

Abstract
The ability of neoplastic cells to recruit blood vasculature is crucial to their survival in the host organism. However, the evidence linking dominant oncogenes to the angiogenic switch remains incomplete. We demonstrate here that Myc, an oncoprotein implicated in many human malignancies, stimulates neovascularization. As an experimental model, we used Rat-1A fibroblasts that form vascular tumors upon transformation by Myc in immunocompromised mice. Our previous work and the use of neutralizing antibodies reveal that in these cells, the angiogenic switch is achieved via down-modulation of thrombospondin-1, a secreted inhibitor of angiogenesis, whereas the levels of vascular endothelial growth factor, a major activator of angiogenesis, remain high and unaffected by Myc. Consistent with this finding, overexpression of Myc confers upon the conditioned media the ability to promote migration of adjacent endothelial cells in vitro and corneal neovascularization in vivo. Furthermore, mobilization of estrogen-dependent Myc in vivo with the appropriate steroid provokes neovascularization of cell implants embedded in Matrigel. These data suggest that Myc is fully competent to trigger the angiogenic switch in vivo and that secondary events may not be required for neovascularization of Myc-induced tumors.
Introduction
Despite enormous interest in the Myc oncoprotein, one of the key molecules in many human cancers, the mechanisms of neoplastic transformation by Myc remain enigmatic. It is generally believed that these mechanisms can be deduced from the putative normal function of Myc, which is to promote entry into the cell cycle (1–3). Indeed, c-myc is transcriptionally inactive in the majority of growth-arrested or terminally differentiated cells but undergoes rapid activation during the course of G0−G1 transition (4, 5). c-Myc is involved in mitogenic signaling (6, 7), and its ectopic activation causes quiescent cells to reenter the cell cycle (8). Conversely, inhibition of c-myc expression using antisense oligonucleotides has been shown to impede cell proliferation (9, 10).
However, even uncontrollably dividing cells would not give rise to tumors unless their survival in the host organism was ensured. One crucial element of their survival is the ability to fight off hypoxia (11). This is achieved via recruitment of the blood vasculature, which provides an unimpeded supply of oxygen (12, 13). Normal adult tissues, however, are not capable of undergoing neovascularization due to the fact that they generally produce more inhibitors of angiogenesis than activators (14). In fibroblasts, for instance, this negative balance is achieved through high levels of Tsp-1, 3 a secreted inhibitor of angiogenesis (15). These high levels are maintained in primary cells because the tsp-1 gene is up-regulated by p53, a tumor suppressor (16). Consequently, should the cell lose both alleles of the p53 gene (which is often the case in tumors), levels of Tsp-1 plunge, and the cell acquires the angiogenic phenotype (17). Biochemical (18) and gene knockout experiments (19) reveal that the related protein Tsp-2 is an inhibitor of angiogenesis as well.
One might assume that dominant oncogenes would also exert profound effects on the expression of angiogenic factors, both positive and negative (20). The prime example of such regulation is activation by Ras of the VEGF (21, 22). There are data suggesting that Src also has the capacity to stimulate secretion of VEGF (23, 24). Src (25) and another oncoprotein, Jun (26), were reported to down-regulate Tsp-1 and Myb-Tsp-2 (27). In some systems, overexpression of Ras correlates with lower levels of Tsp-1 (28) and high levels of matrix metalloproteinases (29) that are required for endothelial cell migration. Neutralizing antibodies against the oncogenic members of the epidermal growth factor receptor family and their cognate ligands have been reported to reduce expression of VEGF and inhibit angiogenesis (30, 31). Finally, expression of several other less-studied oncogenes correlates with the abundant production of matrix metalloproteinases and other proteolytic enzymes that favor angiogenesis (reviewed in Ref. 14). However, in most cases, it is not known whether activation of the oncogene is sufficient for the angiogenic switch or whether a secondary genetic event (e.g., loss of p53) is still required.
