

Abstract
A wide array of recurrent, non-random chromosomal translocations are associated with hematologic malignancies; experimental models have clearly demonstrated that many of these translocations are causal events during malignant transformation. Translocations involving the MLL gene are among the most common of these non-random translocations. Leukemias with MLL translocations have been the topic of intense interest because of the unusual, biphenotypic immunophenotype of these leukemias, because of the unique clinical presentation of some MLL translocations (infant leukemia and therapy-related leukemia), and because of the large number of different chromosomal loci that partner with MLL in these translocations. This review is focused on the potential mechanisms that lead to MLL translocations, and will discuss aberrant VDJ recombination, Alu-mediated recombination, non-homologous end joining, as well as the effect of DNA topoisomerase II poisons and chromatin structure.
Keywords: MLL, chromosomal translocation, infant leukemia, DNA topoisomerase II, non-homologous end-joining
1. Introduction
The study of recurrent, non-random chromosomal translocations has proven to be a rich source of insights into the biology of hematologic malignancies [1,2]. A number of themes concerning these chromosomal translocations has become apparent over the past decades [3]. For instance, particular translocations are associated with particular subtypes of leukemia; the t(15;17) is exclusively associated with acute promyelocytic leukemia (APL) [4], and the t(1;19) is associated only with pre-B cell acute lymphoblastic leukemia (ALL) [5]. Additionally, each gene involved in chromosomal translocations has a preferred partner, with perhaps 2–4 additional, infrequent partners. For instance, E2A is typically fused to PBX1 but less frequently to HLF [6], and RARA is most commonly involved in translocations with PML, but can also be fused to PLZF or NPM [4]. Chromosomal translocations involving the MLL (for Mixed Lineage Leukemia, also called ALL-1, HRX, and Htrx) locus on chromosome 11 band q23 provide exceptions to these two aforementioned generalizations. MLL translocations are associated with a wide array of hematologic malignancy, including acute myelogenous leukemia (AML), T-cell ALL, B lineage ALL, myelodysplastic syndrome (MDS), lymphoblastic lymphoma, and Burkitt’s lymphoma [7–9]. In addition, MLL has been labeled a “promiscuous” oncogene since over 60 partner genes or regions have been identified [9].
The MLL gene was initially cloned by virtue of it’s involvement in t(4;11), t(11;19), and t(9;11) translocations [7,9]. The MLL genomic structure consists of 36 exons distributed over 100 kb, and produces a 12 kb mRNA that encodes a 3968 aa protein with an estimated molecular weight of 430 kD [7]. The MLL protein is widely expressed in the develping embryo, and is expressed at lower levels in most adult tissues. The predicted amino acid sequence of MLL indicates that it is homologous to the trithorax gene of D. melanogaster. Recent experiments have indicated that MLL is normally processed via a cytoplasmic cleavage evetn into a 320 kD amino terminus (MLL-N), and a 180 kD carboxy terminus fragment [7]. A number of protein motifs/domains have been identified in the primary structure of MLL, including AT hooks, a DNA methyltransferase domain, PHD domains, a transactivation domain, and a SET domain. Several lines of evidence indicate that one important function of MLL is in the maintenance of HOX gene expression during embryonic development. Loss of MLL function in flies leads to homeotic transformation, and deletion of MLL in mice leads to embryonic lethality and homeotic transformation [7]. Biochemical analysis of MLL suggests that it normally functions as a transcription regulator, and expression of MLL fusion proteins has been shown to be leukemogenic in mice. A detailed discussion of these aspects is beyond the scope of this review, which will focus on mechanisms that lead to MLL translocations; the reader is referred to several excellent reviews for an overview of these aspects [7,9,10].
2. Clinical observations
2.1 Clinical overview
MLL translocations are among the most common translocations in hematologic maliganacy. Approximately 3–10% of patients with AML have MLL translocations, and 8–10 % of patients with B-lineage ALL have MLL translocations [7,9]. Patients with AML and MLL translocation have an intermediate prognosis compared to all cases of AML, whereas patients with B-lineage ALL and MLL translocations tend to have a poor prognosis. In contrast, although MLL translocations are relatively rarely associated with T-ALL, these patients tend to have a very good prognosis [11]. Two distinct forms of acute leukemia are associated with MLL translocations and will be discussed in more detail below. MLL translocations are exceedingly common in infants with AML and ALL, and have been identified in up to 80% of all infant acute leukemia cases [12]. In addition, MLL translocations are seen in approximately 25% of patients with therapy-related leukemias, particularly those associated with the use of chemotherapeutic agents that target DNA topoisomerase II (topo II) [13].
2.2 Infant leukemia
Current models of malignant transformation suggest that multiple pathways, including those governing cell growth, differentiation, death, and responsiveness to external signals need to be disrupted in order to generate any malignancy [14]. It has been suggested that myeloid leukemias require at least two collaborating mutations, one that impairs blood cell differentiation, and a complementary one that leads to increased proliferation and/or decreased apoptosis [15]. Support for this conceptual framework comes both from clinical observations (patients with APL often have PML-RARA fusions, which impair differentiation, and FLT3 mutations, which lead to increased proliferation), as well as murine bone marrow transduction/transplantation experiments, in which bone marrow cells transduced with both BCR-ABL (which leads to increased proliferation) and NUP98-HOXA9 (which impairs differentiation) were leukemic, whereas cells transduced with only BCR-ABL were not [15]. Further support for this framework comes from the observation that many leukemic fusions can be detected in hematopoietic cells from healthy individuals. For instance, AML1-ETO fusions can be detected in long term survivors who are presumably cured of their disease, and TEL-AML1 fusions can be detected in cord blood from healthy newborns [16]. Moreover, “knock-in” mice that express an AML1-ETO fusion gene do not develop leukemia until they are exposed to mutagens, which presumably cause mutations that complement AML1-ETO [17]. Taken together, these findings support the hypothesis that a single genetic event is not sufficient to cause acute leukemia.
