

Abstract
Nonnucleoside reverse transcriptase inhibitors (NNRTIs) target HIV-1 reverse transcriptase (RT) by binding to a pocket in RT that is close to, but distinct, from the DNA polymerase active site and prevent the synthesis of viral cDNA. NNRTIs, in particular, those that are potent inhibitors of RT polymerase activity, can also act as chemical enhancers of the enzyme's inter-subunit interactions. However, the consequences of this chemical enhancement effect on HIV-1 replication are not understood. Here, we show that the potent NNRTIs efavirenz, TMC120, and TMC125, but not nevirapine or delavirdine, inhibit the late stages of HIV-1 replication. These potent NNRTIs enhanced the intracellular processing of Gag and Gag-Pol polyproteins, and this was associated with a decrease in viral particle production from HIV-1-transfected cells. The increased polyprotein processing is consistent with premature activation of the HIV-1 protease by NNRTI-enhanced Gag-Pol multimerization through the embedded RT sequence. These findings support the view that Gag-Pol multimerization is an important step in viral assembly and demonstrate that regulation of Gag-Pol/Gag-Pol interactions is a novel target for small molecule inhibitors of HIV-1 production. Furthermore, these drugs can serve as useful probes to further understand processes involved in HIV-1 particle assembly and maturation.
Synopsis
HIV-1 encodes reverse transcriptase (RT), an enzyme that is essential for virus replication. Nonnucleoside reverse transcriptase inhibitors (NNRTIs) are allosteric inhibitors of the HIV-1 RT. In HIV-1-infected cells NNRTIs block the RT-catalyzed synthesis of a double-stranded DNA copy of the viral genomic RNA, which is an early step in the virus life cycle. Potent NNRTIs have the novel feature of promoting the interaction between the two RT subunits. However, the importance of this effect on the inhibition of HIV-1 replication has not been defined. In this study, the authors show that potent NNRTIs block an additional step in the virus life cycle. NNRTIs increase the intracellular processing of viral polyproteins called Gag and Gag-Pol that express the HIV-1 structural proteins and viral enzymes. Enhanced polyprotein processing is associated with a decrease in viral particles released from NNRTI-treated cells. NNRTI enhanced polyprotein processing is likely due to the drug binding to RT, expressed as part of the Gag-Pol polyprotein and promoting the interaction between separate Gag-Pol polyproteins. This leads to premature activation of the Gag-Pol embedded HIV-1 protease, resulting in a decrease in full-length viral polyproteins available for assembly and budding from the host cell membrane. This study provides proof-of-concept that small molecules can modulate the interactions between Gag-Pol polyproteins and suggests a new target for the development of HIV-1 antiviral drugs.
Introduction
The HIV-1 reverse transcriptase (RT) is responsible for the conversion of the viral single-stranded genomic RNA into a double-stranded proviral DNA precursor. This process is catalyzed by the RNA- and DNA-dependent polymerase and ribonuclease H activities of the enzyme. HIV-1 RT is an asymmetric dimer that consists of a 66- (p66) and a p66-derived 51-kDa (p51) subunit [1]. The RT heterodimer is the biologically active form of the enzyme; monomeric subunits are devoid of polymerase activity [2,3].
The HIV-1 RT is translated as part of a 160-kDa Gag-Pol polyprotein (Pr160gag-pol), which consists of the gag-encoded structural proteins matrix (MA, p17), capsid (CA, p24), nucleocapsid (NC, p7), and the pol-encoded viral enzymes, the protease (PR, p10), reverse transcriptase (RT, p66/p51), and integrase (IN, p31). A transframe region links the Gag and Pol domains and consists of an N-terminal octapeptide and p6pol [4]. The pol open reading frame partially overlaps with gag and is translated by a ribosomal frameshifting mechanism, which occurs in one out of 20 Gag translation events [5]. This ensures the strict maintenance of a 20:1 ratio of Gag to Gag-Pol that is important for viral assembly, replication, and the production of infectious virions [6]. During or subsequent to virus budding, the viral PR auto-activates and cleaves Gag and Gag-Pol into the structural and viral proteins, which results in the maturation of immature particles to form infectious virions [7].
While HIV-1 PR activation is a critical step in the viral life cycle, the processes required for PR activation in HIV-1-infected cells is not well defined [7,8]. It is thought that Gag-Pol multimerization during viral assembly leads to activation of the HIV-1 PR by dimerization of PR regions on separate Gag-Pol polyproteins, followed by the autocatalytic cleavage and release of a functionally active PR homodimer [7]. Although direct multimerization of Gag-Pol has not been demonstrated biochemically, several domains within Gag-Pol have been shown to influence PR activation including regions that are proximal to the C- and N-termini of PR [9–13]. If Gag-Pol dimerizes, as predicted, then HIV-1 RT, due to its size and propensity to dimerize, is likely to contribute to Gag-Pol dimerization and promote PR activation. In support of this notion, deletions or C-terminal truncations of the RT in the context of Gag-Pol leads to decreased processing of Gag and Gag-Pol and impaired virus maturation [9,11,14]. Therefore, the proper regulation of Gag and Gag-Pol processing is an essential step in the production of mature viral particles.
Nonnucleoside reverse transcriptase inhibitors (NNRTIs) are a chemically diverse group of lipophilic compounds that comprise over 30 different classes and specifically inhibit HIV-1, but not HIV-2 RT [15]. NNRTIs bind to an allosteric pocket in the p66 subunit of the RT and inhibit DNA synthesis reactions by a non-competitive mechanism of action [16,17]. Currently, three NNRTIs, namely nevirapine (NVP) [18], delavirdine (DLV) [19], and efavirenz (EFV) [20] have been approved for the treatment of HIV-1. However, the genetic threshold of resistance for all three of these drugs is low [21,22]. In this regard, the next generation of NNRTIs, such as TMC120-R147681 (dapivirine) and TMC125-R165335 (etravirine), are more promising and are active against a wide selection of NNRTI-resistant viruses [20,23,24]. Whereas EFV, TMC120, and TMC125 are potent inhibitors of HIV-1 replication, NVP and DLV have 50% inhibitory concentrations for HIV-1 replication that are at least 10-fold higher [15,20,23,25].
While inhibitors of HIV-1 RT have been successfully used to treat HIV-1-infected individuals, the emergence of strains with reduced susceptibility to these drugs is an inevitable consequence of their use [26]. Therefore, it is necessary to identify new classes of HIV-1 inhibitors. Protein–protein interactions are necessary for many viral processes that can be targeted by peptides or small molecules [27]. We and others have proposed that modulation of RT dimerization is a potential drug target [2,28]. Since the HIV-1 RT is expressed as part of the Gag-Pol polyprotein, it is conceivable that a molecule that modulates RT subunit interaction can also affect Gag-Pol/Gag-Pol interaction by binding to the embedded RT sequence, which could promote activation of the HIV-1 PR and viral polyprotein processing.
