs do not play a major role in the transcriptional effect of insulin on the human SREBP-1c promoter. The divergence in the mechanism of action of insulin between the rodent and the human genes could eventually rely on species-related differences in promoter sequences and organizations. The two functional neighbouring LXREs are rather well conserved and located at approx. 150 nt upstream of the SRE both in rodents and in humans. In the human gene, however, there are two SRE motifs, both being involved in insulin action as assessed by mutation experiments, whereas in rodents, there is only one SRE and an E-box. Mutation of the SRE abolished insulin action, whereas the E-box did not appear to participate in the effect of insulin [19]. Another important difference that could explain the controversial results is the experimental model used to study SREBP-1c promoter activity. LXRs, notably LXRα, are markedly expressed in liver and in hepatocytes, the cell model used for the studies of the rodent promoters [19,24]. In contrast, LXRs are expressed at rather low levels in HEK-293 cells and in human muscle cells [22]. If the full action of insulin on the SREBP-1c promoter is mediated by the combinatorial activity of LXRE and SRE, as suggested by Cagen et al. [19], it is possible that the relative contribution of these two mechanisms depends on the abundance of LXR and SREBP-1 proteins. Thus we cannot exclude that the LXRs do not play a role in human hepatocytes. However, it is important to consider that insulin exerts an important effect on SREBP-1c expression not only in liver, but also in various tissues, particularly in skeletal muscle [3,13]. Our results strongly suggest therefore that SREBP-1 is the major mediator of insulin action on SREBP-1c promoter in human cells with low level of LXR.
Our results confirmed, using ChIP assays, that both SREBP-1 and SREBP-2 proteins can bind the SREBP-1c promoter of the human srebf-1 gene. However, insulin only increases the binding of SREBP-1. In addition, ectopic expression of SREBP-1a or SREBP-1c nuclear proteins triggers specifically the transcription of SREBP-1c mRNA and the expression of the full-length precursor protein. These results indicate therefore that the initial event in the effect of insulin on SREBP-1c gene expression is likely an action of insulin on the nuclear form of SREBP-1 to increase either its abundance in the nucleus or its transcriptional activity. This first step is immediately followed by induction of SREBP-1c mRNA and protein production, leading thus to an amplification loop. A large number of genes, including FAS, ACC-2 or HKII, are regulated by insulin through an SREBP-1-dependent mechanism [5–7]. It is not known at present whether their induction by insulin is dependent on the stimulation of SREBP-1c gene expression and protein production or whether, as we propose for SREBP-1c, an activation and/or accumulation of the nuclear form of SREBP-1c is sufficient to trigger their expression. It is likely that both mechanisms contribute to the full transcriptional effects of insulin. Previous studies have proposed a direct effect of insulin on SREBP-1 transcriptional activity, particularly for SREBP-1a, which can be phosphorylated by various protein kinases [29]. Mutation of the phosphorylation site of SREBP-1a (Ser117), which is conserved in SREBP-1c, abolished insulin action on LDL receptor promoter [29]. The role of SREBP-1 protein phosphorylation in the action of insulin on SREBP-1c expression is not known.
Accumulation of SREBP-1 protein in the nucleus, independently from the production of newly synthesized protein, could be due either to increased cleavage of the precursor in the ER and its migration to the nucleus or to stabilization of the nuclear form already present in the basal state. It has been demonstrated that the nuclear SREBP-1a and SREBP-2 can be degraded by the proteasome following ubiquitination, and that acetylation by co-activator p300 prevents this process, thus increasing the concentration of the transcription factors in the nucleus [30]. More recently, phosphorylation by GSK3 (glycogen synthase kinase 3) was reported to reduce SREBP-1c transcriptional activity [31], through a phosphorylation-dependent degradation by the ubiquitin–proteasome pathway [32]. Insulin is known to reduce GSK3 activity and thus may stabilize SREBP-1c nuclear content by inhibiting its degradation. The regulation by insulin of SREBP-1c abundance in the nucleus through the GSK3 signalling pathway appears therefore as a very attractive hypothesis, but it still remains to be demonstrated in human cells and in physiological situations.
