

Phenylalanine hydroxylase (PheH),1 tyrosine hydroxylase (TyrH), and tryptophan hydroxylase (TrpH) make up the family of tetrahydropterin dependent aromatic amino acid hydroxylases. Scheme 1 illustrates the general reaction these enzymes catalyze, the hydroxylation of the side chain of an aromatic amino acid utilizing a tetrahydropterin as the source of the two electrons required to reduce the other atom of oxygen to the level of water. The eukaryotic forms of these enzymes have been studied the most due to their physiological importance. PheH is a liver enzyme that catalyzes the catabolism of excess phenylalanine in the diet to tyrosine; the vast majority of cases of phenylketonuria are due to deficiencies in this enzyme (1). TyrH is found in the central nervous system and adrenal gland, where its role is to catalyze the first step in the biosynthesis of catecholamines, the formation of dihydroxyphenylalanine (DOPA) from tyrosine. TrpH is present in the brain where it catalyzes the first step in the biosynthesis of serotonin, the hydroxylation of tryptophan to 5-hydroxytryptophan. PheH is also present in bacteria, although not in Escherichia coli; as of this writing, 17 different bacterial genomes include a PheH ortholog. Only the enzyme from Chromabacterium violaceum has been characterized to any extent.
Structure
The eukaryotic enzymes are all homotetramers. In each case, three distinct structural domains can be identified. Each enzyme contains an N-terminal domain responsible for the regulatory properties. The regulatory domains vary in length from about 100 residues in the case of TrpH to about 160 residues with TyrH. There is a catalytic domain of about 280 amino acids near the C-terminus that contains all of the residues required for catalysis and substrate specificity (2-6); this domain is homologous to the bacterial enzymes. The final C-terminal 40–50 residues make up a tetramerization domain; a critical part of this is a helix of about 25 residues that forms the coiled–coil interaction critical for tetramer formation (7). While the catalytic cores of the rat enzymes are 52% identical, the regulatory domains show very low levels of identity (<14%), consistent with the different regulatory mechanisms of the three enzymes. Three-dimensional structures have been determined for the catalytic domains of all three of the eukaryotic enzymes as well as the intact PheH from C. violaceum, confirming the homology (Figure 1A). Only in the case of PheH has the structure of the regulatory domain been determined (Figure 1B); this shows that the N-terminus physically blocks the entrance to the active site in the substrate-free enzyme (8). The structures of PheH and TrpH were obtained using enzymes lacking the C-terminal residues responsible for tetramer formation, but the structure of the catalytic domain of TyrH used protein containing the C-terminus. The TyrH structures clearly show the C-terminal coiled–coil responsible for tetramer formation. Because of the homology of the catalytic domains of these enzymes, it is possible to construct a model of the intact tetrameric PheH using the PheH and TyrH structures (Figure 2C).
Figure 1.
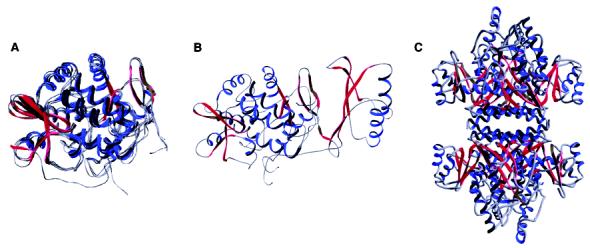
(A) Overlay of the catalytic domains of human PheH (PDB file 1J8U), rat TyrH (PDB file 2TOH), human TrpH (PDB file 1MLW), and C. violaceum PheH (PDB file 1LTV) (B) Combined catalytic and regulatory domains of rat PheH. The structure is based on the PDB file 1PHZ; the protein used for structure determination lacked the C-terminal 24 residues, and the N-terminal 18 residues are not seen in the structure. The orientation is the same as in panel A. (C) Model for the intact rat PheH tetramer. The model was constructed by superimposing the catalytic and regulatory domains of rat PheH shown in panel B with the structure of the tetrameric TyrH catalytic domain (PDB file 2TOH).
Figure 2.
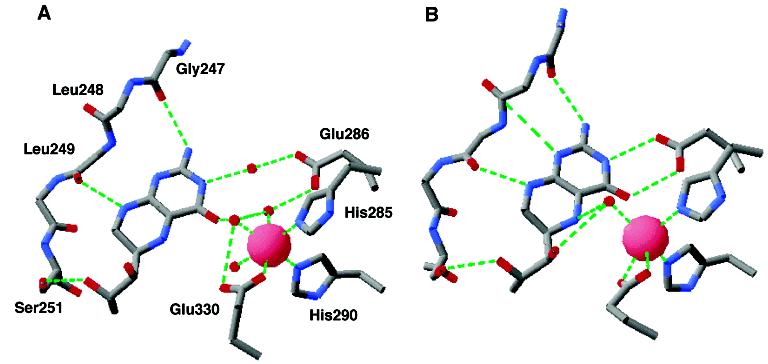
Comparison of the iron and tetrahydrobiopterin binding sites of PheH in the absence (A) and presence (B) of β-thienylalanine. The structures are from the PDB files 1J8U and 1KWO.
Scheme 1.

All three eukaryotic enzymes are regulated in part by the phosphorylation of serine residues in the regulatory domains (9). Phosphorylation of PheH alters the equilibrium between the open and the closed forms of the enzyme (10-12), while phosphorylation of TyrH decreases the inhibition by catecholamines (13); the effects of phosphorylation of TrpH have not been elucidated. With both PheH and TyrH, phosphorylation appears to convert the enzymes between active and inactive forms without altering details of substrate binding or the catalytic mechanism. Because the focus of the present review is the catalytic mechanism, no further discussion of the regulatory properties will be presented here.
The pterin dependent hydroxylases are iron dependent enzymes, requiring one iron atom per subunit for activity (14, 15). When isolated, the iron is typically in the ferric state, while the active form is ferrous (6, 16, 17). The physiological reductant is not known, but tetrahydropterins readily reduce both PheH and TyrH in vitro (16, 18, 19), suggesting that tetrahydrobiopterin is the physiological reductant. There is no evidence that the ferric enzyme is a catalytic intermediate; rather, these enzymes begin and end each catalytic cycle in the ferrous form. The C. violaceum enzyme has been isolated with copper instead of iron (20), and this observation led to the proposal that the bacterial enzyme does not require any metal for activity (21, 22). More recently, C. violaceum PheH was shown to have an absolute requirement for iron for tyrosine formation (23), in agreement with the metal requirements of the eukaryotic enzymes. There have been no other reports to date of a pterin dependent hydroxylase catalyzing amino acid hydroxylation using a metal other than iron.