We and others have previously demonstrated that potent NNRTIs, such as EFV, act as chemical enhancers of HIV-1 RT heterodimerization, whereas other NNRTIs have little (NVP) or no effect (DLV) [29,30]. EFV also significantly enhances p66 homodimerization and the kinetics of processing of a model 90-kDa Pol precursor protein by the Pol-embedded HIV-1 PR [31]. These data prompted us to hypothesize that EFV can affect Gag-Pol processing and possibly the late stages of HIV-1 replication. The current study was designed to address this hypothesis. Here we show that EFV, TMC120, and TMC125, but not NVP or DLV, can enhance intracellular Gag and Gag-Pol processing and decrease viral particle production in HIV-1-transfected 293T and HeLa cells. This is the first time that a small molecule inhibitor has been shown to impact on the late stage of virus replication by enhancing intracellular viral protein processing, and may provide the basis for the design of inhibitors that target this stage of the viral life cycle. Furthermore, these drugs can serve as useful probes to further understand processes involved in viral particle assembly and maturation.
Results
NNRTI Enhancement of p66 Homodimer Formation In Vitro
We have previously demonstrated that the potent NNRTI, EFV, enhances p66 homodimerization in vitro [31]. To determine whether NVP, TMC120, and TMC125 also enhance p66 homodimerization, purified recombinant hexa-histidine tagged p66 (His-p66) was incubated in the presence of a 10-fold molar excess of NNRTI with respect to total protein, and analyzed by size exclusion chromatography (SEC). EFV was used as a control. Consistent with previous studies, EFV shifted the monomer-homodimer equilibrium toward p66 homodimer formation compared to His-p66 incubated in the absence of drug (Figure 1). TMC120 and TMC125 also enhanced p66 homodimerization, although the extent to which the p66 monomer was converted to the p66 homodimer was not as complete as for EFV, since detectable levels of monomers were observed. In contrast, NVP showed the weakest capacity to enhance p66 homodimerization. These data demonstrate that EFV, TMC120, and TMC125 are more potent enhancers of p66 homodimerization compared to NVP.
Figure 1. NNRTI Enhancement of p66 Homodimer Formation In Vitro.

The effect of NNRTIs on the p66 monomer-homodimer equilibrium was determined by incubating His-p66 in the absence and presence of a 10-fold molar excess of drug relative to total protein for 2 h at 4 °C followed by analysis of the quaternary structure by SEC.
EFV Increases Homodimerization of a Model Pol Construct in the Yeast Two-Hybrid System
To determine whether EFV promotes homodimerization of a polyprotein expressing p66 we used the yeast two-hybrid (Y2H) system to assess the effect of EFV on the interactions between “bait” and “prey” fusion constructs expressing a previously described 90-kDa model Pol construct comprising the C-terminus of nucleocapsid (NC), the transframe region, PR, RT, and the first 44 amino acids of IN [31,32]. The model Pol used in these experiments contains a PR active site mutation so that the bait and prey polyproteins are not proteolyzed by the embedded HIV-1 PR. In the absence of drug, the interaction between the Pol-bait and Pol-prey fusions was weak as assessed by measuring β-galactosidase (β-gal) activity. In contrast, when yeast expressing the Pol-bait and Pol-prey fusions were cultured in the presence of increasing concentrations of EFV we observed a dose-dependent increase in β-gal activity which increased 5-fold compared to the untreated control in the presence of 15 μM EFV (Figure 2). Yeast co-transformed with either the Pol-bait and the empty prey plasmid or the Pol-prey and the empty bait plasmid and grown in the absence or presence of EFV did not demonstrate detectable levels of β-gal activity, indicating lack of self-activation of the reporter gene by the model Pol fusion proteins in yeast. These data demonstrate that EFV mediates a concentration-dependent increase in the interaction between two separate model Pol proteins indicating that EFV can promote interactions between p66 RT expressing polyproteins.
Figure 2. Effect of EFV on a Model Pol Construct in the Y2H System.

Yeast strain CTY10-5d expressing a model Pol construct as both bait and prey was assayed for β-gal activity in the presence of EFV. Results are expressed as fold increase in β-gal activity compared to the untreated control. Data represent at least three independent assays and are expressed as the mean ± standard error.
EFV, TMC120, and TMC125 Increase Intracellular Gag-Pol Processing
Since EFV enhances homodimerization of p66 and the model Pol polyprotein, we hypothesized that potent NNRTIs can enhance Gag-Pol/Gag-Pol interactions resulting in increased processing of Gag-Pol due to premature activation of the HIV-1 PR. To investigate this hypothesis we analyzed the intracellular viral protein profile of pDRNL-transfected 293T cells treated with NNRTIs. The concentrations of NNRTIs tested were not cytotoxic (unpublished data) as determined by the CellTiter 96 aqueous non-radioactive cell proliferation assay (Promega, Madison, Wisconsin, United States). Cells treated with EFV, TMC120, and TMC125 demonstrated reduced levels of Pr160gag-pol compared to untreated cells as determined by quantitative Western blot analysis using RT antibodies (Figure 3A). The relative levels of the Gag-Pol processing intermediates p107 (PR-RT-IN) and p76 (PR-RT) were increased compared with p120 (NC-p6-PR-RT-IN), p113 (p6-PR-RT-IN), and p97 (RT-IN) in cells treated with EFV, TMC120, and TMC125 (Figure 3A). These processing intermediates have previously been observed in HIV-1-infected cells [33]. Significantly, we observed a 2- to 2.7-fold increase in the levels of p51 RT with respect to p66 RT in cells treated with EFV, TMC120, and TMC125 compared to the untreated controls (Figure 3A and 3B). In contrast, no significant difference was observed in the Pr160gag-pol processing pattern in cells treated with DLV, NVP, and the nucleoside reverse transcriptase inhibitor, zidovudine (AZT), indicating that enhanced Gag-Pol processing is a property of potent NNRTIs.
Figure 3. EFV, TMC120, and TMC125 Enhance Intracellular Gag and Gag-Pol Processing.

293T cells were transfected with pDRNL and treated with 5 μM of each drug. After 36 h post-transfection, cell lysates were harvested and viral proteins were detected by quantitative Western blot analysis using (A) anti-RT and (C) anti-CA antibodies. AlexFluor680 goat anti-mouse was used as the secondary antibody. Western blots were scanned on an Odyssey Infrared Imager and analyzed using Odyssey software. Quantitation of the intracellular ratio of (B) p51 to p66 and (D) p24 to Pr55Gag. The ratios were calculated from at least three independent experiments and are expressed as the mean ± standard error.
EFV, TMC120, and TMC125 Enhance Intracellular Gag Processing
If enhanced processing of Pr160gag-pol by EFV, TMC120, and TMC125 is due to increased levels of HIV-1 PR activity in the cell, we would expect to see increased processing of Gag. Quantitative Western blot analysis of cell lysates probed with p24 antibodies demonstrated a 3.2- to 3.9-fold increase in the p24 to Pr55gag ratio in cells treated with EFV, TMC120, and TMC125 compared to untreated cells and cells treated with NVP, DLV, and AZT (Figure 3C and 3D). We also observed an increase in total Gag in cells treated with potent NNRTIs suggesting an accumulation of Gag inside the cell. However, consistent with enhanced processing of Pr55gag, we found that the ratio of intracellular Pr55gag to total Gag (including the processing intermediates and p24) in cells treated with potent NNRTIs was 50% of untreated cells or cells treated with the other inhibitors (unpublished data). Paradoxically, we observed an increase in the steady-state levels of the p41 and p25 Gag processing intermediates in cells treated with EFV, TMC120, and TMC125, suggesting an additional effect on Gag processing by these NNRTIs. No significant change in the Gag processing pattern was observed in cells treated with NVP, DLV, and AZT with the p24 to Pr55gag ratios comparable to untreated cells (Figure 3C and 3D). Taken together, these data demonstrate that NNRTIs that enhance p66 homodimerization can enhance the intracellular processing of both HIV-1 Gag and Gag-Pol polyproteins.