An alternative mechanism to increase nuclear SREBP-1 is the regulation of the cleavage process. Indeed, the SREBPs are synthesized as transcriptionally inactive precursors bound to the ER. They undergo proteolytic cleavage in the Golgi apparatus to release the mature transcription factor that can migrate to the nucleus [33]. The regulatory proteins essential for the cleavage process are SCAP (SREBP cleavage-activating protein) and the Insigs (insulin-induced genes). In the ER, SCAP forms a complex with newly synthesized SREBPs precursors and escorts them to the Golgi apparatus, where they are processed by two proteinases [33]. Insig-1 and Insig-2 promote SREBP retention in the ER by interacting with SCAP, thus preventing SREBP translocation to the Golgi apparatus for proteolytic processing [34,35]. Roles of Insig proteins in the regulation of insulin action on SREBP-1c are supported by recent studies [36,37]. Insulin induces Insig-1 expression, whereas it decreases Insig-2, especially the isoform Insig-2a [36,38], suggesting that insulin can modulate SREBP-1 cleavage process. In agreement, it was recently reported that insulin acutely cleaves SREBP-1c precursor in vitro in rat hepatocytes and in vivo in the liver of suckling mice [36]. The in vivo effect was observed 20 min after insulin injection, before the induction of SREBP-1c mRNA, indicating that the appearance of mature SREBP-1c in the nucleus precedes the transcriptional effects of insulin [36].
In summary, the present study demonstrates that the effect of insulin on SREBP-1c gene expression is mediated by the transcription factors SREBP-1, and more probably by SREBP-1c itself. Together with several recent data, our results also indicate that the initial event in the effect of insulin on SREBP-1c gene expression is likely an action of the hormone on the nuclear form of SREBP-1 to increase either its transcriptional activity or its abundance in the nucleus. Further works are now needed to elucidate whether SREBP-1 nuclear accumulation is due to the stabilization of the endogenous protein, the stimulation of the precursor cleavage, or a combination of both mechanisms. Because the regulation by insulin of the expression of a number of SREBP-1 target genes, including SREBP-1c itself, is altered in the muscle and the adipose tissue of Type II diabetic patients [3,7], understanding the mechanisms controlling the nuclear abundance of the mature SREBP-1c in human tissues is now an important issue.
Acknowledgments
We thank Dr Rémi Rabasa-Lhoret (Research Group on Diabetes and Metabolic Regulation, Research Center, Centre Hospitalier de l'Université de Montréal, Montréal, Canada), Dr Fitsum Guebre-Egziabher (Département de Néphrologie, Hôpital E. Herriot and Research Unit JE 2411, University Claude Bernard, Lyon, France) and Dr Delphine Jacquet (INSERM U457, Hôpital Robert Debré, Paris, France) for performing the clamp studies and the tissue biopsies used in the present work. This work was supported in part by research grants from INSERM and INRA. N. D. is the recipient of a doctoral fellowship from the Ministère de l'Enseignement Superieur et de la Recherche, France.