In this context, MLL fusions are unusual in that they display a very brief latent period. Leukemias with MLL fusions have been detected in newborns [18,19], and even in aborted fetuses [20], indicating that the MLL translocations can occur in utero. Moreover, the concordance rate of leukemia in monozygotic twins who have MLL fusions is almost 100% suggesting that the disease is highly penetrant [21]. These observations suggest that either complementary mutations occur very rapidly in cells that have undergone MLL fusions, or that an MLL gene fusion is sufficient to cause malignancy, and does not require additional, complementary events. The observations that infants with MLL fusions often have FLT3 mutations as well as the MLL fusion, and that MLL-AF9 “knock-in” mice develop leukemia only after a protracted latency period argue it is unlikely that cells with MLL fusions do not require additional mutations. Therefore, it seems more likely that complementary mutations occur very rapidly in cells that have undergone MLL fusions. At least two mechanisms have been suggested to explain this hypothesis [10]. It is possible that MLL fusions could lead to a “mutator phenotype” Although there is no direct experimental evidence to support this hypothesis, MLL fusions have been shown to impair DNA double strand break recognition [22] and cell cycle checkpoints [23,24], both of which could lead to increased accumulation of mutations. Alternatively, the insult (for instance, exposure to topo II poisons; see below) that caused the MLL translocation could cause additional mutations, especially if the individual had an inherited deficiency in DNA repair and/or toxin metabolizing enzymes. This possibility is supported by studies which show that certain isoforms of NQO1 [25] or CYP3A4 [26] confer an increased risk of infant or therapy-related leukemia respectively.
2.3 therapy-related leukemia
One of the tragic side-effects of successful cytotoxic chemotherapy treatment of any malignancy is the development of a therapy-related neoplasm. Therapy-related acute myeloid leukemia (t-AML) generally presents either early (within 3 years of cessation of all chemotherapy), or late (seven or more years after completion of therapy). The “early” t-AML often display balanced translocations, especially involving the MLL gene, whereas the “late” t-AML often shown unbalanced rearrangements, and often have an antecedent myelodysplastic syndrome [27]. Although the analysis of t-AML was confounded because most patients had received multi-agent chemotherapy, multivariate analysis strongly implicated topo II poisons, particularly the epipodophyllotoxins etoposide and teniposide, in the generation of balanced MLL translocations in the patients with “early” t-AML [13,28]. The role of topo II poisons in the generation of MLL translocations was reinforced by case reports of t-AML with MLL translocations in patients who received single agent therapy with etoposide [29], and by studies showing that etoposide could generate chromosomal rearrangements in cultured lymphocytes [30]. Data from the initial series of cases with t-AML related to topoisomerase II poisons suggested that the epipodophyllotoxins were more highly associated with t-AML than other topo II poisons, such as the anthracylines, and that t-AML was both dose and schedule dependent [31].
3. Not all gross chromosomal rearrangements involving MLL are translocations
Although most studies of gross chromosomal rearrangements (GCR) involving MLL have focused on chromosomal translocations; chromosomal insertions, duplications, and “jumping” translocations involving MLL have also been described in patients with AML. Insertions of a megabase region of chromosome 11 material, including the 5’ portion of MLL, into chromosome 5q31 leads to production of an MLL-AF5Q31 fusion gene; this insertion has thus far only been reported in infants with AML [32]. A partial tandem duplication (PTD), in which a genomic fragment containing several MLL exons becomes duplicated, is commonly seen in patients with AML, particularly those with trisomy 11 or normal karyotypes [33,34] (fig. 1). As a consequence of this rearrangement, MLL exon 6 sequences become fused to MLL exon 2, generating an MLL “self-fusion” protein. In contrast to the MLL PTD and MLL insertions, jumping translocations involving MLL do not produce an MLL fusion gene. Instead, these jumping translocations result from integration of an amplicon of chromosome 11 on one or more chromosomes, resulting in cells that have multiple additional copies of the MLL gene [35].
Figure 1. MLL partial tandem duplication (PTD).
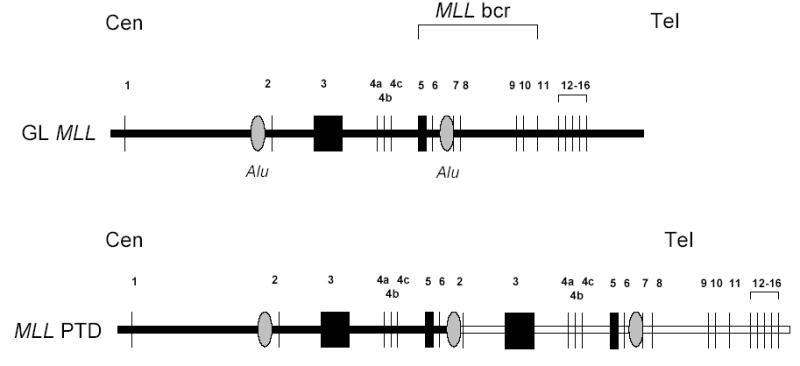
The germline (GL) MLL locus is shown; exons 1–16 are indicated (the remaining 3’ exons are omitted for clarity). Regions of Alu repeats are indicated, as is the MLL breakpoint cluster region (bcr). Centromere to telomere orientation is indicated. The MLL partial tandem duplication (PTD) is depicted.A recombination event occurs between Alu sequences in intron 1 and intron 6, leading to a duplication of exons 2–6 between exons 6 and 7. Exon sizes and intronic distances are not to scale.
4. MLL translocation breakpoints
The first step in identifying possible mechanisms that lead to any chromosomal translocation is simply to localize and characterize the translocation breakpoints. Most (but not all) MLL translocations associated with hematologic malignancy can be mapped to an 8.3 kb breakpoint cluster region (bcr). This region is bounded by BamH1 restriction sites, and encompasses MLL exons originally designated exons 5–11 [36]. Subsequent studies revealed that MLL exon 4 actually consisted of 3 discrete exons (now designated exons 4A, 4B, and 4C) . Therefore, although MLL exon 5 is actually the 7th exon (as per Ensemblv37, http://www.ensembl.org/Homo_sapiens/exonview?db=core;transcript=ENST00000359313), this review will use the older nomenclature to allow direct comparison to the original studies and exon designation.