We have demonstrated previously that overexpression of Myc in avian and rodent fibroblasts results in rapid posttranscriptional down-regulation of the tsp-1 mRNA4 and diminished levels of the Tsp-1 protein in CM (32). This raised the possibility that activation of Myc is sufficient to confer the angiogenic phenotype upon fibroblastoid cells. However, it remained to be seen whether Myc-transformed fibroblasts produced significant amounts of proangiogenic factors and whether the net balance of pro- and antiangiogenic molecules would favor angiogenesis. Furthermore, earlier studies on angiogenesis in reconstituted prostate have failed to detect a contribution of Myc to tumor vascularization in that organ, and this function has been ascribed to Ras (33), an oncoprotein with which Myc is known to cooperate in neoplastic transformation (34). Thus, although many Myc-overexpressing tumors (e.g., Burkitt’s lymphoma) are known to be highly vascular (35), it remained a distinct possibility that their vascularization had been triggered not by Myc per se but rather by secondary mutations that followed the initial transforming event. We set out to determine whether activation of Myc would result in the establishment of the angiogenic phenotype directly, in the absence of an in vivo selection for additional mutations.
To this end, we infected Rat-1A fibroblasts with retroviruses expressing either constitutively active Myc protein (LMycSN) or the chimera between Myc and the mutated estrogen receptor (pBabePuroMycER™). ER™ is refractory to endogenous steroids but can be activated by a synthetic ligand, 4-OHT (36). Rat-1A is a pseudo-normal, immortalized, p53-positive (37) cell line which, unlike primary embryo fibroblasts, is readily transformed by Myc (38). These Myc-expressing cultures were tested for their tumorigenic and angiogenic potentials in an effort to determine whether activation of Myc is sufficient to trigger the angiogenic switch in both in vitro and in vivo settings.
Results
Myc-induced Fibroblastoid Tumors Possess Extensive Vasculature
It is widely accepted that the majority of malignant, aggressively growing tumors develop new vasculature as a means to assure a constant supply of oxygen and nutrients. Some tumors, however, can tolerate surprisingly high levels of hypoxia and remain viable in a relatively avascular environment (39). To confirm that Myc-induced experimental mesenchymal tumors belong to the first group, we analyzed the neoplastic and angiogenic potentials of Rat-1A cells infected with the human c-Myc-encoding retrovirus LMycSN. Rat-1A/LMycSN or parental (Rat-1A) cells (3 × 106) were injected into the flanks of Rag-1−/− immunodeficient mice. After 2 weeks, no palpable tumors developed from Rat-1A cells (Fig. 1A, left specimen); however, their Myc-transformed counterparts formed large, aggressively growing neoplasms exceeding 1 cm in diameter. Gross examination revealed robust vascularization of both the tumor per se and the surrounding fascia (Fig. 1A, right specimen). After histological staining with H&E, more differences became apparent. Whereas the Rat-1A cell masses were composed of fusiform, collagen-producing cells (Fig. 1B), Rat-1A/LMycSN tumors contained poorly differentiated, polygonal, irregularly arranged cells with high mitotic activity (Fig. 1C). Characteristically, these masses were interlaced by very prominent capillaries and venules that were apparently well perfused, as evidenced by the presence of intact RBCs. These blood vessels could have been recruited either by a bulk of Myc-overexpressing cells or by a small number of clones that had acquired the angiogenic phenotype independently of Myc during selection in vivo. To prove that the former scenario is correct, we set out to demonstrate that cultures of unselected Myc-overexpressing fibroblasts, but not the parental cells, secrete factors that promote the formation of blood vessels. To rule out the contribution of random genetic events, all subsequent experiments were performed with pools of LMycSN-infected Rat-1A cells.
Fig. 1.

Gross appearance and histological examination of tumors formed by Rat-1A/LMycSN cells. A, the s.c. tumor formed by Rat-1A/LMycSN cells 10 days after injection into a Rag-1−/− mouse (right specimen). The cell mass representing parental Rat-1A cells is shown on the left. B and C, respectively, histological staining of the above-mentioned specimens with H&E. The white arrows in C point at blood vessels.