EFV Demonstrates No Significant Effect on Virion Gag and Gag-Pol Processing Patterns
To investigate whether the Gag and Gag-Pol processing patterns were also altered in the virus, Western blot analysis was performed on virions released from HIV-1-transfected 293T cells treated with EFV. In contrast to viral proteins in cell lysates, we observed no significant change in the processing pattern of Gag and Gag-Pol in virions from EFV-treated cells compared to untreated cells and cells treated with NVP and AZT. Specifically, we detected no change in the ratios of p24 to Pr55gag (Figure 4A and 4B) and p51 RT to p66 RT (Figure 4C and 4D) in EFV-treated cells compared to untreated controls. These data suggest that enhanced viral polyprotein processing by these drugs are more apparent in the cell rather than in the virion. In contrast, we observed reduced levels of virus produced from cells treated with EFV compared to untreated cells and cells treated with NVP and AZT (Figure 4A and 4C) suggesting that EFV may affect viral particle production.
Figure 4. EFV Does Not Effect Virion Protein Processing.

Viral particles were pelleted from pDRNL-transfected 293T cells treated with 5 μM of each drug, and the Gag and Gag-Pol processing patterns were analyzed by quantitative Western blotting using (A) anti-CA and (C) anti-RT antibodies. Quantitation of the ratio of (B) p24 to Pr55Gag and (D) p51 to p66 from Western blots. The ratios were calculated from at least three independent experiments and are expressed as the mean ± standard error.
Enhanced Gag and Gag-Pol Processing Observed in a Myristoylation Defective Mutant
Our data demonstrate that EFV, TMC120, and TMC125 enhance intracellular Gag and Gag-Pol processing while no significant effect on viral protein processing was detected in the virion in the presence of EFV. To determine whether enhanced processing of Gag and Gag-Pol requires Gag/Gag-Pol localization to the plasma membrane, 293T cells were transfected with pGA2-NL4.3, a myristoylation-defective (myr-) HIV-1 mutant, in the presence of NNRTIs.
Previous studies have shown that myristoylation of HIV-1 Gag is essential for viral assembly and budding at the plasma membrane [34]. Prevention of myristoylation by introduction of a glycine to alanine substitution at the second amino acid in Gag results in the accumulation of Gag and Gag-Pol in cells and no detectable release of viral particles [34]. We observed a dramatic increase in intracellular processing of Pr55gag to p24 (6- to 7.5-fold) and p66 RT to p51 RT (2.0- to 3.3-fold) in cells treated with EFV, TMC120, and TMC125 compared to untreated cells and cells treated with NVP (Figure 5). A faint band corresponding to Gag-Pol was observed in untreated cells and cells treated with NVP, but not in cells treated with potent NNRTIs, consistent with enhanced Gag-Pol processing by potent NNRTIs (unpublished data). A similar enhancement of intracellular Gag and Gag-Pol processing was also observed in HeLa cells treated with potent NNRTIs (unpublished data). As expected, we observed 100% inhibition of viral particle production by Western blot analysis in the culture supernatant of cells transfected with the myr(-) mutant (unpublished data). Enhanced Gag and Gag-Pol processing in cells treated with potent NNRTIs and transfected with either wild-type or the myr(-) HIV-1 mutant support the notion that these drugs enhance viral protein processing within the cell.
Figure 5. Increased Viral Protein Processing by Potent NNRTIs Is Independent of Gag/GagPol Localization to the Plasma Membrane.

Cells were treated as described in Figure 3, except for transfection with pGA2-NL4.3, which expresses a myr(-) defective mutant of HIV-1. All drugs were tested at 5 μM. Cell lysates harvested after 36 h were analyzed by quantitative Western blotting using (A) anti-RT and (C) anti-CA. Quantitation of the ratio of (B) p51 to p66 and (D) p24 to Pr55gag from Western blots was performed using the Odyssey system. The ratios were calculated from at least three independent experiments and are expressed as the mean ± standard error.
Inhibition of Viral Particle Production in the Presence of EFV, TMC120, and TMC125
We have demonstrated that potent NNRTIs can increase intracellular processing of Gag and Gag-Pol, which is consistent with enhanced HIV-1 PR activity in the cell. One consequence of enhanced intracellular processing of Gag and Gag-Pol is that less viral polyproteins are available for assembly and budding from the cell, leading to a decrease in viral particle production. Enhanced Gag and Gag-Pol processing and a block in viral particle production has previously been observed when either Gag-Pol is overexpressed or expressed as a linked PR dimer in cells [6,35]. To explore this possibility, viral proteins were pulse-labeled with Trans[35S]-label in HIV-1-transfected 293T cells to measure viral particle output. EFV treatment resulted in a reduction in virion-associated p24 with the most significant decrease at 5 μM of drug (45% decrease compared to no drug) (Figure 6A and 6B). A similar reduction in viral particles released from cells treated with 5 μM of TMC120 (44% decrease) and 5 μM of TMC125 (33% decrease) was also observed. In contrast, levels of virion-associated p24 from cells treated with NVP or DLV were similar to untreated cells. Reduced viral particle production (60% decrease) was also observed in HIV-1-transfected HeLa cells treated with EFV (Figure 6C and 6D). These data suggest that the enhancement of intracellular viral protein processing mediated by potent NNRTIs leads to a reduction in viral particle production from HIV-1-transfected cells.
Figure 6. Potent NNRTIs Inhibit Viral Particle Production.

Viral particle production in drug-treated 293T cells (A) and in HeLa cells (C). Top panels represent virion-associated p24 and bottom panels are p24 present in cell lysates. Cells were transfected with pDRNL and treated with drug as described in Figure 3, except that after 36 h, cells were metabolically labeled with Trans[35S]-label for 4 h. Cell lysates were immunoprecipitated using anti-CA mAb and viral particles in the supernatant were pelleted through a sucrose cushion. The proteins were separated by SDS-PAGE and visualized and quantified using a phosphorimager. Viral particle production in 293T cells (B) and HeLa cells (D) was quantified as described in Materials and Methods. Data represent at least three independent experiments and are expressed as the mean ± standard error.
Decreased Viral Particle Production in Cells Treated with EFV Is Dependent on a Functional HIV-1 PR
The association between decreased viral particle production and enhancement of intracellular Gag and Gag-Pol processing suggests that EFV promotes the premature activation of the HIV-1 PR in the cell. Hence, inactivating the viral PR would be expected to negate the effect of EFV on viral particle production. To examine this possibility, 293T cells were transfected with PR(-)pNL4.3, which contains an active site mutation (D25A) in the HIV-1 PR, in the absence and presence of NNRTIs [34]. As expected, the PR(-) mutant released immature viral particles expressing Pr55gag (Figure 7A and 7C) and Pr160gag-pol (unpublished data). In cells treated with EFV, no significant reduction in viral particle output was observed, compared to untreated cells and cells treated with NVP (Figure 7A and 7C). These data demonstrate that inhibition of virus production by EFV is dependent on a functional HIV-1 PR.