When studying any chromosomal translocation, it is important to remember that for any translocation to be recognized clinically as a recurrent chromosomal translocation, two criteria must be fulfilled. First, the region involved must undergo a DNA breakage and religation event. Second, the breakage and relegation must confer a growth or survival advantage to the cell. If both criteria are not fulfilled, then the translocation will not be recognized clinically. Translocations between any two non-oncogenic loci, such as those encoding actin and spectrin, may be quite common, but not recognized clinically since they are not leukemogenic, and never become a clonal population of cells. Therefore, the fact that MLL breakpoints in leukemic cells typically localize to this 8.3 kb bcr does not mean a priori that this region is highly susceptible to DNA double strand breakage and chromosomal translocation.
A large number of MLL translocation breakpoints have been cloned and the breakpoints sequenced [37]. The nucleotide sequence at the breakpoints has provided clues as to the potential mechanism(s) leading to the translocation. In general, although the translocations often appear reciprocal, or balanced, at a cytogenetic level, analysis of the nucleotide sequence demonstrates that there are often sub-microscopic deletions, inversions, and amplifications [37].
5. Proposed molecular mechanisms
Several molecular mechanisms have been proposed to account for the recurrent translocations that occur within the MLL bcr. Some of these mechanisms are speculations based on characterization of sequence motifs found at or near translocation breakpoints, whereas other proposed mechanisms have been tested in experimental systems. It should be noted that the proposed mechanisms that are described below are not mutually exclusive, and it seems likely that no single mechanism can explain all MLL translocations.
5.1 Aberrant VDJ recombination
Gu and colleagues noted nucleotide sequences that resembled heptamer and nonamer sequences near the t(4;11) breakpoints in two cell lines derived from patients with B-cell precursor ALL and MLL translocations [38]. They proposed that the VDJ recombination machinery may have been directly involved in the genesis of these translocations. However, although the cryptic heptamer sequences were near the breakpoints (within 20–30 nucleotides) they were not at the breakpoints, and there was no evidence of non-templated nucleotide addition; these findings are in contrast to other examples of illegitimate VDJ recombination [39,40]. Moreover, cloning of additional breakpoints from patients with has not identified cryptic heptamers near the breakpoints.
5.2 Recombination between Alu elements
Alu elements are repetitive sequences that comprise over 10% of the human genome. Intrachromosomal rearrangements between Alu elements have been well documented in meiotic cells (for review, see [41] ). In addition, interchromosomal translocations can be induced in cultured mammalian cells by introducing DNA double strand breaks (DSBs) adjacent to Alu elements [42]. In this system, the majority of the translocations identified were mediated via the single-strand annealing (SSA) pathway. However, although chromosomal translocations caused by interchromosomal recombination between Alu elements in somatic cells has been implicated in complex BCR-ABL translocations [43] and in a single case of AML with an MLL-AF9 translocation [44], these seem to be relatively rare [41]. In contrast the partial tandem duplication of MLL (MLL PTD) is commonly mediated via inter or intra chromosomal recombination between Alu elements within the MLL locus. In seven of nine MLL PTD cases analyzed, the recombination took place between Alu elements within the MLL locus, consistent with repair via the single-strand annealing pathway; the remaining two cases showed one break within an Alu sequence and the second break within a non-Alu sequence [45].
5.3 Recombination mediated via DNA topoisomerase II
The association of t-AML with MLL translocations and topo II poisons has led to the hypothesis that these translocations are directly caused by topo II poisons. This is an attractive hypothesis, since topo II normally functions as a homodimeric enzyme that catalyzes the relaxation of supercoiled DNA. It does so in a three-step reaction, consisting of DNA double-strand cleavage, strand passage, and strand religation. In the first step, each monomer of topo II introduces a 4-bp staggered nick in double stranded DNA. This reaction results in a normally short-lived intermediate known as the cleavable complex. Topo II poisons stabilize the cleavable complex, which is subsequently recognized by the cell as damaged DNA. Upon recognition of this damaged DNA, the cell is eliminated through an apoptotic pathway [46]. Given that chromosomal translocations are the result of DNA double strand cleavage and religation events, it seems reasonable to think that topo II, which is known to catalyze DNA cleavage and religation reactions, would be a likely candidate for directing chromosomal translocations.
Although preferred sites of topo II cleavage in vivo do not consistently correlate with preferred cleavage sites in vitro [47], an MLL translocation breakpoint cloned from a patient with AML and a t(9;11) was noted to be a preferred site for epipodophylotoxin-induced topo II cleavage in vitro [48], leading to the suggestion that cleavage at this site triggered the translocation. Since topo II normally functions as a homodimer, and the transient cleavable complex contains topo II monomers covalently bound to the phosphodiester backbone, it has been proposed that perfect (ie, no net gain or loss of genetic material) or near-perfect (gain or loss of 4 or fewer nucleotides) reciprocal translocations could occur via an exchange of the topo II subunits and covalently linked chromosomal DNA (figure 2) [49]. This model is supported by observations that DNA recombination events consistent with this hypothesis have been detected in vitro, using purified topo II and lambda phage DNA substrates [50]. In addition, a near-perfect reciprocal chromosomal translocation was detected in Chinese hamster ovary cells treated with the topo II poison amsacrine [49]. More recently, chromosomal translocations showing this type of perfect or near-perfect interchromosomal exchange have been identified at the NUP98 [51] and MLL [52] loci in patients with t-AML following multiagent chemotherapy regimens that included one or more topo II poisons. However, it should be noted that these cases are probably the exception rather than the rule, as more extensive surveys of MLL translocation breakpoints, from either de novo leukemia patients [37] or t-AML patients [53], do not contain large numbers of samples with these near-perfect reciprocal interchromosomal exchanges. Interestingly, preliminary studies have suggested that individuals with common polymorphisms of enzymes involved in drug metabolism (CYP3A and GSTP1) may be more likely to develop t-AML, perhaps due to ineffective elimination of topo II poisons and/or active metabolites [26,54].