The location of the iron atom allowed the active site to be readily identified in the first structure of a pterin dependent hydroxylase to be determined, that of the catalytic domain of rat TyrH (24). The iron atom lies at the bottom of 10 Å deep cleft in the enzyme surface. It is bound to three amino acid residues, His331, His336, and Glu376.2 His 331 and His336 had previously been identified as metal ligands in TyrH by site-directed mutagenesis (25), and the corresponding residues in PheH, His285 and 290, had been identified as metal ligands in that enzyme (26, 27). The presence of the third acidic residue had been predicted by Cox et al. (28) based on spectroscopic studies of catecholate complexes of PheH. This arrangement of ligands, two histidines and one acidic residue, has been seen in a number of metalloproteins with divergent functions (29). In contrast to the other proteins with this metal-binding motif, such as the α-ketoglutarate dependent enzymes and the intra- and extradiol dioxygenases, there is no evidence that a substrate becomes a metal ligand during the reaction. In the absence of substrates, up to three water molecules make up the remaining ligands to the metal in all three eukaryotic enzymes (30-32), resulting in a distorted octahedral arrangement of ligands (Figure 2A). The actual number of water molecules varies with the structure (33, 34). In the C. violaceum PheH structure, the glutamate (Glu184) binds in bidentate fashion, and there are only two water molecules bound. While the replacement of a metal ligand in TyrH or PheH with alanine or serine yields inactive enzyme (25, 26), TyrH in which His336 has been replaced with glutamate or glutamine still retains some activity as a tyrosine hydroxylase (35).
The kinetic mechanism, which provides a framework for discussion of the order of the individual steps in catalysis, has been determined for TyrH (36) and for the bacterial PheH (37, 38). For both enzymes, all three substrates must be bound before catalysis occurs. However, there is disagreement regarding the order of substrate binding. In the case of rat TyrH, the order is 6-methyltetrahydropterin first, followed by oxygen in rapid equilibrium, and then tyrosine (36). In the case of bacterial PheH, Pember et al. (37) reported that oxygen bound first, followed by a random binding of 6,7-dimethyltetrahydropterin and phenylalanine. In contrast, Volner et al. (38), using an identical approach, concluded that the binding was ordered and that the order was dimethyltetrahydropterin, phenylalanine, and oxygen. The reasons for this discrepancy in the PheH mechanism are not clear. Less complete studies with TrpH are consistent with a random order for the binding of amino acid and tetrahydropterin (39). Substrate inhibition at high concentrations of the amino acid has been reported for all three enzymes (36, 37, 39); this phenomenon can occur in an ordered kinetic mechanism if the second substrate binds to the free enzyme, hindering binding of the first. The data for binding of amino acid and tetrahydropterin suggest that the order of binding is somewhat random for all these enzymes, with the different enzymes showing different degrees of preference for binding of the pterin before the amino acid.
It is clear that an enzyme–tetrahydropterin complex can form since structures are available for all three enzymes with a pterin bound. Complexes in which the iron is in the ferrous state and a tetrahydropterin is the ligand are the most relevant to catalysis; these are available only for PheH. Figure 2A shows the interactions between tetrahydrobiopterin and PheH in the binary complex of rat PheH (33). Most of the interactions involve the pyrimidine ring of the pterin, consistent with the observation that triaminopyrimidines will function in place of tetrahydropterins as substrates for PheH (40). The N(1)–N(8) side of the pterin ring packs against a flexible loop consisting of residues 247–251. Several of the interactions with this loop involve the peptide backbone rather than amino acid side chains. The only exception is Ser251, which forms a hydrogen bond with the dihydroxypropyl side chain of biopterin; this residue is not conserved in the other hydroxylases, and a variety of 6-substituted tetrahydropterins are effective substrates for all these enzymes (41). The carboxylate of Glu286 provides the only electrostatic interaction between the protein and the pterin; in the absence of the amino acid substrate, this interaction is through two water molecules. The importance of this residue in tetrahydropterin binding has been confirmed by site-directed mutagenesis of both PheH and TyrH (42, 43). In addition to the interactions shown in Figure 2A, there is a stacking interaction between Phe254 and the pterin (44).
No structure is yet available with only an amino acid bound. However, a structure is available for the catalytic domain of human PheH with both tetrahydrobiopterin and β-thienylalanine bound (45). β-Thienylalanine has been shown to bind both human and rat PheH and activate tetrahydropterin consumption (46, 47), but whether this amino acid is hydroxylated has not been established directly. Figure 3 shows the interactions between the amino acid substrate and the protein in the ternary complex. The carboxylate of β-thienylalanine is bound to the side chain of Arg270; this importance of this electrostatic interaction has been demonstrated by the decrease of 4700-fold in the V/K value for the amino acid substrate when the corresponding residue in TyrH is mutated to lysine (43). The carboxylate also forms hydrogen bonds with the side chain of Ser349 and the amide nitrogen of Thr278. The substrate amino group forms hydrogen bonds with the side chain hydroxyl of Thr278 and two water molecules (not shown). The substrate side chain is held in a hydrophobic cage made up the side chains of mainly aromatic amino acids, including Trp326, Phe331, and Pro281, and the backbone of Gly346. In addition, the substrate aromatic moiety is stacked over the side chain of His285, one of the metal ligands. The iron atom is 4.2–4.3 Å from the ε carbons of β-thienylalanine.
Figure 3.
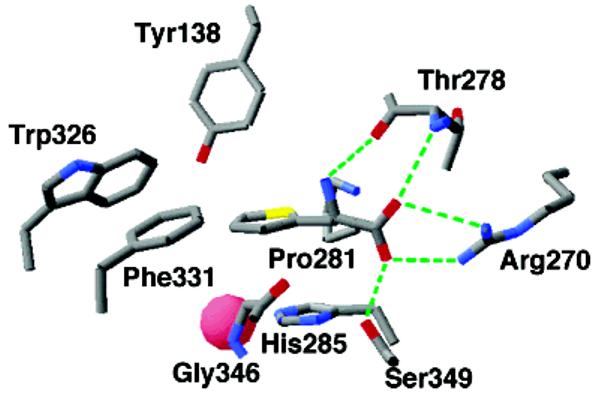
Amino acid substrate binding site of PheH. The structure is from the PDB file 1KWO.
In addition to providing structural evidence for the binding site of the amino acid substrate, this structure shows several differences from those of enzymes with only a pterin bound. While Glu330 has a monodentate interaction with the iron in the structures of the eukaryotic hydroxylases that do not contain an amino acid substrate, in the ternary PheH complex, Glu330 binds the iron in a bidentate fashion (Figure 2B). As noted previously, similar bidentate binding of this glutamate has also been reported for the structure of the C. violaceum PheH both in the presence and in the absence of pterin (34). The position of the pterin is altered in the ternary complex as compared to the various binary complexes with pterin (Figure 2B). The 247–251 loop moves 2.6 Å closer to the iron, so that the pterin C(4a) is only 4.5 Å from the iron as compared to distances of about 6 Å in the binary complexes. Glu330 now forms direct interactions with the pyrimidine ring of the pterin rather than the water mediated bonds seen in the binary complex. Only one water molecule remains bound to the iron in the ternary complex. Spectroscopic studies of rat PheH with phenylalanine and 6-methyl-5-deazatetrahydropterin bound confirm the decrease in the number of metal ligands in the ternary complex (48). It appears that both substrates must be bound before the change in the arrangement of the metal ligands occurs since spectroscopic studies of the ferrous iron in rat PheH do not show a change to a pentacoordinate site when phenylalanine alone binds (49).