Figure 7. Inhibition of Viral Particle Production by EFV Is Dependent on a Functional HIV-1 PR and Is Mediated by the Drug Binding to p66 RT.

293 T cells were transfected with (A) PR(-)pNL4.3 that contains an active site mutation in the HIV-1 PR or (B) K103NpNL4.3 that prevents EFV binding to RT. Cells were transfected and treated with Trans[35S]-label as described in Figure 6. Top panels represent virion-associated Gag or p24 and bottom panels represent Gag or Gag processing products present in cell lysates. Quantitation of viral particle production in 293T cells transfected with (C) PR(-)pNL4.3 and (D) K103NpNL4.3. Data represent at least three independent experiments and are expressed as the mean ± standard error.
EFV Does Not Inhibit Viral Production of EFV-Resistant HIV-1
Our previous studies suggest a correlation between enhancement of p66 homodimerization by EFV and its capacity to increase the processing kinetics of a model HIV-1 Pol construct [31]. Since p66 RT is expressed as part of the Gag-Pol polyprotein, we investigated whether EFV mediates its effect on viral particle production by binding to the p66 subunit in the context of Gag-Pol. 293T cells were transfected with K103NpNL4.3, which expresses the K103N mutation in the RT that confers resistance to EFV by preventing drug binding to the HIV-1 RT [36]. In cells treated with EFV, no significant reduction in viral particle production was observed compared to untreated cells and cells treated with NVP (Figure 7B and 7D). These data suggest that in order for EFV to inhibit viral particle production it must bind to p66 RT in the context of Gag-Pol.
EFV Has No Effect on Viral Particle Production of Moloney Murine Leukemia Virus
NNRTIs are highly specific to the HIV-1 RT, and therefore effects on viral particle production by EFV should be specific to HIV-1-transfected cells [15]. To confirm that EFV does not affect viral particle production of other retroviruses, 293T cells were transfected with an infectious molecular clone of Moloney murine leukemia virus (Mo-MuLV) in the presence of drug. We found no difference in viral particle production for EFV- and NVP-treated cells compared to the no-drug control (Figure 8). These data demonstrate that the effect of EFV on viral particle output is specific and not due to cytotoxicity mediated by this lipophillic NNRTI.
Figure 8. Effect of EFV on Mo-MuLV Particle Production.
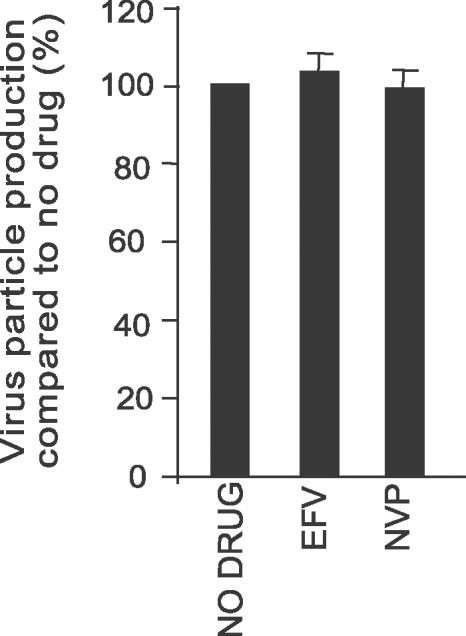
293 T cells were transfected with Mo-MuLV proviral DNA and treated as described in Figure 6, except that cell lysates were immunoprecipitated with a Mo-MuLV CA antibody. All drugs were tested at 5 μM. Viral particle production was quantified as described in Materials and Methods. Data represent at least three independent experiments and are expressed as the mean ± standard error.
Discussion
In this study, we report that potent NNRTIs EFV, TMC120, and TMC125 enhance p66 RT homodimerization and that this property correlates with the capacity of these NNRTIs to inhibit the late stages of HIV-1 replication. This effect was not observed with NVP, which has little effect on p66 homodimerization in vitro, or with AZT, a drug that belongs to the separate nucleoside analog class of RT inhibitors. EFV promoted homodimerization of a model Pol construct in the Y2H system, indicating that this drug binds to p66 in the context of a larger Pol polyprotein. Pol multimerization, leading to HIV-1 PR homodimerization and its activation, is the likely mechanism for our previous data demonstrating that EFV enhances processing of a model Pol polyprotein [31]. Potent NNRTIs also enhanced intracellular processing of Gag-Pol and Gag, and this correlated with reduced viral particle production. In addition, enhanced polyprotein processing did not require localization of Gag/Gag-Pol to the plasma membrane. The effect was dependent on a functional HIV-1 PR, as EFV did not decrease viral particle production in cells transfected with a PR(-) mutant of HIV-1. EFV also failed to decrease viral particle production of HIV-1 expressing the K103N mutation in the RT gene that confers EFV resistance indicating that the effect is specific to the drug binding to the HIV-1 RT in the context of Gag-Pol. We observed no evidence of cytotoxicity at the drug concentrations tested, and consistent with this observation we found that EFV did not reduce the output of Mo-MuLV particles from 293T cells. Therefore, in addition to the established effects of NNRTIs on inhibiting HIV-1 RT enzyme function at an early step in the virus life cycle, our data demonstrate that potent NNRTIs target the late stages of HIV-1 replication and that Gag-Pol multimerization is an effective target for the development of small molecule inhibitors against HIV-1.
What is the mechanism of increased HIV-1 polyprotein and reduced viral particle production in cells treated with highly potent NNRTIs? Our data are consistent with the explanation that potent NNRTIs specifically bind to p66 in the context of Gag-Pol in the cytoplasm of the cell, thereby promoting Gag-Pol/Gag-Pol interactions leading to premature activation of the HIV-1 PR. The PR cleaved from Gag-Pol is then available for cleavage of Gag, explaining why the effect on processing was not confined to Gag-Pol. The ultimate consequence of enhanced intracellular processing of Gag and Gag-Pol is that there are less unprocessed polyproteins in the cell available for assembly and budding at the plasma membrane leading to a reduction in viral particle production. Premature activation of the HIV-1 PR leading to a block in viral particle production has been previously described when either Gag-Pol is overexpressed [6] or expressed as a linked PR dimer in cells [35]. Our study demonstrates for the first time that a small molecule inhibitor can enhance HIV-1 polyprotein processing, leading to inhibition of viral particle production. In addition, our study underscores the importance of the RT domain and its propensity to dimerize in promoting Gag-Pol/Gag-Pol interactions and activation of the HIV-1 PR.
The levels of enhancement of intracellular Gag and Gag-Pol processing as quantified by the increase in ratios of p24 to Pr55gag (for Gag) and p51 RT to p66 RT (for Gag-Pol) were in the range of 3.2- to 3.9-fold and 2- to 2.7-fold, respectively. The decrease in viral particle output for potent NNRTIs in the same cells as determined by pulse-labeling (Figure 6) was 33%–44%. Calculation of the enhancement of processing relies on the assumption that antibody detection of precursor and processing products are equivalent on a molar level. This could account for the slightly greater effects observed for polyprotein processing compared to viral particle output. Nevertheless, our data are consistent with the assertion that NNRTI enhancement of Gag and Gag-Pol processing is associated with a decrease in viral particle production.