Figure 2. Chromosomal translocation consistent with a DNA topoisomerase II subunit exchange.
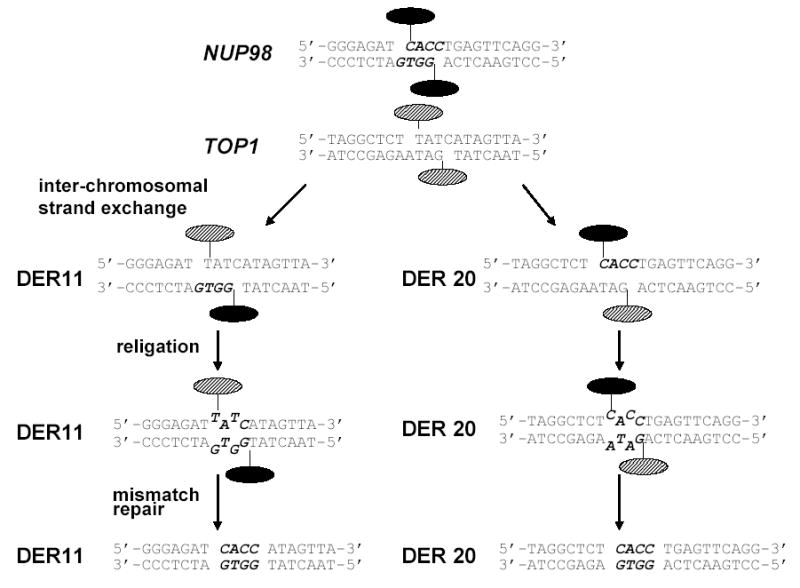
The germline NUP98, TOP1, and both derivative chromosomes from a patient with t-AML were cloned and sequenced [51]. In this model, topo II monomers initially bound to NUP98 are indicated with solid ovals; monomers initially bound to TOP1 are indicated with cross-hatched ovals. A 4-bp staggered DNA DSB occurs as indicated, and an inter-chromosomal stand exchange occurs, with the NUP98 and TOP1 sequences aligning as indicated. Religation occurs as indicated, leaving 2 nucleotide mismatches (T-G on the der 11; C-A on the der 20). Repair of the mismatches leads to the final sequences shown as the der 11 and der 20. Note that the 4 nucleotides in bold italics (CACC) are present at the breakpoint on both the der 11 and der 20 chromosomes.
5.4 Translocation mediated via non-homologous end joining
Marschalek and colleagues have cloned a large number of MLL translocation breakpoints, primarily from patients with a t(4;11) translocation, and shown that the breakpoints often display features consistent with DNA repair via non-homologous end joining (NHEJ) [37,55]. These features include short deletions, inversions, insertions of “filler” DNA or direct repeats, and microhomology at the breakpoints. These features were found in both infant and adult patients with t(4;11) translocations involving MLL. They conclude that the translocations were likely initiated by DNA double or single-strand breaks, followed by an error prone repair process (such as NHEJ).
5.5 Site-specific MLL cleavage within the breakpoint cluster region
As discussed above, almost all MLL translocations are localized to a 8.3 kb breakpoint cluster region (bcr). This bcr can be further subdivided into a centromeric and telomeric portion. The MLL breakpoints in cases of infant leukemia and t-AML tend to be clustered near the telomeric portion of the bcr, whereas the breakpoints in patients with de novo AML tend to be near the centromeric portion of the bcr [37,56]. A number of years ago, site-specific cleavage of the MLL bcr was noted to occur in patients, cell lines, and naked DNA following treatment with topo II poisons [57,58]. Intriguingly, this cleavage site mapped close to the telomeric portion of the MLL bcr, near a sequence that was an 18/18 bp match for a consensus topo II cleavage site, leading to the hypothesis that this cleavage may be an initial step in chromosomal translocation involving the MLL gene [57].
However, it seems likely that this site is a marker for a region that is generally susceptible to DNA DSB. Subsequent studies showed that cleavage at this site could be induced by DNAse I, or more generally initiated during the high molecular weight (HMW) DNA fragmentation that occurs in cells undergoing apoptotic cell death [58,59]. As a cell undergoes apoptosis, prior to the oligonucelosomal degradation of genomic DNA, high molecular weight (HMW) DNA is broken into fragments of ~50 kb [60]. It has been proposed that these fragments are produced by excision of chromosomal “loops” via cleavage at scaffold/matrix attachment regions (S/MARs) [61–63]. In this context, it is important to note that this MLL cleavage site maps to a S/MAR, leading to the proposal that this MLL cleavage site is located at the base of a S/MAR, and is in a more “open” configuration, and thus more susceptible to cleavage induced by a variety of agents, including apoptotic nucleases [58]. This cleavage is clearly directed by sequences within the MLL bcr, and not more long-range effects, since episomes containing the MLL bcr are cleaved at the same site when cells containing the episomes undergo apoptosis [64].
Taken together, these observations have led several investigators to speculate that an “aborted” apoptosis program could lead to GCRs following abnormal repair at this cleavage site [58,65,66]. Although apoptosis has traditionally been thought of as irreversible once initiated, studies in C. elegans suggest that apoptosis might be reversible [67–69]. Intriguingly, two recent studies have used inverse PCR to identify PCR products consistent with GCRs produced by improper repair of DNA double strand cleavage at this site caused by treatment with fas ligand or etoposide [70,71].
6. Summary
Although rare cases may be mediated via SSA involving Alu elements, or topo II subunit exchange, nucleotide sequence data supports the contention that most of the leukemogenic translocations involving MLL are mediated by inappropriate repair of DNA DSBs via an “error-prone” NHEJ pathway. However, the proximate cause of the DNA DSBs remains unclear. Epidemiologic data suggests that the MLL bcr may be more susceptible to DNA DSBs, and a site within the MLL bcr that is generally susceptible to DNA DSBs has been identified. It remains unclear whether the MLL bcr is remarkably susceptible to DNA DSBs (ie, one of the very most susceptible regions throughout the entire genome), whether the MLL bcr is more susceptible to aberrant NHEJ repair of DNA DSBs, or whether neither of these are true, and the frequent presence of MLL translocations in leukemic samples is simply due to a remarkable growth/survival advantage conferred by MLL fusion proteins.