In addition to the changes in positions of active site residues and an overall compression of the entire catalytic domain, the ternary complex shows a major conformational change in a mobile loop at the active site opening. In the ternary complex, the loop containing residues 131–150 has moved to close down over the active site (Figure 4). This moves Tyr138 from an exposed position on the surface to being buried in the active site, where it packs against several residues that make up the hydrophobic pocket for the amino acid side chain (Figure 3). All of the eukaryotic aromatic amino acid hydroxylases require that the amino acid be bound before any reaction occurs between oxygen and tetrahydropterin. The combination of changes seen in the ternary complex, the movement of the pterin, the change in iron ligation, and the formation of the amino acid binding site, provide a ready structural explanation for this requirement for amino acid binding before catalysis is initiated. The importance of this loop movement and other structural changes for the bacterial PheHs is less clear since the mobile loop is not conserved in the bacterial enzymes.
Figure 4.
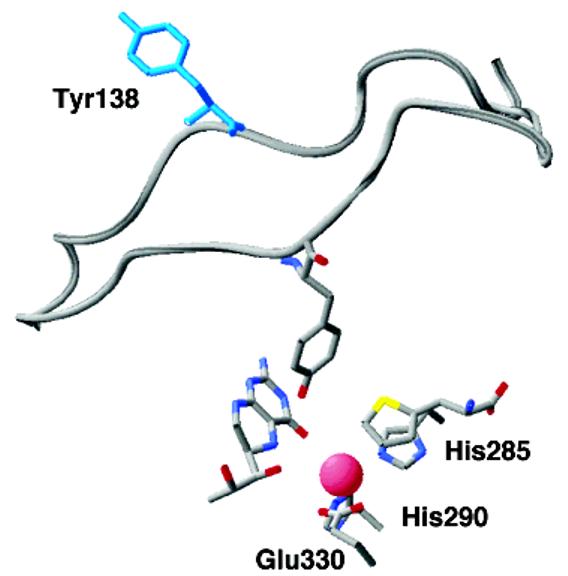
Movement of the 130–150 loop in PheH upon binding of β-thienylalanine to the tetrahydrobiopterin bound enzyme. The loop with Tyr138 in blue is from the binary complex (PDB file 1J8U); the positions of the active site iron, the substrates, and the metal ligands are from the ternary complex (PDB file 1KWO).
Substrate Specificity
While most of the variation among the eukaryotic enzymes occurs in the regulatory domains, the substrate specificities are determined by residues in the catalytic domains (5). Each of the three eukaryotic enzymes is able to hydroxylate all three aromatic amino acids to some extent. For rat PheH, the V/K value for phenylalanine is 105-fold greater than the V/K value for tyrosine (50) but comparable to that for tryptophan (51). For TrpH, the V/K value for tryptophan is only 12-fold that for phenylalanine, but the enzyme shows a preference for tryptophan over tyrosine of at least 5000-fold (6). TyrH is the least specific of the three enzymes, in that the V/K value for tyrosine is only 10-fold that for phenylalanine (50) and 30-fold that for tryptophan (52). The site of hydroxylation in each case is determined by the substrate, in that all three enzymes form tyrosine, DOPA, or 5-hydroxytryptophan from phenylalanine, tyrosine, or tryptophan, respectively. There are some exceptions to the rigid regiospecificity; TyrH forms a 25:1 mixture of tyrosine and 3-hydroxyphenylalanine from phenylalanine (53), and TrpH will hydroxylate 2-azaisotryptophan at both the 5- and the 6-carbons (54). PheH has also been reported to hydroxylate norleucine, methionine, and cyclohexylalanine (21, 46).
As noted previously, the aromatic side chain of the amino acid substrate is surrounded by a hydrophobic cage. The residues that make up this cage are obvious choices for determinants of the substrate specificities. The residues that make up this cage in PheH are Trp326, Phe331, Pro281, His285, and Gly346. All but Trp326 are conserved in both the bacterial and the eukaryotic enzymes, with the minor exception that Phe331 is tyrosine in some bacterial enzymes; Trp326 is replaced with phenylalanine in TrpH. The obvious possibility that this residue is involved in discriminating between the larger indole and the smaller phenyl or phenolic side chains has been examined by site-directed mutagenesis. Mutation of this phenylalanine in TrpH to tryptophan decreases the preference for tryptophan over phenylalanine as a substrate, so that the V/K values for the two amino acids are comparable (51, 52). Mutation of Trp326 of PheH to phenylalanine yields a mutant protein with a 30-fold preference for tryptophan over phenylalanine (51), but the same mutation in TyrH has almost no effect on the relative V/K values for the three aromatic amino acids (52). Much more dramatic effects on substrate specificity have been found upon mutagenesis of second sphere residues. Val379 of PheH, which packs against Tyr138 in the ternary complex, is replaced by Asp425 in TyrH. D425V TyrH shows a preference for phenylalanine over tyrosine of 8000-fold, while the double mutant, V379D/H264Q PheH, shows a 3000-fold decrease in its preference for phenylalanine over tyrosine (50). These results suggest that the substrate specificity of these enzymes is determined by the packing of second sphere residues against the aromatic cage surrounding the substrate.
Scheme 2.

Mechanism of Oxygen Activation
While our knowledge of the structures of these proteins is most advanced in the case of PheH, details of the catalytic mechanism have been worked out with all three. The homology of the catalytic domains of the pterin dependent hydroxylases suggests that they share a common catalytic mechanism. For the present discussion, it will be assumed that this is indeed the case, so that experimental results for all three enzymes can be used to develop a common mechanistic scheme. Scheme 2 summarizes the present understanding of the chemical mechanism of these enzymes. The reaction can be divided into two partial reactions, formation of the hydroxylating intermediate and oxygen transfer to the amino acid substrate. In neither has an intermediate been trapped for direct structural analysis. However, a variety of kinetic and computational approaches is providing insights into the catalytic details.
As noted previously, activity requires that the iron be ferrous. Tetrahydropterins have been shown to reduce the ferric iron in both PheH (16, 18) and TyrH (19). The mechanism of the reduction has not been elucidated. While formally one molecule of tetrahydropterin should be able to reduce two iron atoms, at least in the case of PheH the stoichiometry depends on the experimental conditions. There is no evidence that formation of the ferric enzyme occurs during turnover. Indeed, in the case of TyrH, no EPR-detectable intermediates are seen during catalysis (19). Thus, for mechanistic discussions, the ferrous enzyme can be considered the resting form in the absence of substrates.
A 4a-hydroxypterin (Scheme 1) is a product of the reaction catalyzed by all three enzymes (55-58). Molecular oxygen is the source of the oxygen atom incorporated into the pterin product (59) as well as the oxygen atom incorporated into the amino acid (60). This requires that there be a reaction between an oxygen species and the pterin during catalysis, so that the pterin must participate in catalysis to a greater extent than simply supplying electrons. Studies of the autoxidation of tetrahydropterin provide a possible model for the enzymatic reaction between oxygen and tetrahydropterin. The products of the autoxidation reaction are H2O2 and quinonoid dihydropterin (61, 62). Scheme 3 summarizes the mechanism proposed by Eberlein et al. (63) for this reaction. The first and rate-limiting step is single electron transfer from the pterin to oxygen to form superoxide and a pterin cation radical. Radical collapse generates a peroxypterin; loss of peroxide from the peroxypterin leaves the dihydropterin. The proposed radical and peroxypterin intermediates have not been directly detected during the autoxidation reaction. The intermediacy of a peroxypterin is supported by the observation that a stable peroxypterin can be formed by reacting a 5-deazatetrahydropterin with singlet oxygen (64), by the production of H2O2 as an autoxidation product, and by the direct observation of a similar 4a-peroxyflavin in the reaction of reduced flavoprotein hydroxylases with oxygen (65). Eberlein et al. (63) proposed the intermediacy of a pterin cation radical based upon analyses of the thermodynamics of the autoxidation reaction and the pH insensitivity of the reaction. There is no solvent isotope effect on the autoxidation reaction (66), consistent with rate-limiting single electron transfer to form the pterin cation radical.