While we observed increased intracellular processing of Gag and Gag-Pol in the presence of potent NNRTIs, this effect was not detected in viral particles that were released in the culture supernatant. Our experiments with a myr (-) HIV-1 mutant clearly demonstrated a dramatic effect on enhanced processing of Pr55gag to p24 and p66 RT to p51 RT in 239T and HeLa cells treated with EFV, TMC120, and TMC125 compared to untreated cells. These data indicate that enhanced polyprotein processing occurs in the absence of Gag and Gag-Pol localization to the plasma membrane as previously described for myr (-) mutants of HIV-1 [34].
The impact of potent NNRTIs on the inhibition of viral particle production was observed at concentrations of inhibitor (5 μM) that are two to three orders of magnitude higher than levels required to inhibit HIV-1 replication in cell culture based assays (0.001–0.084 μM) [15,23,25]. Nevertheless, the median plasma levels of EFV in drug-treated individuals is 7 μM indicating that the effect of the drug is at physiological concentrations [37]. Furthermore, the intracellular concentrations of EFV in peripheral blood mononuclear cells from drug-treated patients are similar to the total plasma concentrations of the drug [38]. Therefore, it is possible that affects on the late stage of HIV-1 replication may contribute in part to the inhibitory activity of these drugs in vivo, although this remains to be determined.
The large differences in potency of the drugs for inhibition of RT compared to viral particle production can be explained by the relative affinity of the drugs for the mature RT heterodimer and the proposed target for the late effect, p66 RT in the context Gag-Pol. While the structure of the NNRTI binding site is well characterized in the RT heterodimer, its structure in the context of the Gag-Pol polyprotein is unknown. It is possible that while potent NNRTIs can bind to p66 in Gag-Pol, the binding is weaker than its interaction with the RT heterodimer, thereby explaining the requirement for much higher concentrations of drug to observe an effect on viral particle production compared to inhibition of RT activity.
In cells treated with potent NNRTIs we observed enhanced processing of Pr55gag to p24 CA. Paradoxically, we also observed evidence of less efficient processing of Gag by the presence of an accumulation of the p41 and p25 Gag processing intermediates, which is reminiscent of a p6 late domain defect [39]. An accumulation of total Gag was also observed in cells treated with potent NNRTIs consistent with a decrease in viral particle output. The first cleavage in Gag is between p2/NC, which would release the p6 late domain that is required for viral particle egress from the cell [40]. It is possible that a proportion of Gag molecules that have reached the plasma membrane lack p6, thus conferring a late domain effect. Electron microscopy analysis of EFV-treated HeLa cells demonstrated a reduction in viral particle production compared to untreated cells and a higher proportion of immature particles attached to the plasma membrane (unpublished data). These were generally single immature particles with stalks still attached to the plasma membrane, which were few in number but still greater in EFV-treated compared to untreated cells. While the main defect we observed both by electron microscopy and biochemical analyses was a reduction in viral output, our data also suggest a secondary late domain defect, which could be attributed to enhanced Gag processing by potent NNRTIs.
This is the first report of small molecule inhibitors that can inhibit assembly and viral particle production by mediating enhanced intracellular processing of the HIV-1 Gag and Gag-Pol polyproteins. In contrast, there are several reports of small molecules or peptides that decrease virus processing and maturation, including the HIV-1 PR inhibitors [41,42], a 12-mer peptide that targets the CA domain of Gag [43], CAP-1, which targets CA [44], and 3-O-(3′,3′-dimethylsuccinyl)-betulinic acid (DSB) which targets the cleavage of the CA precursor resulting in a block in viral core condensation [45,46]. Our studies provide proof-of-concept that a small molecule inhibitor can induce premature processing of Gag and Gag-Pol in the cell. The development of screens to detect agents that enhance Gag-Pol/Gag-Pol interactions may lead to the identification of more potent inducers of premature processing of HIV-1 polyproteins. Such a screen could be enhanced by incorporating mutations in the RT that are known to confer resistance to the current NNRTIs in order to select for agents that will be active against drug resistant strains. Furthermore, these potent NNRTIs can be used as probes to dissect the role of Gag-Pol multimerization in HIV-1 PR activation and viral assembly.
Materials and Methods
Drugs and reagents.
EFV, NVP, and AZT were obtained from the AIDS Research and Reference Reagent Program, National Institute of Allergy and Infectious Diseases, National Institutes of Health. DLV was purchased from Biomol (Plymouth Meeting, Pennsylvania, United States). TMC120 and TMC125 were supplied by Tibotec Pharmaceuticals (Mechelen, Belgium). The HIV-1 RT monoclonal antibody (mAb) 11G10 was provided by Dag Helland (University of Bergen, Norway) [47]. MAb to HIV-1 capsid (p24) was obtained from the NIH AIDS Research and Reference Reagent Program (contributed by B. Chesebro). Mo-MuLV CA antibody (NCI Number 79S-804) was provided by S. Goff (Columbia University, New York, United States).
SEC.
Recombinant p66 expressed with an N-terminal His-p66 was purified by Ni-NTA and DEAE-Sepharose chromatography as previously described. His-p66 was diluted in running buffer (50 mM sodium phosphate [pH 7.8] and 150 mM NaCl) to a final concentration of 3.8 μM in the absence and presence of a 10-fold molar excess of NNRTI relative to total protein. Protein was resolved by SEC after 2 h incubation on ice using a Superdex 200 column (Amersham Biosciences, Little Chalfont, United Kingdom) and fast protein liquid chromatography (FPLC) as previously described [31].
DNA plasmid constructs.
The plasmid pDRNL contains the NL4.3 infectious molecular clone of HIV-1 [48]. The plasmid pNCS, containing the infectious Mo-MuLV clone, was kindly provided by S. Goff.
PR(-)pNL4–3, which expresses the D25N PR active site mutation, was constructed as previously described [49]. K103NpNL4.3, expressing the K103N mutation in the RT gene, was constructed by PCR stitch mutagenesis. Primers F1-sense (5′-GCTTCAGGTTTGGGGAAGAG-3′) and F1-antisense (5′-CCAGTACTGTTACTGATTTGTTCTGTTTTAACCCTGCAGGATGTGG-3′) were used to amplify a 700-bp F1 fragment. Primers F2-sense (5′-GCAGGGTTAAAACAGAACAAATCAGTAACAGTACTGGATGTGGGCG-3′) and F2-antisense (5′-GCTACACTGGAATATTGC-3′) were used to amplify a 200-bp F2 fragment. F1 and F2 fragments were joined by PCR extension as previously described. The resulting amplimers were cloned into the Bcl I and Bst Z171 sites of pDRNL. The pGA2-NL4.3 clone expressing the G2A mutation in HIV-1 Gag was generated by stitch PCR. The primers F3-sense (5′-GGAGAGAGATGGTTGCGAGAGCGTCGGTATTAAGCG-3′) and F3-antisense (5′-TTTGGTCCTTGTCTTATGTCCAGAATGC-3′) were used to amplify an 878-bp fragment. Primers F4-sense (5′-CTCTAGCAGTGGCGCCCGAACAGGGAC-3′) and F4-antisense (5′-CGACGCTCTCGCAACCATCTCTCTCCTTCTAGCC-3′) were used to amplify a 182-bp fragment. The PCR fragments were joined by PCR extension and then cloned into the BssH II and Spe I sites in pDRNL. The Y2H constructs pGADNOT-Pol and pSH2–1-Pol express a previously described model 90-kDa HIV-1 Pol polyprotein [32]. The region representing the model Pol (corresponding to nucleotides 2083–4367 of HXB2) was amplified by PCR from pGEM:Pol using the primers 5′pol-B (5′-GTCGACTTATTTTAGCTGACATTTATC-3′) and 3′pol-B (5′-GGATCCAAGCCAACTTTTTTAGGGAAG-3′). The Bam HI-Sal I digested amplimers were cloned into the corresponding sites in pSH2–1 [50] and pGADNOT [50] to yield pGADNOT-Pol and pSH2–1-Pol, respectively. For all constructs, the introduced mutation was confirmed by sequencing and no spontaneous mutations were detected.