Acknowledgments
I would like to thank Drs. Ilan R. Kirsch, W. Michael Kuehl, Thomas Ried, Martin Stanulla, Rolf Marschalek, Carolyn Felix, Harish Ahuja, Andrew Vaughan, Tamas Varga, and Janet D. Rowley for many fascinating and fruitful discussions of chromosomal translocations in general and MLL translocations in particular. This work was supported by the Intramural Research Program of the NIH, NCI.
References
- 1.Rowley JD. Chromosome translocations: dangerous liaisons revisited. Nat Rev Cancer. 2001;1:245–250. doi: 10.1038/35106108. [DOI] [PubMed] [Google Scholar]
- 2.Rabbitts TH. Chromosomal translocation master genes, mouse models and experimental therapeutics. Oncogene. 2001;20:5763–5777. doi: 10.1038/sj.onc.1204597. [DOI] [PubMed] [Google Scholar]
- 3.Aplan PD. Causes of oncogenic chromosomal translocation. Trends Genet. 2006;22:46–55. doi: 10.1016/j.tig.2005.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chou WC, Dang CV. Acute promyelocytic leukemia: recent advances in therapy and molecular basis of response to arsenic therapies. Curr Opin Hematol. 2005;12:1–6. doi: 10.1097/01.moh.0000148552.93303.45. [DOI] [PubMed] [Google Scholar]
- 5.Hunger SP. Chromosomal translocations involving the E2A gene in acute lymphoblastic leukemia: clinical features and molecular pathogenesis. Blood. 1996;87:1211–1224. [PubMed] [Google Scholar]
- 6.Hunger SP, Li S, Fall MZ, Naumovski L, Cleary ML. The proto-oncogene HLF and the related basic leucine zipper protein TEF display highly similar DNA-binding and transcriptional regulatory properties. Blood. 1996;87:4607–4617. [PubMed] [Google Scholar]
- 7.Hess JL. MLL: a histone methyltransferase disrupted in leukemia. Trends Mol Med. 2004;10:500–507. doi: 10.1016/j.molmed.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 8.Thirman MJ, Gill HJ, Burnett RC, Mbangkollo D, McCabe NR, Kobayashi H, Zieminvanderpoel S, Kaneko Y, Morgan R, Sandberg AA, Chaganti RSK, Larson RA, Lebeau MM, Diaz MO, Rowley JD. Rearrangement of the MLL gene in acute lymphoblastic and acute myeloid leukemias with 11q23 chromosomal translocations. New England Journal of Medicine. 1993;329:909–914. doi: 10.1056/NEJM199309233291302. [DOI] [PubMed] [Google Scholar]
- 9.Daser A, Rabbitts TH. The versatile mixed lineage leukaemia gene MLL and its many associations in leukaemogenesis. Semin Cancer Biol. 2005;15:175–188. doi: 10.1016/j.semcancer.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 10.Eguchi M, Eguchi-Ishimae M, Greaves M. The role of the MLL gene in infant leukemia. Int J Hematol. 2003;78:390–401. doi: 10.1007/BF02983811. [DOI] [PubMed] [Google Scholar]
- 11.Rubnitz JE, Camitta BM, Mahmoud H, Raimondi SC, Carroll AJ, Borowitz MJ, Shuster JJ, Link MP, Pullen DJ, Downing JR, Behm FG, Pui CH. Childhood acute lymphoblastic leukemia with the MLL-ENL fusion and t(11;19)(q23;p13.3) translocation. J Clin Oncol. 1999;17:191–196. doi: 10.1200/JCO.1999.17.1.191. [DOI] [PubMed] [Google Scholar]
- 12.Nagayama J, Tomizawa D, Koh K, Nagatoshi Y, Hotta N, Kishimoto T, Takahashi Y, Kuno T, Sugita K, Sato T, Kato K, Ogawa A, Nakahata T, Mizutani S, Horibe K, Ishii E. Infants with acute lymphoblastic leukemia and a germline MLL gene are highly curable with use of chemotherapy alone: results from the Japan Infant Leukemia Study Group. Blood. 2006 doi: 10.1182/blood-2005-11-4728. [DOI] [PubMed] [Google Scholar]
- 13.Pedersen-Bjergaard J, Andersen MK, Johansson B. Balanced chromosome aberrations in leukemias following chemotherapy with DNA-topoisomerase II inhibitors. J Clin Oncol. 1998;16:1897–1898. doi: 10.1200/JCO.1998.16.5.1897. [DOI] [PubMed] [Google Scholar]
- 14.Hahn WC, Weinberg RA. Rules for making human tumor cells. N Engl J Med. 2002;347:1593–1603. doi: 10.1056/NEJMra021902. [DOI] [PubMed] [Google Scholar]
- 15.Gilliland DG, Tallman MS. Focus on acute leukemias. Cancer Cell. 2002;1:417–420. doi: 10.1016/s1535-6108(02)00081-8. [DOI] [PubMed] [Google Scholar]
- 16.Greaves M. Pre-natal origins of childhood leukemia. Rev Clin Exp Hematol. 2003;7:233–245. [PubMed] [Google Scholar]
- 17.Higuchi M, O’Brien D, Kumaravelu P, Lenny N, Yeoh EJ, Downing JR. Expression of a conditional AML1-ETO oncogene bypasses embryonic lethality and establishes a murine model of human t(8;21) acute myeloid leukemia. Cancer Cell. 2002;1:63–74. doi: 10.1016/s1535-6108(02)00016-8. [DOI] [PubMed] [Google Scholar]
- 18.Abe R, Ryan D, Cecalupo A, Cohen H, Sandberg AA. Cytogenetic findings in congenital leukemia: case report and review of the literature. Cancer Genet Cytogenet. 1983;9:139–144. doi: 10.1016/0165-4608(83)90034-1. [DOI] [PubMed] [Google Scholar]
- 19.Ridge SA, Cabrera ME, Ford AM, Tapia S, Risueno C, Labra S, Barriga F, Greaves MF. Rapid intraclonal switch of lineage dominance in congenital leukaemia with a MLL gene rearrangement. Leukemia. 1995;9:2023–2026. [PubMed] [Google Scholar]