References
- 1.O'Brien R. M., Granner D. K. Regulation of gene expression by insulin. Physiol. Rev. 1996;76:1109–1161. doi: 10.1152/physrev.1996.76.4.1109. [DOI] [PubMed] [Google Scholar]
- 2.Rome S., Clement K., Rabasa-Lhoret R., Loizon E., Poitou C., Barsh G. S., Riou J. P., Laville M., Vidal H. Microarray profiling of human skeletal muscle reveals that insulin regulates approximately 800 genes during a hyperinsulinemic clamp. J. Biol. Chem. 2003;278:18063–18068. doi: 10.1074/jbc.M300293200. [DOI] [PubMed] [Google Scholar]
- 3.Ducluzeau P. H., Perretti N., Laville M., Andreelli F., Vega N., Riou J. P., Vidal H. Regulation by insulin of gene expression in human skeletal muscle and adipose tissue. Evidence for specific defects in type 2 diabetes. Diabetes. 2001;50:1134–1142. doi: 10.2337/diabetes.50.5.1134. [DOI] [PubMed] [Google Scholar]
- 4.Patti M. E. Gene expression in humans with diabetes and prediabetes: what have we learned about diabetes pathophysiology? Curr. Opin. Clin. Nutr. Metab. Care. 2004;7:383–390. doi: 10.1097/01.mco.0000134359.23288.72. [DOI] [PubMed] [Google Scholar]
- 5.Latasa M. J., Griffin M. J., Moon Y. S., Kang C., Sul H. S. Occupancy and function of the -150 sterol regulatory element and -65 E-box in nutritional regulation of the fatty acid synthase gene in living animals. Mol. Cell. Biol. 2003;23:5896–5907. doi: 10.1128/MCB.23.16.5896-5907.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oh S. Y., Park S. K., Kim J. W., Ahn Y. H., Park S. W., Kim K. S. Acetyl-CoA carboxylase beta gene is regulated by sterol regulatory element-binding protein-1 in liver. J. Biol. Chem. 2003;278:28410–28417. doi: 10.1074/jbc.M300553200. [DOI] [PubMed] [Google Scholar]
- 7.Gosmain Y., Lefai E., Ryser S., Roques M., Vidal H. Sterol regulatory element-binding protein-1 mediates the effect of insulin on hexokinase II gene expression in human muscle cells. Diabetes. 2004;53:321–329. doi: 10.2337/diabetes.53.2.321. [DOI] [PubMed] [Google Scholar]
- 8.Osborne T. F. Sterol regulatory element-binding proteins (SREBPs): key regulators of nutritional homeostasis and insulin action. J. Biol. Chem. 2000;275:32379–32382. doi: 10.1074/jbc.R000017200. [DOI] [PubMed] [Google Scholar]
- 9.Foufelle F., Ferre P. New perspectives in the regulation of hepatic glycolytic and lipogenic genes by insulin and glucose: a role for the transcription factor sterol regulatory element binding protein-1c. Biochem. J. 2002;366:377–391. doi: 10.1042/BJ20020430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brown M. S., Goldstein J. S. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc. Natl. Acad. Sci. U.S.A. 1999;96:11041–11048. doi: 10.1073/pnas.96.20.11041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown M. S., Goldstein J. L. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89:331–340. doi: 10.1016/s0092-8674(00)80213-5. [DOI] [PubMed] [Google Scholar]
- 12.Amemiya-Kudo M., Shimano H., Hasty A. H., Yahagi N., Yoshikawa T., Matsuzaka T., Okazaki H., Tamura Y., Iizuka Y., Ohashi K., et al. Transcriptional activities of nuclear SREBP-1a, -1c, and -2 to different target promoters of lipogenic and cholesterogenic genes. J. Lipid Res. 2002;43:1220–1235. [PubMed] [Google Scholar]
- 13.Gosmain Y., Dif N., Berbe V., Loizon E., Rieusset J., Vidal H., Lefai E. Regulation of SREBP-1 expression and transcriptional action on HKII and FAS genes during fasting and refeeding in rat tissues. J. Lipid Res. 2005;46:697–705. doi: 10.1194/jlr.M400261-JLR200. [DOI] [PubMed] [Google Scholar]