Scheme 3.

The precedent of the autoxidation reaction and the observed intermediacy of 4a-peroxyflavins in flavoproteincatalyzed hydroxylation reactions make a similar 4a-peroxypterin an attractive intermediate for the pterin dependent hydroxylases. Support for a peroxypterin intermediate comes from studies with tyrosine as a substrate for PheH. As noted previously, tyrosine is hydroxylated very slowly by PheH. This is in part due to the unproductive consumption of tetrahydropterin and oxygen by the enzyme without concomitant amino acid hydroxylation (67). Hydrogen peroxide is produced by this unproductive reaction, consistent with the breakdown of a peroxypterin intermediate (68). In addition, C. violaceum PheH is reported to form hydrogen peroxide in the presence of phenylalanine if the metal-free enzyme is used (23).
While the intermediacy of a peroxypterin in the enzyme-catalyzed reactions is thus reasonable, the mechanism of its formation remains unsettled. The reaction of TyrH with oxygen has been probed using 18O kinetic isotope effects. With this enzyme, the rate-limiting step in turnover appears to be the formation of the hydroxylating intermediate since the kcat value is relatively insensitive to the identity of the amino acid substrate (69). There is an 18O isotope effect on the V/K value for oxygen of 1.0175 (70). This establishes that there is a change in the bond order to oxygen in the rate-determining step. The value is too large to be due to the formation of neutral superoxide or hydrogen peroxide but is compatible with the formation of superoxide or peroxide anion (71). Thus, the 18O kinetic isotope effect is consistent with an enzymatic reaction that resembles the mechanism for the autoxidation reaction in Scheme 3.
Scheme 4.

One critical difference between the enzyme-catalyzed reaction and the autoxidation reaction is that the enzyme has a metal in the active site; this raises the possibility that the iron is explicitly involved in the reaction between oxygen and tetrahydropterin in the active site. A mechanism consistent with the data is shown in Scheme 4. Binding of oxygen to the Fe(II) atom would generate a complex equivalent to Fe(III)O2−. This could then attack the C(4a) position of the tetrahydropterin to form the peroxypterin. Such a mechanism would avoid the spin-forbidden direct reaction of triplet oxygen with the tetrahydropterin. The oxygen isotope effects for TyrH are too small to be due only to binding of oxygen to Fe(II) (71). They can be rationalized by equilibrium binding of oxygen to the iron followed by an irreversible step involving either electron transfer from the pterin to the oxygen to form Fe(II)O2− or by nucleophilic attack of the pterin on the iron-bound oxygen to form the Fe(II) μ-peroxypterin species shown in Scheme 2. In the absence of direct detection of the intermediates proposed for the two different mechanisms, one criterion for clarifying the role of the iron in the initial reaction between tetrahydropterin is to determine whether these enzymes can catalyze a reaction between tetrahydropterin and oxygen in the absence of iron. The metal-free PheH from C. violaceum has been reported to catalyze the oxidation of dimethyltetrahydropterin to quinonoid dihydropterin and hydrogen peroxide at about 5% the turnover of the iron-containing enzyme (23), suggesting that the metal is not absolutely required. In contrast, several mutant forms of TyrH in which the metal affinity has been decreased by mutation of the one of the metal ligands still show an absolute requirement for metal to catalyze any tetrahydropterin consumption (35). The metal requirement of the eukaryotic enzyme may reflect a need for bound metal to effect the conformational change that occurs when both amino acid and pterin are bound before any reaction between pterin and oxygen can occur rather than direct involvement of the metal in the reaction. Alternatively, the reaction of the bacterial enzyme may be due to a low level of catalysis of tetrahydropterin oxidation by enzyme containing a metal other than iron. When His336 in TyrH is mutated to glutamine, the enzyme retains substantial hydroxylase activity with an absolute need for iron. However, in the presence of Co(II), this mutant protein will catalyze tyrosine dependent tetrahydropterin oxidation at 14% the rate seen with Fe(II), although it does not catalyze DOPA formation (72). The iron-free C. violaceum PheH was not metal-free but contained substoichiometric amounts of nickel and copper (23).
The role of the iron in the reaction between oxygen and tetrahydropterin has also been addressed computationally. Bassan et al. (73) used hybrid density functional theory and a truncated model of the PheH site based on the binary structure with tetrahydrobiopterin to analyze the mechanism of that enzyme. They were unable to find a mechanism for oxygen reacting with the pterin in the absence of the metal. Instead, the calculations indicated that the favored pathway resembled the mechanism of Scheme 4, with initial binding of oxygen to Fe(II) to form a species better described as Fe(III)O2−. This then reacted with the pterin to form the Fe(II) μ-peroxypterin without any detectable intermediates. Energetically, the highest barrier was for the reaction of Fe(III)O2− with the pterin; this is consistent with the prediction from the 18O isotope effects. While the calculations thus seem to support the mechanism of Scheme 4, they are based on the structure of the binary complex with tetrahydrobiopterin. As discussed above, there are significant changes in the metal ligands and the interaction of the tetrahydropterin with the protein when the amino acid is bound; the effect of these changes on the computations remains to be resolved.
A peroxypterin formed from the reaction of oxygen and the tetrahydropterin is one candidate for the hydroxylating intermediate (74). The flavoprotein phenol hydroxylases catalyze a reaction which is formally identical to the reaction of TyrH, and the hydroxylating intermediate in these enzymes has been identified as a 4a-peroxyflavin (65). However, the flavoprotein hydroxylases require that the aromatic ring of the substrate be activated by a hydroxyl, amino or thiol moiety (74). In contrast, all three pterin dependent enzymes can hydroxylate the unactivated phenyl ring of phenylalanine (6, 75) and, as discussed below, can also catalyze benzylic hydroxylation. This suggests that the hydroxylating intermediate in the latter enzymes is more reactive than a simple peroxypterin. One clear difference between the pterin and flavin dependent systems is the requirement of the former for iron. Based on the iron requirement and the need for a more reactive species, Dix et al. (59) proposed that an Fe(II) μ-peroxypterin was either the hydroxylating intermediate or a precursor to it. As discussed above, the experimental and computational data support such a species as an intermediate. However, for all three enzymes, a peroxypterin, by itself or as an iron ligand, has been ruled out as the actual hydroxylating intermediate. Direct hydroxylation by a peroxypterin requires that cleavage of the peroxy OO bond and addition of oxygen to the amino acid substrate be concerted. As a result, the amount of hydroxypterin produced must agree with the amount of amino acid hydroxylated. When tyrosine is a substrate for either PheH or TrpH, a large excess (>100-fold) of tetrahydropterin is consumed as compared to the amount of DOPA formed, while 20–30% of the tetrahydropterin is converted to the hydroxypterin product (58, 67). In the case of TyrH, the S395A enzyme catalyzes the formation of the hydroxypterin product at a rate not substantially different from the wild-type enzyme, but only 1–2% of this is accompanied by formation of DOPA (76). These results establish that OO bond cleavage can occur without hydroxylation of the amino acid substrate. The most straightforward explanation is that cleavage of the OO bond occurs in the formation of the actual hydroxylating intermediate, ruling out a peroxypterin as the actual oxygen donor to the amino acid substrate. Further support for the need to cleave the OO bond of a peroxy intermediate to form the actual hydroxylating intermediate comes from the properties of TyrH H336Q (72). The decreased metal affinity of this enzyme allowed the enzyme to be reconstituted with Co(II); the cobalt-containing enzyme was unable to catalyze amino acid hydroxylation but was able to catalyze tetrahydropterin oxidation in the presence of tyrosine. This result is consistent with the ability to form Co-peroxide but not Co(IV).