- 20.Hunger SP, McGavran L, Meltesen L, Parker NB, Kassenbrock CK, Bitter MA. Oncogenesis in utero: fetal death due to acute myelogenous leukaemia with an MLL translocation. Br J Haematol. 1998;103:539–542. doi: 10.1046/j.1365-2141.1998.00994.x. [DOI] [PubMed] [Google Scholar]
- 21.Greaves MF, Maia AT, Wiemels JL, Ford AM. Leukemia in twins: lessons in natural history. Blood. 2003;102:2321–2333. doi: 10.1182/blood-2002-12-3817. [DOI] [PubMed] [Google Scholar]
- 22.Adler HT, Chinery R, Wu DY, Kussick SJ, Payne JM, Fornace AJ, Jr, Tkachuk DC. Leukemic HRX fusion proteins inhibit GADD34-induced apoptosis and associate with the GADD34 and hSNF5/INI1 proteins. Mol Cell Biol. 1999;19:7050–7060. doi: 10.1128/mcb.19.10.7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wiederschain D, Kawai H, Gu J, Shilatifard A, Yuan ZM. Molecular basis of p53 functional inactivation by the leukemic protein MLL-ELL. Mol Cell Biol. 2003;23:4230–4246. doi: 10.1128/MCB.23.12.4230-4246.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Caslini C, Shilatifard A, Yang L, Hess JL. The amino terminus of the mixed lineage leukemia protein (MLL) promotes cell cycle arrest and monocytic differentiation. Proc Natl Acad Sci U S A. 2000;97:2797–2802. doi: 10.1073/pnas.040574897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiemels JL, Pagnamenta A, Taylor GM, Eden OB, Alexander FE, Greaves MF. A lack of a functional NAD(P)H:quinone oxidoreductase allele is selectively associated with pediatric leukemias that have MLL fusions. United Kingdom Childhood Cancer Study Investigators. Cancer Res. 1999;59:4095–4099. [PubMed] [Google Scholar]
- 26.Felix CA, Walker AH, Lange BJ, Williams TM, Winick NJ, Cheung NK, Lovett BD, Nowell PC, Blair IA, Rebbeck TR. Association of CYP3A4 genotype with treatment-related leukemia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:13176–13181. doi: 10.1073/pnas.95.22.13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pedersen-Bjergaard J, Pedersen M, Roulston D, Philip P. Different genetic pathways in leukemogenesis for patients presenting with therapy-related myelodysplasia and therapy-related acute myeloid leukemia. Blood. 1995;86:3542–3552. [PubMed] [Google Scholar]
- 28.Smith MA, McCaffrey RP, Karp JE. The secondary leukemias: challenges and research directions. Journal of the National Cancer Institute. 1996;88:407–418. doi: 10.1093/jnci/88.7.407. [DOI] [PubMed] [Google Scholar]
- 29.Haupt R, Fears TR, Rosso P, Colella R, Loiacono G, de Terlizzi M, Mancini A, Comelli A, Indolfi P, Donfrancesco A, et al. Increased risk of secondary leukemia after single-agent treatment with etoposide for Langerhans’ cell histiocytosis. Pediatr Hematol Oncol. 1994;11:499–507. doi: 10.3109/08880019409141688. [DOI] [PubMed] [Google Scholar]
- 30.Maraschin J, Dutrillaux B, Aurias A. Chromosome aberrations induced by etoposide (VP-16) are not random. International Journal of Cancer. 1990;46:808–812. doi: 10.1002/ijc.2910460511. [DOI] [PubMed] [Google Scholar]
- 31.Smith MA, Rubinstein L, Ungerleider RS. Therapy-related acute myeloid leukemia following treatment with epipodophyllotoxins: estimating the risks. [Review] [131 refs] Medical & Pediatric Oncology. 1994;23:86–98. doi: 10.1002/mpo.2950230205. [DOI] [PubMed] [Google Scholar]
- 32.Deveney R, Chervinsky DS, Jani-Sait SN, Grossi M, Aplan PD. Insertion of MLL sequences into chromosome band 5q31 results in an MLL-AF5Q31 fusion and is a rare but recurrent abnormality associated with infant leukemia. Genes Chromosomes Cancer. 2003;37:326–331. doi: 10.1002/gcc.10224. [DOI] [PubMed] [Google Scholar]
- 33.Caligiuri MA, Strout MP, Schichman SA, Mrozek K, Arthur DC, Herzig GP, Baer MR, Schiffer CA, Heinonen K, Knuutila S, Nousiainen T, Ruutu T, Block AW, Schulman P, Pedersen-Bjergaard J, Croce CM, Bloomfield CD. Partial tandem duplication of ALL1 as a recurrent molecular defect in acute myeloid leukemia with trisomy 11. Cancer Res. 1996;56:1418–1425. [PubMed] [Google Scholar]
- 34.Caligiuri MA, Strout MP, Lawrence D, Arthur DC, Baer MR, Yu F, Knuutila S, Mrozek K, Oberkircher AR, Marcucci G, de la Chapelle A, Elonen E, Block AW, Rao PN, Herzig GP, Powell BL, Ruutu T, Schiffer CA, Bloomfield CD. Rearrangement of ALL1 (MLL) in acute myeloid leukemia with normal cytogenetics. Cancer Res. 1998;58:55–59. [PubMed] [Google Scholar]
- 35.Felix CA, Megonigal MD, Chervinsky DS, Leonard DG, Tsuchida N, Kakati S, Block AM, Fisher J, Grossi M, Salhany KI, Jani-Sait SN, Aplan PD. Association of germline p53 mutation with MLL segmental jumping translocation in treatment-related leukemia. Blood. 1998;91:4451–4456. [PubMed] [Google Scholar]
- 36.Gu Y, Alder H, Nakamura T, Schichman SA, Prasad R, Canaani O, Saito H, Croce CM, Canaani E. Sequence analysis of the breakpoint cluster region in the ALL-1 gene involved in acute leukemia. Cancer Res. 1994;54:2326–2330. [PubMed] [Google Scholar]