- 14.Shimomura I., Bashmakov Y., Ikemoto S., Horton J. D., Brown M. S., Goldstein J. L. Insulin selectively increases SREBP-1c mRNA in the livers of rats with streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. U.S.A. 1999;96:13656–13661. doi: 10.1073/pnas.96.24.13656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guillet-Deniau I., Mieulet V., Le Lay S., Achouri Y., Carré D., Girard J., Foufelle F., Ferré P. Sterol regulatory element binding protein-1c expression and action in rat muscles: insulin-like effects on the control of glycolytic and lipogenic enzymes and UCP3 gene expression. Diabetes. 2002;51:1722–1728. doi: 10.2337/diabetes.51.6.1722. [DOI] [PubMed] [Google Scholar]
- 16.Azzout-Marniche D., Becard D., Guichard C., Foretz M., Ferre P., Foufelle F. Insulin effects on sterol regulatory-element-binding protein-1c (SREBP-1c) transcriptional activity in rat hepatocytes. Biochem. J. 2000;350:389–393. [PMC free article] [PubMed] [Google Scholar]
- 17.Amemiya-Kudo M., Shimano H., Yoshikawa T., Yahagi N., Hasty A. H., Okazaki H., Tamura Y., Shionoiri F., Iizuka Y., Ohashi K., et al. Promoter analysis of the mouse sterol regulatory element-binding protein-1c gene. J. Biol. Chem. 2000;275:31078–31085. doi: 10.1074/jbc.M005353200. [DOI] [PubMed] [Google Scholar]
- 18.Repa J. J., Liang G., Ou J., Bashmakov Y., Lobaccaro J. M., Shimomura I., Shan B., Brown M. S., Goldstein J. L., Mangelsdorf D. J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000;14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cagen L. M., Deng X., Wilcox H. G., Park E. A., Raghow R., Elam M. B. Insulin activates the rat sterol-regulatory-element-binding protein 1c (SREBP-1c) promoter through the combinatorial actions of SREBP, LXR, Sp-1 and NF-Y cis-acting elements. Biochem. J. 2005;385:207–216. doi: 10.1042/BJ20040162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schultz J. R., Tu H., Luk A., Repa J. J., Medina J. C., Li L., Schwendner S., Wang S., Thoolen M., Mangelsdorf D. J., et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yoshikawa T., Shimano H., Amemiya-Kudo M., Yahagi N., Hasty A. H., Matsuzaka T., Okazaki H., Tamura Y., Iizuka Y., Ohashi K., et al. Identification of liver X receptor-retinoid X receptor as an activator of the sterol regulatory element-binding protein 1c gene promoter. Mol. Cell. Biol. 2001;21:2991–3000. doi: 10.1128/MCB.21.9.2991-3000.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cozzone D., Debard C., Dif N., Ricard N, Disse E., Vouillarmet J., Rabasa-Lhoret R., Laville M., Pruneau D., Rieusset J., et al. Activation of liver X receptors promotes lipid accumulation but does not alter insulin action in human skeletal muscle cells. Diabetologia. 2006;49:990–999. doi: 10.1007/s00125-006-0140-8. [DOI] [PubMed] [Google Scholar]
- 23.DeBose-Boyd R. A., Ou J., Goldstein J. L., Brown M. S. Expression of sterol regulatory element-binding protein 1c (SREBP-1c) mRNA in rat hepatoma cells requires endogenous LXR ligands. Proc. Natl. Acad. Sci. U.S.A. 2001;98:1477–1482. doi: 10.1073/pnas.98.4.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen G., Liang G., Ou J., Goldstein J. L., Brown M. S. Central role for liver X receptor in insulin-mediated activation of Srebp-1c transcription and stimulation of fatty acid synthesis in liver. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11245–11250. doi: 10.1073/pnas.0404297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chomczynski P., Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal. Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 26.Zabarovsky E. R., Gizatullin R., Podowski R. M., Zabarovska V. V., Xie L., Muravenko O. V., Kozyrev S., Petrenko L., Skobeleva N., Li J., et al. NotI clones in the analysis of the human genome. Nucleic Acids Res. 2000;28:1635–1639. doi: 10.1093/nar/28.7.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chaussade C., Pirola L., Bonnafous S., Blondeau F., Brenz-Verca S., Tronchere H., Portis F., Rusconi S., Payrastre B., Laporte J., Van Obberghen E. Expression of myotubularin by an adenoviral vector demonstrates its function as a phosphatidylinositol 3-phosphate [PtdIns(3)P] phosphatase in muscle cell lines: involvement of PtdIns(3)P in insulin-stimulated glucose transport. Mol. Endocrinol. 2003;17:2448–2460. doi: 10.1210/me.2003-0261. [DOI] [PubMed] [Google Scholar]