CM from Myc-overexpressing Cells Promote Endothelial Cell Migration and Corneal Neovascularization
To determine whether CM from Myc-transformed cells positively influence blood vessel formation, these media have to be devoid of serum because the latter contains large, uncontrollable quantities of various pro- and antiangiogenic factors including Tsp-1. However, Myc-overexpressing fibroblasts promptly undergo apoptosis when deprived of serum (40, 41). To circumvent this problem, we tested several commercially available serum-free media and found that cells constitutively expressing Myc survive best in FGM-2 medium from Clonetics (Walkersville, MD). To corroborate this notion, cells fed with FGM-2 for 24–48 h were subjected to bright-field microscopy and TUNEL assays, as described in “Materials and Methods.” As evidenced by phase-contrast microphotographs in Fig. 2, E and F, FGM-2-fed Rat-1A and Rat-1A/LMycSN cultures remained viable and were morphologically similar to cultures maintained in a serum-containing growth medium (DMEM; Fig. 2, A and B). In contrast, in serum-free DMEM, only Rat-1A cultures remained viable (Fig. 2C), but virtually all Rat-1A/LMycSN cells rounded off and detached from the plate (Fig. 2D). To confirm that no significant apoptosis took place in FGM-2 medium, we performed TUNEL assays that detect breaks in the genomic DNA characteristic of apoptosis. As illustrated by the flow cytometry profiles in Fig. 2, Rat-1A cultures remain largely apoptosis free under any conditions (Fig. 2, A′, C′, and E′), as evidenced by low percentages of FITC-labeled apoptotic cells (the fraction designated M1) and high percentages of unlabeled viable cells (the fraction designated M2). In contrast, Rat-1A/LMycSN cells only remained viable in DMEM + 10% FCS and FGM-2 media (Fig. 2, B′ and F′) where less than 10% of cells were in the M1 fraction. The same culture deteriorated rapidly in serum-free DMEM (Fig. 2, D′) where 64% of cells were in the M1 fraction 24 h after the switch. We thus concluded that CM from FGM-2-fed cells would represent mainly live cells and can be used in angiogenesis experiments.
Fig. 2.
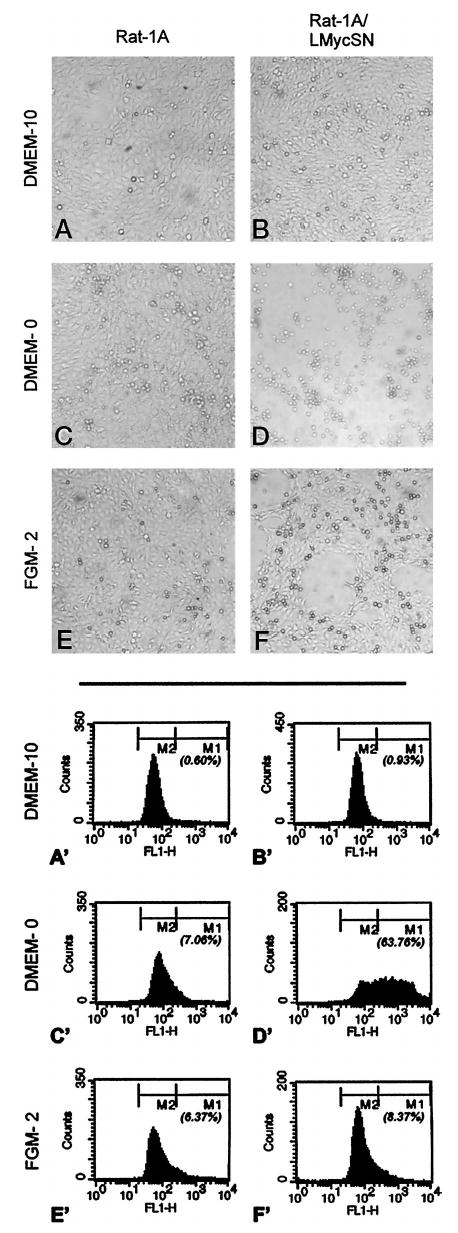
Analysis of cell viability on culturing in different media. A–F, phase-contrast microphotographs of Rat-1A and Rat-1A/LMycSN cells incubated in DMEM supplemented with 10% (DMEM-10) or 0% FCS (DMEM-0) or in FGM-2. A′–F′, flow cytometry profiles of the same cultures on fluorescent staining for DNA breaks. M1 and M2 populations refer to apoptotic and viable cells, respectively; numbers below M1 refer to the percentages of apoptotic cells.