- 37.Reichel M, Gillert E, Angermuller S, Hensel JP, Heidel F, Lode M, Leis T, Biondi A, Haas OA, Strehl S, Panzer-Grumayer ER, Griesinger F, Beck JD, Greil J, Fey GH, Uckun FM, Marschalek R. Biased distribution of chromosomal breakpoints involving the MLL gene in infants versus children and adults with t(4;11) ALL. Oncogene. 2001;20:2900–2907. doi: 10.1038/sj.onc.1204401. [DOI] [PubMed] [Google Scholar]
- 38.Gu Y, Cimino G, Alder H, Nakamura T, Prasad R, Canaani O, Moir DT, Jones C, Nowell PC, Croce CM, et al. The (4;11)(q21;q23) chromosome translocations in acute leukemias involve the VDJ recombinase. Proc Natl Acad Sci U S A. 1992;89:10464–10468. doi: 10.1073/pnas.89.21.10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aplan PD, Lombardi DP, Ginsberg AM, Cossman J, Bertness Vl, Kirsch IR. Disruption of the human SCL locus by "illegitimate" V-(D)-J recombinase activity. Science. 1990;250:1426–1429. doi: 10.1126/science.2255914. [DOI] [PubMed] [Google Scholar]
- 40.Tycko B, Sklar J. Chromosomal translocations in lymphoid neoplasia: a reappraisal of the recombinase model. Cancer Cells. 1990;2:1–8. [PubMed] [Google Scholar]
- 41.Kolomietz E, Meyn MS, Pandita A, Squire JA. The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer. 2002;35:97–112. doi: 10.1002/gcc.10111. [DOI] [PubMed] [Google Scholar]
- 42.Elliott B, Richardson C, Jasin M. Chromosomal translocation mechanisms at intronic alu elements in mammalian cells. Mol Cell. 2005;17:885–894. doi: 10.1016/j.molcel.2005.02.028. [DOI] [PubMed] [Google Scholar]
- 43.Jeffs AR, Benjes SM, Smith TL, Sowerby SJ, Morris CM. The Bcr gene recombines preferentially with Alu elements in complex Bcr-Abl translocations of chronic myeloid leukaemia. Human Molecular Genetics. 1998;7:767–776. doi: 10.1093/hmg/7.5.767. [DOI] [PubMed] [Google Scholar]
- 44.Super HG, Strissel PL, Sobulo OM, Burian D, Reshmi SC, Roe B, Zeleznik-Le NJ, Diaz MO, Rowley JD. Identification of complex genomic breakpoint junctions in the t(9;11) MLL-AF9 fusion gene in acute leukemia. Genes Chromosomes Cancer. 1997;20:185–195. [PubMed] [Google Scholar]
- 45.Strout MP, Marcucci G, Bloomfield CD, Caligiuri MA. The partial tandem duplication of ALL1 (MLL) is consistently generated by Alu-mediated homologous recombination in acute myeloid leukemia. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:2390–2395. doi: 10.1073/pnas.95.5.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burden DA, Osheroff N. Mechanism of action of eukaryotic topoisomerase II and drugs targeted to the enzyme. Biochimica et Biophysica Acta. 1998;1400:139–154. doi: 10.1016/s0167-4781(98)00132-8. [DOI] [PubMed] [Google Scholar]
- 47.Kas E, Laemmli UK. In vivo topoisomerase II cleavage of the Drosophila histone and satellite III repeats: DNA sequence and structural characteristics. Embo J. 1992;11:705–716. doi: 10.1002/j.1460-2075.1992.tb05103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Felix CA, Lange BJ, Hosler MR, Fertala J, Bjornsti MA. Chromosome band 11q23 translocation breakpoints are DNA topoisomerase II cleavage sites. Cancer Res. 1995;55:4287–4292. [PubMed] [Google Scholar]
- 49.Zhou RH, Wang P, Zou Y, Jackson-Cook CK, Povirk LF. A precise interchromosomal reciprocal exchange between hot spots for cleavable complex formation by topoisomerase II in amsacrine-treated Chinese hamster ovary cells. Cancer Research. 1997;57:4699–4702. [PubMed] [Google Scholar]
- 50.Bae Ys, Kawasaki I, Ikeda H, Liu Lf. Illegitimate recombination mediated by calf thymus DNA topoisomerase II in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1988;85:2076–2080. doi: 10.1073/pnas.85.7.2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahuja HG, Felix CA, Aplan PD. Potential role for DNA topoisomerase II poisons in the generation of t(11;20)(p15;q11) translocations. Genes Chromosomes Cancer. 2000;29:96–105. doi: 10.1002/1098-2264(2000)9999:9999<::aid-gcc1013>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 52.Whitmarsh RJ, Saginario C, Zhuo Y, Hilgenfeld E, Rappaport EF, Megonigal MD, Carroll M, Liu M, Osheroff N, Cheung NK, Slater DJ, Ried T, Knutsen T, Blair IA, Felix CA. Reciprocal DNA topoisomerase II cleavage events at 5′-TATTA-3′ sequences in MLL and AF-9 create homologous single-stranded overhangs that anneal to form der(11) and der(9) genomic breakpoint junctions in treatment-related AML without further processing. Oncogene. 2003;22:8448–8459. doi: 10.1038/sj.onc.1207052. [DOI] [PubMed] [Google Scholar]
- 53.Langer T, Metzler M, Reinhardt D, Viehmann S, Borkhardt A, Reichel M, Stanulla M, Schrappe M, Creutzig U, Ritter J, Leis T, Jacobs U, Harbott J, Beck JD, Rascher W, Repp R. Analysis of t(9;11) chromosomal breakpoint sequences in childhood acute leukemia: almost identical MLL breakpoints in therapy-related AML after treatment without etoposides. Genes Chromosomes Cancer. 2003;36:393–401. doi: 10.1002/gcc.10167. [DOI] [PubMed] [Google Scholar]