The intermediacy of the Fe(II) μ-peroxypterin initially proposed by Dix et al. (59) is consistent with several experimental observations. As discussed previously, computations support such a species as the product of the iron-catalyzed reaction of molecular oxygen and the tetrahydropterin in the active site (73). In addition, in the structure of the ternary complex of PheH with tetrahydrobiopterin and β-thienylalanine, C(4a) of the pterin is 4.5 Å from the iron (45). This distance is compatible with a peroxy bridge between these two atoms if one assumes that oxygen binds in place of the remaining water molecule bound to the iron. Heterolytic cleavage of the OO bond of such an intermediate would generate the hydroxypterin product directly while forming a species at the formal oxidation level of Fe(IV)O. A ferryl oxo species is a reasonable candidate for the actual hydroxylating intermediate. Similar species have been proposed as the active oxygen intermediates in several other non-heme iron monooxygenases (77). Very recently, an intermediate with spectroscopic properties consistent with high spin Fe(IV)O was detected in the reaction of the non-heme iron enzyme taurine/α-ketoglutarate dioxygenase (78); while the mechanisms of the α-ketoglutarate dependent hydroxylases differ in detail from that of the pterin dependent enzymes (79), in both, the iron is bound by a two histidineone carboxylate triad (80). Biomimetic studies have established the ability of Fe(IV)O species to catalyze aromatic hydroxylation, sulfoxidation, and epoxidation reactions (81-84).
Mechanism of Hydroxylation
Studies of the mechanism of oxygen addition to the amino acid substrate are consistent with an electrophilic hydroxylating intermediate such as Fe(IV)O. Probing the mechanism of oxygen addition to the amino acid substrate has been complicated by the fact that other first-order steps in the reaction are slower for both PheH and TyrH (46, 69), so that changes in steady state kinetic parameters with alternate or isotopically labeled substrates do not directly reflect changes in the rate constant for this chemical step. In the case of TyrH, this complication has been circumvented by analyzing product partitioning rather than steady-state kinetics. The kcat value for this enzyme is relatively constant for a number of ring-substituted phenylalanines (69). However, changes in the electron-donating ability of the substituent at the 4-position of the aromatic ring alter the efficiency of the reaction (85). This is consistent with a kinetic model (Scheme 5) in which the rate of formation of the Fe(IV)O intermediate is insensitive to the reactivity of the amino acid substrate, but its subsequent fate is determined by partitioning between productive hydroxylation and unproductive breakdown. When the relative amount of hydroxylated amino acid formed was determined for a series of 4-substituted phenylalanines, there was a good correlation between the relative rate constants for hydroxylation and the σ values of the substituents, with an average ρ value of −5 for tetrahydrobiopterin and 6-methyltetrahydropterin (85). This result was interpreted as evidence for a cationic transition state for oxygen addition to the aromatic ring. An electrophilic aromatic substitution reaction for hydroxylation involving the cationic species shown in Scheme 2 was proposed based on these data. The proposed cationic intermediate is consistent with the observation that these enzymes exhibit NIH shifts, 1,2 shifts of the substituent at the site of hydroxylation to the adjacent ring carbon (86, 87). The extent of the shift decreases with the electronegativity of the substituent, consistent with a requirement for the substituent to participate in the three-centered transition state for the 1,2 shift (85).
Scheme 5.

Support for electrophilic aromatic substitution as the mechanism of oxygen addition also comes from kinetic isotope effects with TrpH. In contrast to the situation with PheH and TyrH, the rate-limiting step in TrpH turnover is oxygen addition to the aromatic ring of the substrate (54). In the hydroxylation of tryptophan, the NIH shift is from C5 to C4 only (58). With 5-2H-tryptophan as substrate for TrpH, there is an inverse isotope effect on the kcat value of 0.93, while no effect is seen with 4-2H-tryptophan as substrate (58). This is consistent with rehybridization from sp2 to sp3 at the site of oxygen addition but no change at the adjacent carbon to which the NIH shift occurs (58). With TyrH, isotope effects on the hydroxylation step can be measured by using mutant enzymes in which most of the tetrahydropterin is consumed unproductively. Analogously to the mechanism of Scheme 5, if part of this unproductive turnover is due to breakdown of the Fe(IV)O intermediate, isotope effects on the hydroxylation step will be manifest. With two mutant enzymes that meet this criterion, E326A and H336E TyrH, inverse isotope effects with an average value of 0.89 have been found.3 The inverse isotope effects for both TrpH and TyrH are consistent with the mechanism of hydroxylation shown in Scheme 2. The lack of an effect with 4-2H-tryptophan as a substrate for TrpH rules out an alternate proposal in which oxygen adds across the 4,5-bond directly to form an arene oxide (58). Further evidence against an arene oxide intermediate has come from studies of isotope effects on product partitioning when phenylalanine is used as a substrate for TyrH (53). In this case, both tyrosine and 3-hydroxyphenylalanine are formed. The amount of tyrosine decreases with 4-2H-phenylalanine as the substrate, but there is no change in the amount of 3-hydroxyphenylalanine. Similarly, the amount of 3-hydroxyphenylalanine formed decreases with 3-2H-phenylalanine, but the amount of tyrosine formed is unaffected. Partitioning of an arene oxide intermediate would result in compensating increases in the other product, so that such a species cannot be an obligatory intermediate along the catalytic pathway to tyrosine.
Scheme 6.