Cell culture and transfection.
293T and HeLa cells were maintained in Dulbecco's modified Eagle medium containing 10% fetal calf serum (DMEM-10). Cells (2 × 106) were seeded into 10 cm in diameter tissue culture plates 24 h before transfection. Cells were transfected with 10 μg of HIV-1 or Mo-MuLV plasmid and 2 μg of pEGFP (expressing enhanced fluorescent green protein) using a calcium phosphate procedure as previously described [51]. Cells were pretreated with drug immediately prior to transfection and drugs were maintained in the culture up to the time of harvesting cells and culture supernatant.
Preparation of viral and cell lysates.
At 36 h post-transfection, cell-free culture supernatants were collected and centrifuged through a 20% w/v sucrose cushion (100,000g, 1 h, 4 °C) using a SW41 rotor (Beckman L-90 ultracentrifuge, Fullerton, California, United States). Viral pellets were solubilized in 80 μl of 2X sodium dodecyl sulfate (SDS) loading buffer and heated to 95 °C for 3 min prior to Western blot analysis [51]. Cells were washed in ice-cold phosphate buffered saline without magnesium and calcium PBS(-) and centrifuged (1,000g, 5 min, 4 °C). Pellets were lysed in 500 μl TNEN buffer (55 mM Tris [pH 8.0], 75 mM NaCl, 10 mM EDTA, 0.5 % IGEPAL, 1.0 μg/ml leupeptin, 1.0 μg/ml aprotinin, 1.0 μg/ml pepstatin) for Western blot analysis or in 500 μl RIPA buffer (150 mM NaCl, 1.0% IGEPAL, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris [pH 8.0], 1.0 μg/ml leupeptin, 1.0 μg/ml aprotinin, 1.0 μg/ml pepstatin) for immunoprecipitation. Cell debris was removed by centrifugation (20,000g, 20 min, 4 °C) and clarified lysate was mixed with equal volumes of 2X SDS sample buffer and boiled. Virion- and cell-associated viral proteins, normalized for equivalent levels of EGFP, were resolved on a 10% SDS-polyacrylamide (SDS-PAGE) gel. Resolved proteins were transferred to Hybond C-extra (Amersham) followed by quantitative Western blot analysis.
Quantitative Western blot analysis.
Quantitative Western blot analysis was performed using the Odyssey Infrared Imaging System according to manufacturer's instructions (LI-COR, Lincoln, Nebraska, United States). For detection, membranes were blocked with a 1:1 v/v of Odyssey buffer (LI-COR) and PBS(-) for 1 h at room temperature followed by a 1-h incubation with anti-HIV CA p24 mAb or anti-RT 11G10 mAb diluted 1:500 and 1:100, respectively in Odyssey buffer containing 0.2% Tween 20. Membranes were washed in PBS(-) containing 0.1% Tween 20 and incubated for 1 h with a 1:10,000 dilution of AlexaFluor 680 goat anti-mouse IgG (Molecular Probes, Eugene, Oregon, United States) in Odyssey buffer containing 0.2% Tween 20 and 0.02% SDS. Viral proteins were visualized using the Odyssey Infrared Imager (LI-COR) and molecular analysis of band intensities was quantified using the Odyssey Application Software Version 1.2 (LI-COR) by measuring the integrated intensity (I.I) (also referred to as pixel volume). I.I is proportional to the amount of dye-labeled antibodies bound to the membrane.
Viral particle production.
At 36 h post-transfection, cells were starved for 30 min in DMEM lacking methionine and cysteine followed by a 4-h pulse with 125 μCi of Trans[35S]-label per ml (1175.0 Ci/mmol, PerkinElmer, Wellesley, California, United States) in the presence and absence of drug. After radio-labeling, viral particles were pelleted through a 20% w/v sucrose cushion and cells were collected, washed with PBS(-), and lysed in RIPA buffer. Cell extracts were immunoprecipitated with 5 μg of p24 mAb and 20 μl of Protein G PLUS agarose (Santa Cruz Biotechnology, Santa Cruz, California, United States). Virion proteins were resolved by 10% SDS-PAGE, and the band intensities quantified using the Bio Imaging Analyzer (Fuji Photo Film Company, Tokyo, Japan). Virus production was calculated as the amount of virion-associated Gag (p55 + p24/p25) divided by the total (cell and virion) Gag [52].
Yeast two-hybrid assay.
The yeast strain, CTY10-5d, was transformed with “bait” and “prey” constructs as previously described [53]. Quantitation of protein–protein interactions was determined by measuring β-gal activity in yeast lysates from at least three independent transformants using ortho-nitrophenyl-D-galactopyranoside (ONPG) as the substrate, as previously described [53].
Supporting Information
Accession Numbers
The GenBank (http://www.ncbi.nlm.nih.gov/Genbank) accession number for NL4.3 is U26942.
Acknowledgments
We thank Dag Helland for the supply of RT antibodies, Andy Poumbourios for the supply of p24 antibodies, Stephen Goff for Mo-MuLV CA antibody, and the National Institutes of Health AIDS Research Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH for the supply of efavirenz, nevirapine, and zidovudine.
Abbreviations
- AZT
zidovudine
- β-gal
β-galactosidase
- DLV
delavirdine
- EFV
efavirenz
- His-p66
hexa-histidine tagged p66
- IN
integrase
- Mo-MuLV
Moloney murine leukemia virus
- myr(-)
myristoylation-defective
- NC
nucleocapsid
- NNRTI
nonnucleoside reverse transcriptase inhibitor
- NVP
nevirapine
- PR
protease
- RT
reverse transcriptase
- SEC
size exclusion chromatography
- Y2H
yeast two-hybrid
Footnotes
Competing interests. Marie-Pierre de Bethune is an employee of Tibotec Pharmaceuticals, which provided TMC120 and TMC125.
Author contributions. AF and GT conceived and designed the experiments. AF and KLM performed the experiments. AF and GT analyzed the data. JM, NSC, and MPdB contributed reagents/materials/analysis tools. AF, NSC, and GT wrote the paper.
Funding. GT was supported by the National Health and Medical Research Council career development award 235102 and NHMRC project grants 235030 and 381705. KLM was supported by NHMRC project grants 235030 and 381705. AF was supported by the Burnet Institute Postgraduate Award and Monash University. NSC was supported by the National Institutes of Health R01 GM68406-01A1. JM was supported by NHMRC and the Pfizer Foundation.