- 54.Allan JM, Wild CP, Rollinson S, Willett EV, Moorman AV, Dovey GJ, Roddam PL, Roman E, Cartwright RA, Morgan GJ. Polymorphism in glutathione S-transferase P1 is associated with susceptibility to chemotherapy-induced leukemia. Proc Natl Acad Sci U S A. 2001;98:11592–11597. doi: 10.1073/pnas.191211198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Reichel M, Gillert E, Nilson I, Siegler G, Greil J, Fey GH, Marschalek R. Fine structure of translocation breakpoints in leukemic blasts with chromosomal translocation t(4;11): the DNA damage-repair model of translocation. Oncogene. 1998;17:3035–3044. doi: 10.1038/sj.onc.1202229. [DOI] [PubMed] [Google Scholar]
- 56.Broeker PL, Super HG, Thirman MJ, Pomykala H, Yonebayashi Y, Tanabe S, Zeleznik-Le N, Rowley JD. Distribution of 11q23 breakpoints within the MLL breakpoint cluster region in de novo acute leukemia and in treatment-related acute myeloid leukemia: correlation with scaffold attachment regions and topoisomerase II consensus binding sites. Blood. 1996;87:1912–1922. [PubMed] [Google Scholar]
- 57.Aplan PD, Chervinsky DS, Stanulla M, Burhans WC. Site-specific DNA cleavage within the MLL breakpoint cluster region induced by topoisomerase II inhibitors. Blood. 1996;87:2649–2658. [PubMed] [Google Scholar]
- 58.Stanulla M, Wang J, Chervinsky DS, Thandla S, Aplan PD. DNA cleavage within the MLL breakpoint cluster region is a specific event which occurs as part of higher-order chromatin fragmentation during the initial stages of apoptosis. Molecular & Cellular Biology. 1997;17:4070–4079. doi: 10.1128/mcb.17.7.4070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Strissel PL, Strick R, Rowley JD, Zeleznik-Le NJ. An in vivo topoisomerase II cleavage site and a DNase I hypersensitive site colocalize near exon 9 in the MLL breakpoint cluster region. Blood. 1998;92:3793–3803. [PubMed] [Google Scholar]
- 60.Oberhammer F, Wilson Jw, Dive C, Morris Id, Hickman Ja, Wakeling Ae, Walker Pr, Sikorska M. Apoptotic death in epithelial cells: cleavage of DNA to 300 and/or 50 kb fragments prior to or in the absence of internucleosomal fragmentation. EMBO Journal. 1993;12:3679–3684. doi: 10.1002/j.1460-2075.1993.tb06042.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Razin Sv, Gromova II, Iarovaia Ov. Specificity and functional significance of DNA interaction with the nuclear matrix: new approaches to clarify the old questions. [Review] [142 refs] International Review of Cytology. 1995;162B doi: 10.1016/s0074-7696(08)62623-6. [DOI] [PubMed] [Google Scholar]
- 62.Bortner CD, Oldenburg NBE, Cidlowski JA. The role of DNA fragmentation in apoptosis. Trends in Cell Biology. 1995;5:21–26. doi: 10.1016/s0962-8924(00)88932-1. [DOI] [PubMed] [Google Scholar]
- 63.Bode J, Schlake T, Rios-Ramirez M, Mielke C, Stengert M, Kay V, Klehr-Wirth D. Scaffold/matrix-attached regions: structural properties creating transcriptionally active loci. International Review of Cytology. 1995;162A doi: 10.1016/s0074-7696(08)61235-8. [DOI] [PubMed] [Google Scholar]
- 64.Stanulla M, Chhalliyil P, Wang J, Jani-Sait SN, Aplan PD. Mechanisms of MLL gene rearrangement: site-specific DNA cleavage within the breakpoint cluster region is independent of chromosomal context. Hum Mol Genet. 2001;10:2481–2491. doi: 10.1093/hmg/10.22.2481. [DOI] [PubMed] [Google Scholar]
- 65.Bode J, Benham C, Ernst E, Knopp A, Marschalek R, Strick R, Strissel P. Fatal connections: when DNA ends meet on the nuclear matrix. J Cell Biochem Suppl Suppl. 2000;35:3–22. doi: 10.1002/1097-4644(2000)79:35+<3::aid-jcb1121>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 66.Vaughan AT, Betti CJ, Villalobos MJ. Surviving apoptosis. Apoptosis. 2002;7:173–177. doi: 10.1023/a:1014374717773. [DOI] [PubMed] [Google Scholar]
- 67.Hoeppner DJ, Hengartner MO, Schnabel R. Engulfment genes cooperate with ced-3 to promote cell death in Caenorhabditis elegans. Nature. 2001;412:202–206. doi: 10.1038/35084103. [DOI] [PubMed] [Google Scholar]
- 68.Green DR, Beere HM. Apoptosis. Mostly dead. Nature. 2001;412:133–135. doi: 10.1038/35084313. [DOI] [PubMed] [Google Scholar]
- 69.Reddien PW, Cameron S, Horvitz HR. Phagocytosis promotes programmed cell death in C. elegans. Nature. 2001;412:198–202. doi: 10.1038/35084096. [DOI] [PubMed] [Google Scholar]
- 70.Betti CJ, Villalobos MJ, Diaz MO, Vaughan AT. Apoptotic stimuli initiate MLL-AF9 translocations that are transcribed in cells capable of division. Cancer Res. 2003;63:1377–1381. [PubMed] [Google Scholar]
- 71.Libura J, Slater DJ, Felix CA, Richardson C. Therapy-related acute myeloid leukemia-like MLL rearrangements are induced by etoposide in primary human CD34+ cells and remain stable after clonal expansion. Blood. 2005;105:2124–2131. doi: 10.1182/blood-2004-07-2683. [DOI] [PubMed] [Google Scholar]