To determine the ability of CM from Myc-overexpressing cells to influence the growth of new blood vessels, we performed a corneal neovascularization assay. In this assay, CM are introduced into a polyvinyl sponge that is then coated with Hydron and inserted into a surgically made pocket in the mouse cornea. The cornea is normally avascular, but should CM contain more proangiogenic than antiangiogenic factors, blood capillaries would grow from the limbus toward the sponge. New vessels can be visualized within 2–3 days, and their progress can be monitored. At the time the experiment is terminated, usually 7–10 days after implanting the sponge, animals are injected with high molecular weight FITC-conjugated dextran, and the corneas are examined microscopically for patent vessels. For each type of CM, at least six grafts were analyzed in two independent experiments. We found that the media from Rat-1A cells failed to promote angiogenesis, whereas the media from the same cells transformed by Myc were as angiogenic as the sponges containing bFGF, a known angiogenic factor (Fig. 3). We hypothesized that the angiogenic phenotype of Rat-1A/LMycSN cells was due to down-regulation of Tsp-1. One way to corroborate this idea was to demonstrate that the addition of a neutralizing anti-Tsp-1 antibody to the Rat-1A CM makes the medium angiogenic, as does activation of Myc. To ensure that the antibody does not diffuse out during the course of the experiment, we elected to perform a more expeditious endothelial cell chemotaxis assay because substances known to promote angiogenesis in vivo are usually able to promote the migration of endothelial cells comprising blood vessels (42).
Fig. 3.

Stimulation of corneal neovascularization by CM from Myc-expressing cells. Left column, phase-contrast microphotographs of dissected corneas; right column, fluorescence by FITC-dextran injected i.v. and marking perfused blood vessels. Sponges containing CM from Rat-1A and Rat-1A/LMycSN cells were implanted in two animals (mouse #1 and mouse #2) and elicited consistent responses (negative in the case of Rat-1A cells and positive in the case of Rat-1A/LMycSN cells). A total number of six sponges were analyzed for each CM in two independent experiments yielding similar results. bFGF was used as a control proangiogenic molecule.
In the first set of experiments, the media were assayed for the presence of factors promoting the migration of bovine adrenal capillary endothelial cells, as described in “Materials and Methods.” The indicator endothelial cells were seeded onto a porous membrane whose opposite surface was washed with the test medium. Migration of endothelial cells through the pores toward the medium and onto the opposite surface of the membrane was indicative of the angiogenic properties of the medium. As shown in Fig. 4A (left panel), CM from Rat-1A cells (□, Rat-1A cells) does not promote migration any better than the control medium containing only BSA (dotted reference line). This is due to the presence of an inhibitor because bFGF, a strong angiogenic factor in itself (▨, no cells), was not active when mixed with Rat-1A CM (▨, Rat-1A cells). This inhibitor is likely to be Tsp-1 because the inclusion of a neutralizing anti-Tsp-1 antibody, either alone ( ) or in combination with bFGF (■) fully restored migration. The same effect was achieved by substituting CM from Rat-1A cells with CM from Rat-1A/LMycSN cells (Fig. 4A, right panel). Thus, down-regulation of Tsp-1 is likely to underlie the angiogenic phenotype of Rat-1A/LMycSN cells.