References
- di Marzo Veronese F, Copeland TD, DeVico AL, Rahman R, Oroszlan S, et al. Characterization of highly immunogenic p66/p51 as the reverse transcriptase of HTLV-III/LAV. Science. 1986;231:1289–1291. doi: 10.1126/science.2418504. [DOI] [PubMed] [Google Scholar]
- Restle T, Muller B, Goody RS. Dimerization of human immunodeficiency virus type 1 reverse transcriptase. A target for chemotherapeutic intervention. J Biol Chem. 1990;265:8986–8988. [PubMed] [Google Scholar]
- Tachedjian G, Radzio J, Sluis-Cremer N. Relationship between enzyme activity and dimeric structure of recombinant HIV-1 reverse transcriptase. Proteins. 2005;60:5–13. doi: 10.1002/prot.20480. [DOI] [PubMed] [Google Scholar]
- Louis JM, Clore GM, Gronenborn AM. Autoprocessing of HIV-1 protease is tightly coupled to protein folding. Nat Struct Biol. 1999;6:868–875. doi: 10.1038/12327. [DOI] [PubMed] [Google Scholar]
- Jacks T, Power MD, Masiarz FR, Luciw PA, Barr PJ, et al. Characterization of ribosomal frameshifting in HIV-1 gag-pol expression. Nature. 1988;331:280–283. doi: 10.1038/331280a0. [DOI] [PubMed] [Google Scholar]
- Park J, Morrow CD. Overexpression of the gag-pol precursor from human immunodeficiency virus type 1 proviral genomes results in efficient proteolytic processing in the absence of virion production. J Virol. 1991;65:5111–5117. doi: 10.1128/jvi.65.9.5111-5117.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt VM. Proteolytic processing and particle maturation. Curr Top Microbiol Immunol. 1996;214:95–131. doi: 10.1007/978-3-642-80145-7_4. [DOI] [PubMed] [Google Scholar]
- Louis JM, Nashed NT, Parris KD, Kimmel AR, Jerina DM. Kinetics and mechanism of autoprocessing of human immunodeficiency virus type 1 protease from an analog of the Gag-Pol polyprotein. Proc Natl Acad Sci U S A. 1994;91:7970–7974. doi: 10.1073/pnas.91.17.7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao WH, Wang CT. Characterization of human immunodeficiency virus type 1 Pr160 gag-pol mutants with truncations downstream of the protease domain. Virology. 2004;329:180–188. doi: 10.1016/j.virol.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Zybarth G, Carter C. Domains upstream of the protease (PR) in human immunodeficiency virus type 1 Gag-Pol influence PR autoprocessing. J Virol. 1995;69:3878–3884. doi: 10.1128/jvi.69.6.3878-3884.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quillent C, Borman AM, Paulous S, Dauguet C, Clavel F. Extensive regions of pol are required for efficient human immunodeficiency virus polyprotein processing and particle maturation. Virology. 1996;219:29–36. doi: 10.1006/viro.1996.0219. [DOI] [PubMed] [Google Scholar]
- Partin K, Zybarth G, Ehrlich L, DeCrombrugghe M, Wimmer E, et al. Deletion of sequences upstream of the proteinase improves the proteolytic processing of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1991;88:4776–4780. doi: 10.1073/pnas.88.11.4776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit SC, Gulnik S, Everitt L, Kaplan AH. The dimer interfaces of protease and extra-protease domains influence the activation of protease and the specificity of GagPol cleavage. J Virol. 2003;77:366–374. doi: 10.1128/JVI.77.1.366-374.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherry E, Liang C, Rong L, Quan Y, Inouye P, et al. Characterization of human immunodeficiency virus type-1 (HIV-1) particles that express protease-reverse transcriptase fusion proteins. J Mol Biol. 1998;284:43–56. doi: 10.1006/jmbi.1998.1968. [DOI] [PubMed] [Google Scholar]
- De Clercq E. The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antiviral Res. 1998;38:153–179. doi: 10.1016/s0166-3542(98)00025-4. [DOI] [PubMed] [Google Scholar]
- Esnouf R, Ren J, Ross C, Jones Y, Stammers D, et al. Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat Struct Biol. 1995;2:303–308. doi: 10.1038/nsb0495-303. [DOI] [PubMed] [Google Scholar]
- Spence RA, Kati WM, Anderson KS, Johnson KA. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science. 1995;267:988–993. doi: 10.1126/science.7532321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merluzzi VJ, Hargrave KD, Labadia M, Grozinger K, Skoog M, et al. Inhibition of HIV-1 replication by a nonnucleoside reverse transcriptase inhibitor. Science. 1990;250:1411–1413. doi: 10.1126/science.1701568. [DOI] [PubMed] [Google Scholar]
- Romero DL, Busso M, Tan CK, Reusser F, Palmer JR, et al. Nonnucleoside reverse transcriptase inhibitors that potently and specifically block human immunodeficiency virus type 1 replication. Proc Natl Acad Sci U S A. 1991;88:8806–8810. doi: 10.1073/pnas.88.19.8806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SD, Britcher SF, Tran LO, Payne LS, Lumma WC, et al. L-743, 726 (DMP-266): A novel, highly potent nonnucleoside inhibitor of the human immunodeficiency virus type 1 reverse transcriptase. Antimicrob Agents Chemother. 1995;39:2602–2605. doi: 10.1128/aac.39.12.2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richman DD, Havlir D, Corbeil J, Looney D, Ignacio C, et al. Nevirapine resistance mutations of human immunodeficiency virus type 1 selected during therapy. J Virol. 1994;68:1660–1666. doi: 10.1128/jvi.68.3.1660-1666.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardana VV, Emini EA, Gotlib L, Graham DJ, Lineberger DW, et al. Functional analysis of HIV-1 reverse transcriptase amino acids involved in resistance to multiple nonnucleoside inhibitors. J Biol Chem. 1992;267:17526–17530. [PubMed] [Google Scholar]
- Andries K, Azijn H, Thielemans T, Ludovici D, Kukla M, et al. TMC125, a novel next-generation nonnucleoside reverse transcriptase inhibitor active against nonnucleoside reverse transcriptase inhibitor-resistant human immunodeficiency virus type 1. Antimicrob Agents Chemother. 2004;48:4680–4686. doi: 10.1128/AAC.48.12.4680-4686.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludovici DW, De Corte BL, Kukla MJ, Ye H, Ho CY, et al. Evolution of anti-HIV drug candidates. Part 3: Diarylpyrimidine (DAPY) analogs. Bioorg Med Chem Lett. 2001;11:2235–2239. doi: 10.1016/s0960-894x(01)00412-7. [DOI] [PubMed] [Google Scholar]
- Van Herrewege Y, Michiels J, Van Roey J, Fransen K, Kestens L, et al. In vitro evaluation of nonnucleoside reverse transcriptase inhibitors UC-781 and TMC120-R147681 as human immunodeficiency virus microbicides. Antimicrob Agents Chemother. 2004;48:337–339. doi: 10.1128/AAC.48.1.337-339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavel F, Hance AJ. HIV drug resistance. N Engl J Med. 2004;350:1023–1035. doi: 10.1056/NEJMra025195. [DOI] [PubMed] [Google Scholar]