A Fe(IV)O intermediate for the pterin dependent enzymes would be expected to have reactivity comparable to the heme-based cytochrome P450 systems. Other enzymes for which such an intermediate has been invoked (e.g., iso-penicillin synthase and the α-ketoglutarate dependent hydroxylases) are capable of aliphatic hydroxylation (77). Consistent with such an expectation, all three hydroxylases have been shown to catalyze benzylic hydroxylation of methylated aromatic amino acids (54, 85, 88). In addition, PheH has been reported to catalyze aliphatic hydroxylation (21, 46) and epoxidation (23). Only in the case of benzylic hydroxylation by TyrH has the mechanism been investigated in detail. Analysis of the products when 4-methylphenylalanine containing one, two, or three deuterium atoms in the methyl group was used as a substrate allowed determination of the intrinsic primary and secondary deuterium isotope effects for the hydroxylation step (89). The secondary isotope effect of 1.2 demonstrated that there is significant rehybridization of the methyl carbon in the transition state for hydroxylation; such a result is most consistent with the formation of a benzylic radical intermediate by abstraction of a hydrogen atom from the methyl group by the electrophilic Fe(IV)O species, followed by radical rebound of a hydroxyl radical to form the hydroxylated product (Scheme 6). The primary isotope effect for this reaction was 9.6; together with the large secondary effect, this value suggests that there is a significant contribution of quantum mechanical tunneling to the reaction. This mechanism is similar to that proposed for aliphatic hydroxylation by the cytochrome P450 family (90), demonstrating the comparable reactivity of the hydroxylating intermediates of the non-heme pterin dependent enzymes and the heme dependent hydroxylases.
Recent computation studies also provide support for an Fe(IV)O hydroxylating intermediate and provide further insight into the mechanism. Bassan et al. (91) used hybrid density functional theory to examine the mechanisms of aromatic and benzylic hydroxylation by the aromatic amino acid hydroxylases. Two different Fe(IV)O models were used, with both having a monodentate formate, two imidazoles, and two water molecules as ligands. Different results were obtained if one of the water molecules was modeled as hydroxide or water. A model using water was more consistent with a cationic intermediate for aromatic hydroxylation, while that with hydroxide led to a radical intermediate. With either model, an arene oxide was not on the pathway for aromatic hydroxylation. The HO-Fe(IV)O model was found competent to carry out benzylic hydroxylation via the rebound mechanism of Scheme 6.
Conclusion
In recent years, a variety of experimental approaches, including biomimetic systems, structural analyses, enzymology, and computational approaches has provided substantial support for the mechanism of Scheme 2 for the aromatic amino acid hydroxylases. Clearly, much remains to be done. There is as yet no direct evidence for the individual intermediates in the proposed mechanism, and the role of the protein in directing the site of hydroxylation is far from clear. The contribution of the protein conformational changes seen in the crystal structures to gating the formation of the hydroxylating intermediate also remains to be established.
Footnotes
This work was supported in part by grants from the NIH (GM 47291) and the Robert A. Welch Foundation (A-1245).
Abbreviations: PheH, phenylalanine hydroxylase; TyrH, tyrosine hydroxylase; TrpH, tryptophan hydroxylase; DOPA, dihydroxyphenylalanine.
Amino acid residue numbers refer to the individual proteins. The catalytic domains of the three eukaryotic hydroxylases are readily aligned with no gaps. As a result of the different lengths of their N-terminal domains, residues in TyrH are 46 residues farther from the N-terminus than the respective residues in PheH, while residues in TrpH are 13 residues closer to the N-terminus than in PheH. Human and rat PheH differ in that the human enzyme has one additional residue at the C-terminus, so that the numbering for active site residues is the same for each. Chromabacterium violaceum PheH lacks the N-terminal regulatory domain and contains both short insertions and deletions as compared to the catalytic domains of the eukaryotic enzymes, so that there is no simple arithmetic difference in residue numbering.
Frantom, P. A., and Fitzpatrick, P. F., manuscript in preparation.
REFERENCES
- 1.Eisensmith RC, Woo SLC. Mol. Biol. Med. 1991;8:3–18. [PubMed] [Google Scholar]
- 2.Ledley FD, DiLella AG, Kwok SCM, Woo SLC. Biochemistry. 1985;24:3389–3394. doi: 10.1021/bi00335a001. [DOI] [PubMed] [Google Scholar]
- 3.Grenett HE, Ledley FD, Reed LL, Woo SLC. Proc. Natl. Acad. Sci. U.S.A. 1987;84:5530–5534. doi: 10.1073/pnas.84.16.5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Daubner SC, Lohse DL, Fitzpatrick PF. Protein Sci. 1993;2:1452–1460. doi: 10.1002/pro.5560020909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daubner SC, Hillas PJ, Fitzpatrick PF. Biochemistry. 1997;36:11574–11582. doi: 10.1021/bi9711137. [DOI] [PubMed] [Google Scholar]
- 6.Moran GR, Daubner SC, Fitzpatrick PF. J. Biol. Chem. 1998;273:12259–12266. doi: 10.1074/jbc.273.20.12259. [DOI] [PubMed] [Google Scholar]
- 7.Lohse DL, Fitzpatrick PF. Biochem. Biophys. Res. Commun. 1993;197:1543–1548. doi: 10.1006/bbrc.1993.2653. [DOI] [PubMed] [Google Scholar]