- 28.Kast-Woelbern H. R., Dana S. L., Cesario R. M., Sun L., de Grandpre L. Y., Brooks M. E., Osburn D. L., Reifel-Miller A., Klausing K., Leibowitz M. D. Rosiglitazone induction of Insig-1 in white adipose tissue reveals a novel interplay of peroxisome proliferator-activated receptor gamma and sterol regulatory element-binding protein in the regulation of adipogenesis. J. Biol. Chem. 2004;279:23908–23915. doi: 10.1074/jbc.M403145200. [DOI] [PubMed] [Google Scholar]
- 29.Roth G., Kotzka J., Kremer L., Lehr S., Lohaus C., Meyer H. E., Krone W., Muller-Wieland D. MAP kinases Erk1/2 phosphorylate sterol regulatory element-binding protein (SREBP)-1a at serine 117 in vitro. J. Biol. Chem. 2000;275:33302–33307. doi: 10.1074/jbc.M005425200. [DOI] [PubMed] [Google Scholar]
- 30.Giandomenico V., Simonsson M., Gronroos E., Ericsson J. Coactivator-dependent acetylation stabilizes members of the SREBP family of transcription factors. Mol. Cell. Biol. 2003;23:2587–2599. doi: 10.1128/MCB.23.7.2587-2599.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim K. H., Song M. J., Yoo E. J., Choe S. S., Park S. D., Kim J. B. Regulatory role of glycogen synthase kinase 3 for transcriptional activity of ADD1/SREBP1c. J. Biol. Chem. 2004;279:51999–52006. doi: 10.1074/jbc.M405522200. [DOI] [PubMed] [Google Scholar]
- 32.Sundqvist A., Bengoechea-Alonso M. T., Ye X., Lukiyanchuk V., Jin J., Harper J. W., Ericsson J. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7) Cell. Metab. 2005;1:379–391. doi: 10.1016/j.cmet.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 33.Sakai J., Duncan E. A., Rawson R. B., Hua X., Brown M. S., Goldstein J. L. Sterol-regulated release of SREBP-2 from cell membranes requires two sequential cleavages, one within a transmembrane segment. Cell. 1996;85:1037–1046. doi: 10.1016/s0092-8674(00)81304-5. [DOI] [PubMed] [Google Scholar]
- 34.Yang T., Espenshade P. J., Wright M. E., Yabe D., Gong Y., Aebersold R., Goldstein J. L., Brown M. S. Crucial step in cholesterol homeostasis: sterols promote binding of SCAP to INSIG-1, a membrane protein that facilitates retention of SREBPs in ER. Cell. 2002;110:489–500. doi: 10.1016/s0092-8674(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 35.Yabe D., Brown M. S., Goldstein J. L. Insig-2, a second endoplasmic reticulum protein that binds SCAP and blocks export of sterol regulatory element-binding proteins. Proc. Natl. Acad. Sci. U.S.A. 2002;99:12753–12758. doi: 10.1073/pnas.162488899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hegarty B. D., Bobard A., Hainault I., Ferre P., Bossard P., Foufelle F. Distinct roles of insulin and liver X receptor in the induction and cleavage of sterol regulatory element-binding protein-1c. Proc. Natl. Acad. Sci. U.S.A. 2005;102:791–796. doi: 10.1073/pnas.0405067102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Engelking L. J., Kuriyama H., Hammer R. E., Horton J. D., Brown M. S., Goldstein J. L., Liang G. Overexpression of Insig-1 in the livers of transgenic mice inhibits SREBP processing and reduces insulin-stimulated lipogenesis. J. Clin. Invest. 2004;113:1168–1175. doi: 10.1172/JCI20978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yabe D., Komuro R., Liang G., Goldstein J. L., Brown M. S. Liver-specific mRNA for Insig-2 down-regulated by insulin: implications for fatty acid synthesis. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3155–3160. doi: 10.1073/pnas.0130116100. [DOI] [PMC free article] [PubMed] [Google Scholar]