- Loregian A, Marsden HS, Palu G. Protein-protein interactions as targets for antiviral chemotherapy. Rev Med Virol. 2002;12:239–262. doi: 10.1002/rmv.356. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Sluis-Cremer N, Tachedjian G. Dimerization of human immunodeficiency virus type 1 reverse transcriptase as an antiviral target. Curr Pharm Des. 2006;12:1879–1894. doi: 10.2174/138161206776873590. [DOI] [PubMed] [Google Scholar]
- Tachedjian G, Orlova M, Sarafianos SG, Arnold E, Goff SP. Nonnucleoside reverse transcriptase inhibitors are chemical enhancers of dimerization of the HIV type 1 reverse transcriptase. Proc Natl Acad Sci U S A. 2001;98:7188–7193. doi: 10.1073/pnas.121055998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venezia CF, Howard KJ, Ignatov ME, Holladay LA, Barkley MD. Effects of efavirenz binding on the subunit equilibria of HIV-1 reverse transcriptase. Biochemistry. 2006;45:2779–2789. doi: 10.1021/bi051915z. [DOI] [PubMed] [Google Scholar]
- Tachedjian G, Moore KL, Goff SP, Sluis-Cremer N. Efavirenz enhances the proteolytic processing of an HIV-1 pol polyprotein precursor and reverse transcriptase homodimer formation. FEBS Lett. 2005;579:379–384. doi: 10.1016/j.febslet.2004.11.099. [DOI] [PubMed] [Google Scholar]
- Sluis-Cremer N, Arion D, Abram ME, Parniak MA. Proteolytic processing of an HIV-1 pol polyprotein precursor: Insights into the mechanism of reverse transcriptase p66/p51 heterodimer formation. Int J Biochem Cell Biol. 2004;36:1836–1847. doi: 10.1016/j.biocel.2004.02.020. [DOI] [PubMed] [Google Scholar]
- Lindhofer H, von der Helm K, Nitschko H. In vivo processing of Pr160gag-pol from human immunodeficiency virus type 1 (HIV) in acutely infected, cultured human T-lymphocytes. Virology. 1995;214:624–627. doi: 10.1006/viro.1995.0074. [DOI] [PubMed] [Google Scholar]
- Gottlinger HG, Sodroski JG, Haseltine WA. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1989;86:5781–5785. doi: 10.1073/pnas.86.15.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krausslich HG. Human immunodeficiency virus proteinase dimer as component of the viral polyprotein prevents particle assembly and viral infectivity. Proc Natl Acad Sci U S A. 1991;88:3213–3217. doi: 10.1073/pnas.88.8.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiou Y, Ding J, Das K, Clark AD, Jr, Boyer PL, et al. The Lys103Asn mutation of HIV-1 RT: A novel mechanism of drug resistance. J Mol Biol. 2001;309:437–445. doi: 10.1006/jmbi.2001.4648. [DOI] [PubMed] [Google Scholar]
- Marzolini C, Telenti A, Decosterd LA, Greub G, Biollaz J, et al. Efavirenz plasma levels can predict treatment failure and central nervous system side effects in HIV-1-infected patients. AIDS. 2001;15:71–75. doi: 10.1097/00002030-200101050-00011. [DOI] [PubMed] [Google Scholar]
- Almond LM, Hoggard PG, Edirisinghe D, Khoo SH, Back DJ. Intracellular and plasma pharmacokinetics of efavirenz in HIV-infected individuals. J Antimicrob Chemother. 2005;56:738–744. doi: 10.1093/jac/dki308. [DOI] [PubMed] [Google Scholar]
- Gottlinger HG, Dorfman T, Sodroski JG, Haseltine WA. Effect of mutations affecting the p6 gag protein on human immunodeficiency virus particle release. Proc Natl Acad Sci U S A. 1991;88:3195–3199. doi: 10.1073/pnas.88.8.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit SC, Moody MD, Wehbie RS, Kaplan AH, Nantermet PV, et al. The p2 domain of human immunodeficiency virus type 1 Gag regulates sequential proteolytic processing and is required to produce fully infectious virions. J Virol. 1994;68:8017–8027. doi: 10.1128/jvi.68.12.8017-8027.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashorn P, McQuade TJ, Thaisrivongs S, Tomasselli AG, Tarpley WG, et al. An inhibitor of the protease blocks maturation of human and simian immunodeficiency viruses and spread of infection. Proc Natl Acad Sci U S A. 1990;87:7472–7476. doi: 10.1073/pnas.87.19.7472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayner MM, Cordova BC, Meade RP, Aldrich PE, Jadhav PK, et al. DMP 323, a nonpeptide cyclic urea inhibitor of human immunodeficiency virus (HIV) protease, specifically and persistently blocks intracellular processing of HIV gag polyprotein. Antimicrob Agents Chemother. 1994;38:1635–1640. doi: 10.1128/aac.38.7.1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sticht J, Humbert M, Findlow S, Bodem J, Muller B, et al. A peptide inhibitor of HIV-1 assembly in vitro. Nat Struct Mol Biol. 2005;12:671–677. doi: 10.1038/nsmb964. [DOI] [PubMed] [Google Scholar]
- Tang C, Loeliger E, Kinde I, Kyere S, Mayo K, et al. Antiviral inhibition of the HIV-1 capsid protein. J Mol Biol. 2003;327:1013–1020. doi: 10.1016/s0022-2836(03)00289-4. [DOI] [PubMed] [Google Scholar]
- Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, et al. PA-457: A potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc Natl Acad Sci U S A. 2003;100:13555–13560. doi: 10.1073/pnas.2234683100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Yuan X, Dismuke D, Forshey BM, Lundquist C, et al. Small-molecule inhibition of human immunodeficiency virus type 1 replication by specific targeting of the final step of virion maturation. J Virol. 2004;78:922–929. doi: 10.1128/JVI.78.2.922-929.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szilvay AM, Nornes S, Haugan IR, Olsen L, Prasad VR, et al. Epitope mapping of HIV-1 reverse transcriptase with monoclonal antibodies that inhibit polymerase and RNase H activities. J Acquir Immune Defic Syndr. 1992;5:647–657. [PubMed] [Google Scholar]
- Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, et al. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxton P, Tachedjian G, Mak J. Analysis of the contribution of reverse transcriptase and integrase proteins to retroviral RNA dimer conformation. J Virol. 2005;79:6338–6348. doi: 10.1128/JVI.79.10.6338-6348.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachedjian G, Aronson HE, Goff SP. Analysis of mutations and suppressors affecting interactions between the subunits of the HIV type 1 reverse transcriptase. Proc Natl Acad Sci U S A. 2000;97:6334–6339. doi: 10.1073/pnas.97.12.6334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapling J, Moore KL, Sonza S, Mak J, Tachedjian G. Mutations that abrogate human immunodeficiency virus type 1 reverse transcriptase dimerization affect maturation of the reverse transcriptase heterodimer. J Virol. 2005;79:10247–10257. doi: 10.1128/JVI.79.16.10247-10257.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shehu-Xhilaga M, Ablan S, Demirov DG, Chen C, Montelaro RC, et al. Late domain-dependent inhibition of equine infectious anemia virus budding. J Virol. 2004;78:724–732. doi: 10.1128/JVI.78.2.724-732.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachedjian G, Aronson HE, de los Santos M, Seehra J, McCoy JM, et al. Role of residues in the tryptophan repeat motif for HIV-1 reverse transcriptase dimerization. J Mol Biol. 2003;326:381–396. doi: 10.1016/s0022-2836(02)01433-x. [DOI] [PubMed] [Google Scholar]