- 8.Kobe G, Jennings IG, House CM, Feil SC, Michell BJ, Tiganis T, Parker MW, Cotton RGH, Kemp BE. Protein Sci. 1997;6:1352–1357. doi: 10.1002/pro.5560060626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fitzpatrick PF. Annu. Rev. Biochem. 1999;68:355–381. doi: 10.1146/annurev.biochem.68.1.355. [DOI] [PubMed] [Google Scholar]
- 10.Shiman R, Gray DW, Hill MA. J. Biol. Chem. 1994;269:24637–24646. [PubMed] [Google Scholar]
- 11.Shiman R, Xia T, Hill MA, Gray DW. J. Biol. Chem. 1994;269:24647–24656. [PubMed] [Google Scholar]
- 12.Xia T, Gray DW, Shiman R. J. Biol. Chem. 1994;269:24657–24665. [PubMed] [Google Scholar]
- 13.Ramsey AJ, Fitzpatrick PF. Biochemistry. 1998;37:8980–8986. doi: 10.1021/bi980582l. [DOI] [PubMed] [Google Scholar]
- 14.Gottschall DW, Dietrich RF, Benkovic SJ. J. Biol. Chem. 1982;257:845–849. [PubMed] [Google Scholar]
- 15.Haavik J, Le Bourdelles B, Martinez A, Flatmark T, Mallet J. Eur. J. Biochem. 1991;19:371–378. doi: 10.1111/j.1432-1033.1991.tb16133.x. [DOI] [PubMed] [Google Scholar]
- 16.Wallick DE, Bloom LM, Gaffney BJ, Benkovic SJ. Biochemistry. 1984;23:1295–1302. doi: 10.1021/bi00301a043. [DOI] [PubMed] [Google Scholar]
- 17.Fitzpatrick PF. Biochem. Biophys. Res. Commun. 1989;161:211–215. doi: 10.1016/0006-291x(89)91582-9. [DOI] [PubMed] [Google Scholar]
- 18.Marota JJA, Shiman R. Biochemistry. 1984;23:1303–1311. doi: 10.1021/bi00301a044. [DOI] [PubMed] [Google Scholar]
- 19.Ramsey AJ, Hillas PJ, Fitzpatrick PF. J. Biol. Chem. 1996;271:24395–24400. doi: 10.1074/jbc.271.40.24395. [DOI] [PubMed] [Google Scholar]
- 20.Pember SO, Villafranca JJ, Benkovic SJ. Biochemistry. 1986;25:6611–6619. doi: 10.1021/bi00369a042. [DOI] [PubMed] [Google Scholar]
- 21.Carr RT, Balasubramanian S, Hawkins PCD, Benkovic SJ. Biochemistry. 1995;34:7525–7532. doi: 10.1021/bi00022a028. [DOI] [PubMed] [Google Scholar]
- 22.Carr RT, Benkovic SJ. Biochemistry. 1993;32:14132–14138. doi: 10.1021/bi00214a009. [DOI] [PubMed] [Google Scholar]
- 23.Chen D, Frey P. J. Biol. Chem. 1998;273:25594–25601. doi: 10.1074/jbc.273.40.25594. [DOI] [PubMed] [Google Scholar]
- 24.Goodwill KE, Sabatier C, Marks C, Raag R, Fitzpatrick PF, Stevens RC. Nat. Struct. Biol. 1997;4:578–585. doi: 10.1038/nsb0797-578. [DOI] [PubMed] [Google Scholar]
- 25.Ramsey AJ, Daubner SC, Ehrlich JI, Fitzpatrick PF. Protein Sci. 1995;4:2082–2086. doi: 10.1002/pro.5560041013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gibbs BS, Wojchowski D, Benkovic SJ. J. Biol. Chem. 1993;268:8046–8052. [PubMed] [Google Scholar]
- 27.Balasubramanian S, Carr RT, Bender CJ, Peisach J, Benkovic SJ. Biochemistry. 1994;33:8532–8537. doi: 10.1021/bi00194a019. [DOI] [PubMed] [Google Scholar]
- 28.Cox DD, Benkovic SJ, Bloom LM, Bradley FC, Nelson MJ, Que L, Jr., Wallick DE. J. Am. Chem. Soc. 1988;110:2026–2032. [Google Scholar]
- 29.Hegg EL, Que L. Eur. J. Biochem. 1997;250:625–629. doi: 10.1111/j.1432-1033.1997.t01-1-00625.x. [DOI] [PubMed] [Google Scholar]
- 30.Erlandsen H, Fusetti F, Martinez A, Hough E, Flatmark T, Stevens RC. Nat. Struct. Biol. 1997;4:995–1000. doi: 10.1038/nsb1297-995. [DOI] [PubMed] [Google Scholar]
- 31.Schunemann V, Meier C, Meyer-Klaucke W, Winkler H, Trautwein AX, Kappskog PM, Toska K, Haavik J. J. Inorg. Biochem. 1999;4:223–231. doi: 10.1007/s007750050308. [DOI] [PubMed] [Google Scholar]
- 32.Wang L, Erlandsen H, Haavik J, Knappskog PM, Stevens RC. Biochemistry. 2002;41:12569–12574. doi: 10.1021/bi026561f. [DOI] [PubMed] [Google Scholar]
- 33.Andersen OA, Flatmark T, Hough E. J. Mol. Biol. 2001;314:279–291. doi: 10.1006/jmbi.2001.5061. [DOI] [PubMed] [Google Scholar]
- 34.Erlandsen H, Kim JY, Patch MG, Han A, Volner A, Abu-Omar MM, Stevens RC. J. Mol. Biol. 2002;320:645–661. doi: 10.1016/s0022-2836(02)00496-5. [DOI] [PubMed] [Google Scholar]
- 35.Fitzpatrick P, Ralph EC, Ellis HR, Willmon OJ, Daubner SC. Biochemistry. 2003;42:2081–2088. doi: 10.1021/bi0271493. [DOI] [PubMed] [Google Scholar]
- 36.Fitzpatrick PF. Biochemistry. 1991;30:3658–3662. doi: 10.1021/bi00229a010. [DOI] [PubMed] [Google Scholar]
- 37.Pember SO, Johnson KA, Villafranca JJ, Benkovic SJ. Biochemistry. 1989;28:2124–2130. doi: 10.1021/bi00431a024. [DOI] [PubMed] [Google Scholar]
- 38.Volner A, Zoidakis J, Abu-Omar MM. J. Biol. Inorg. Chem. 2003;8:121–128. doi: 10.1007/s00775-002-0395-6. [DOI] [PubMed] [Google Scholar]
- 39.Fitzpatrick PF. In: Advances in Enzymology and Related Areas of Molecular Biology. Purich DL, editor. John Wiley & Sons, Inc.; New York: 2000. pp. 235–294. [DOI] [PubMed] [Google Scholar]
- 40.Kaufman S. J. Biol. Chem. 1979;254:5150–5154. [PubMed] [Google Scholar]
- 41.Shiman R. In: Folates and Pterins. Blakley RL, Benkovic SJ, editors. Vol. 2. John Wiley & Sons; New York: 1985. pp. 179–249. [Google Scholar]
- 42.Dickson PW, Jennings IG, Cotton RGH. J. Biol. Chem. 1994;269:20369–20375. [PubMed] [Google Scholar]
- 43.Daubner SC, Fitzpatrick PF. Biochemistry. 1999;38:4448–4454. doi: 10.1021/bi983012u. [DOI] [PubMed] [Google Scholar]
- 44.Ellis HR, Daubner SC, McCulloch RI, Fitzpatrick PF. Biochemistry. 1999;38:10909–10914. doi: 10.1021/bi991160u. [DOI] [PubMed] [Google Scholar]
- 45.Andersen OA, Flatmark T, Hough E. J. Mol. Biol. 2002;320:1095–1108. doi: 10.1016/s0022-2836(02)00560-0. [DOI] [PubMed] [Google Scholar]
- 46.Kaufman S, Mason K. J. Biol. Chem. 1982;257:14667–14678. [PubMed] [Google Scholar]
- 47.Stokka AJ, Flatmark T. In: Chemistry and Biology of Pteridines and Folates. Milstien S, Kapatos G, Levine RA, Shane B, editors. Kluwer Academic Publishers; Boston: 2001. pp. 109–126. [Google Scholar]
- 48.Wasinger EC, Mitic N, Hedman B, Caradonna J, Solomon EI, Hodgson KO. Biochemistry. 2002;41:6211–6217. doi: 10.1021/bi0121510. [DOI] [PubMed] [Google Scholar]
- 49.Loeb KE, Westre TE, Kappock TJ, Mitic N, Glasfeld E, Caradonna JP, Hedman B, Hodgson KO, Solomon EI. J. Am. Chem. Soc. 1997;119:1901–1915. [Google Scholar]
- 50.Daubner SC, Melendez J, Fitzpatrick PF. Biochemistry. 2000;39:9652–9661. doi: 10.1021/bi000493k. [DOI] [PubMed] [Google Scholar]
- 51.McKinney J, Teigen K, Frøystein NA, Salaün C, Knappskog PM, Haavik J, Martínez A. Biochemistry. 2001;40:15591–15601. doi: 10.1021/bi015722x. [DOI] [PubMed] [Google Scholar]
- 52.Daubner SC, Moran GR, Fitzpatrick PF. Biochem. Biophys. Res. Commun. 2002;292:639–641. doi: 10.1006/bbrc.2002.6719. [DOI] [PubMed] [Google Scholar]
- 53.Fitzpatrick PF. J. Am. Chem. Soc. 1994;116:1133–1134. [Google Scholar]
- 54.Moran GR, Phillips RS, Fitzpatrick PF. Biochemistry. 1999;38:16283–16289. doi: 10.1021/bi991983j. [DOI] [PubMed] [Google Scholar]
- 55.Lazarus RA, Dietrich RF, Wallick DE, Benkovic SJ. Biochemistry. 1981;20:6834–6841. doi: 10.1021/bi00527a015. [DOI] [PubMed] [Google Scholar]
- 56.Dix TA, Kuhn DM, Benkovic SJ. Biochemistry. 1987;26:3354–3361. doi: 10.1021/bi00386a016. [DOI] [PubMed] [Google Scholar]
- 57.Haavik J, Flatmark T. Eur. J. Biochem. 1987;168:21–26. doi: 10.1111/j.1432-1033.1987.tb13381.x. [DOI] [PubMed] [Google Scholar]
- 58.Moran GR, Derecskei-Kovacs A, Hillas PJ, Fitzpatrick PF. J. Am. Chem. Soc. 2000;122:4535–4541. [Google Scholar]
- 59.Dix TA, Bollag G, Domanico PL, Benkovic SJ. Biochemistry. 1985;24:2955–2958. doi: 10.1021/bi00333a022. [DOI] [PubMed] [Google Scholar]
- 60.Kaufman S, Bridgers WF, Eisenberg F, Friedman S. Biochem. Biophys. Res. Commun. 1962;9:497–502. doi: 10.1016/0006-291x(62)90115-8. [DOI] [PubMed] [Google Scholar]
- 61.Mager HIX, Addink R, Berends W. Recl. Trav. Chim. Pays-Bas. 1967;86:833–850. [Google Scholar]
- 62.Benkovic SJ, Sammons D, Armarego WLF, Waring P, Inners R. J. Am. Chem. Soc. 1985;107:3706–3712. [Google Scholar]
- 63.Eberlein GA, Bruice TC, Lazarus RA, Henrie R, Benkovic SJ. J. Am. Chem. Soc. 1984;106:7916–7924. [Google Scholar]
- 64.Moad G, Luthy CL, Benkovic PA, Benkovic SJ. J. Am. Chem. Soc. 1979;101:6068–6076. [Google Scholar]
- 65.Massey V. J. Biol. Chem. 1994;269:22459–22462. [PubMed] [Google Scholar]
- 66.Blair JA, Pearson AJ. J. C. S. Perkin Trans. 1974;2:80–88. [Google Scholar]
- 67.Davis MD, Kaufman S. J. Biol. Chem. 1989;264:8585–8596. [PubMed] [Google Scholar]
- 68.Davis MD, Kaufman S. Arch. Biochem. Biophys. 1993;304:9–16. doi: 10.1006/abbi.1993.1315. [DOI] [PubMed] [Google Scholar]
- 69.Fitzpatrick PF. Biochemistry. 1991;30:6386–6391. doi: 10.1021/bi00240a006. [DOI] [PubMed] [Google Scholar]
- 70.Francisco WA, Tian G, Fitzpatrick PF, Klinman JP. J. Am. Chem. Soc. 1998;120:4057–4062. [Google Scholar]
- 71.Tian G, Klinman JP. J. Am. Chem. Soc. 1993;115:8891–8897. [Google Scholar]
- 72.Ellis HR, McCusker KP, Fitzpatrick P. Arch. Biochem. Biophys. 2002;408:305–307. doi: 10.1016/s0003-9861(02)00568-4. [DOI] [PubMed] [Google Scholar]
- 73.Bassan A, Blomberg MRA, Siegbahn PEM. Chem.–Eur. J. 2003;9:106–115. [Google Scholar]
- 74.Massey V, Hemmerich P. In: The Enzymes. 3rd ed. Boyer PD, editor. Vol. 12. Academic Press, Inc.; New York: 1975. pp. 191–252. [Google Scholar]
- 75.Tong JH, D'Iorio A, Benoiton NL. Biochem. Biophys. Res. Commun. 1971;44:229–236. doi: 10.1016/s0006-291x(71)80183-3. [DOI] [PubMed] [Google Scholar]
- 76.Ellis HR, Daubner SC, Fitzpatrick PF. Biochemistry. 2000;39:4174–4181. doi: 10.1021/bi9928546. [DOI] [PubMed] [Google Scholar]
- 77.Que L, Jr., Ho RYN. Chem. Rev. 1996;96:2607–2624. doi: 10.1021/cr960039f. [DOI] [PubMed] [Google Scholar]
- 78.Price JC, Barr EW, Tirupati B, Bollinger JM, Krebs C. Biochemistry. 2003;42:7497–7508. doi: 10.1021/bi030011f. [DOI] [PubMed] [Google Scholar]
- 79.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee S-K, Lehnert N, Neese F, Skulan AJ, Yang Y-S, Zhou J. Chem. Rev. 2000;100:235–349. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 80.Schofield CJ, Zhang Z. Curr. Opin. Struct. Biol. 1999;9:722–731. doi: 10.1016/s0959-440x(99)00036-6. [DOI] [PubMed] [Google Scholar]
- 81.Grapperhaus CA, Mienert B, Bill E, Weyhermuller T, Wieghardt K. Inorg. Chem. 2000;39:5306–5317. doi: 10.1021/ic0005238. [DOI] [PubMed] [Google Scholar]
- 82.Lange SJ, Miyake H, Que L., Jr. J. Am. Chem. Soc. 1999;121:6330–6331. [Google Scholar]
- 83.Rohde J, In J, Lim MH, Brennessel WW, Bukowski MR, Stubna A, Münck E, Nam W, Que L., Jr. Science. 2003;299:1037–1039. doi: 10.1126/science.299.5609.1037. [DOI] [PubMed] [Google Scholar]
- 84.Lim MH, Rohde J, Stubna A, Bukowski MR, Costas M, Ho RYN, Munck E, Nam W, Que L., Jr. Proc. Natl. Acad. Sci. U.S.A. 2003;100:3665–3670. doi: 10.1073/pnas.0636830100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hillas PJ, Fitzpatrick PF. Biochemistry. 1996;35:6969–6975. doi: 10.1021/bi9606861. [DOI] [PubMed] [Google Scholar]
- 86.Guroff G, Levitt M, Daly J, Udenfriend S. Biochem. Biophys. Res. Commun. 1966;25:253–259. doi: 10.1016/0006-291x(66)90589-4. [DOI] [PubMed] [Google Scholar]
- 87.Renson JD, Daly J, Weissbach H, Witkop B, Udenfriend S. Biochem. Biophys. Res. Commun. 1966;25:504–513. [Google Scholar]
- 88.Daly J, Guroff G. Arch. Biochem. Biophys. 1968;125:136–141. doi: 10.1016/0003-9861(68)90647-4. [DOI] [PubMed] [Google Scholar]
- 89.Frantom PA, Pongdee R, Sulikowski GA, Fitzpatrick PA. J. Am. Chem. Soc. 2002;124:4202–4203. doi: 10.1021/ja025602s. [DOI] [PubMed] [Google Scholar]
- 90.Sono M, Roach MP, Coulter ED, Dawson JH. Chem. Rev. 1996;96:2841–2888. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 91.Bassan A, Blomberg MRA, Siegbahn PEM. Chem.–Eur. J. 2003;9:4055–4067. doi: 10.1002/chem.200304768. [DOI] [PubMed] [Google Scholar]