Fig. 4.
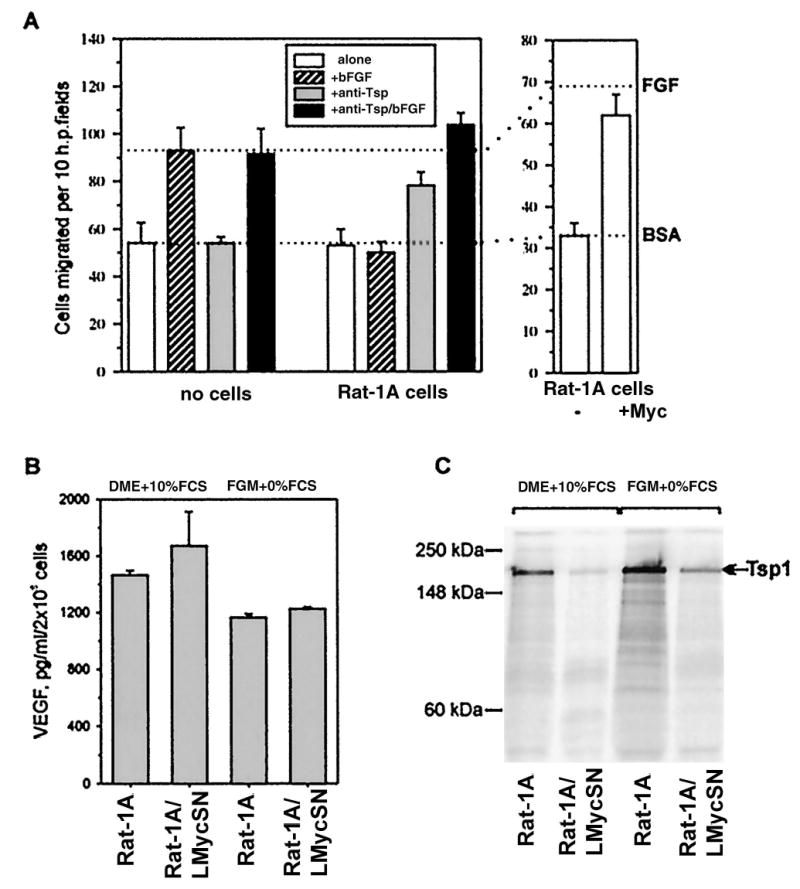
The effect of Myc on the expression of pro- and antiangiogenic factors in Rat-1A fibroblasts. A, endothelial cell chemotaxis assay performed with CM from Rat-1A and Rat-1A/LMycSN cells, as described in “Materials and Methods.” CM were used at a total protein concentration of 20 μg/ml. Supplemental reagents included the anti-Tsp-1 monoclonal antibody A4.1 (20 μg/ml) and bFGF (10 ng/ml). Serum-free DMEM supplemented with BSA was used as a negative control. Dotted line labeled BSA, background migration toward media containing BSA alone. Dotted line labeled bFGF, migration toward media containing both BSA and bFGF, a strong angiogenic factor. B, murine VEGF ELISA performed on media conditioned by Rat-1A (bars 1 and 3 ) and Rat-1A/LMycSN (bars 2 and 4 ) cells cultured in either complete (bars 1 and 2 ) or serum-free (bars 3 and 4 ) media. C, radioimmunoprecipitation of the Tsp-1 protein from media conditioned by Rat-1A (Lanes 1 and 3 ) and Rat-1A/LMycSN (Lanes 2 and 4 ) cells cultured in either complete (Lanes 1 and 2 ) or serum-free (Lanes 3 and 4 ) media. Migration of MultiMark Multi-Colored Protein Standards (Novel Experimental Technologies, San Diego, CA) is indicated on the left. Due to extensive glycosylation, Tsp-1 has an apparent molecular mass of approximately 170 kDa, as indicated by the arrow on the right.
To rule out the possibility that the angiogenic phenotype is dictated by up-regulation of a proangiogenic molecule, we compared the levels of the VEGF in CM from Rat-1A and Rat-1A/LMycSN cells. VEGF and bFGF are two major proangiogenic factors expressed by cultured fibroblasts; however, the steady-state levels of bFGF can be misleading because this protein is usually bound to the extracellular matrix and may not be biologically active. Using a rodent VEGF-specific ELISA, we observed, in all CM tested, high levels of VEGF [>1 ng/ml media conditioned by 2 × 106 cells (Fig. 4B)]. Even before concentrating, these levels exceed the ED50 for VEGF (43) and thus can account for the angiogenic potential of Rat-1A cells deprived of Tsp-1. Importantly, no significant differences between Rat-1 and Rat-1A/LMycSN cells were observed. In contrast, levels of Tsp-1 were sharply reduced in Myc-transformed cells, as evidenced by the immunoprecipitation experiment (Fig. 4C). This down-regulation was apparent in cells maintained in either complete medium or FGM-2. All these findings were consistent with the idea that it is down-regulation of the tsp-1 gene by Myc that triggers the angiogenic switch in Rat-1A cells.
Transient Activation of Myc in Vivo Results in the Acquisition of the Angiogenic Phenotype
Having demonstrated that overexpression of Myc in cultured fibroblasts renders them angiogenic, we set out to determine whether activation of Myc in vivo has the same effect and whether Myc is all that is necessary to induce angiogenesis. Both questions could be addressed by transient activation of the conditional mutant of Myc (MycER), whose activity can be regulated in vivo by treatment with 4-OHT (44). To facilitate the delivery of 4-OHT to MycER-expressing cells, we chose to perform the assay where test cells are embedded in Matrigel and injected between the dermis and the peritoneum of the mouse (45), a site that, unlike the cornea, is rich in preexisting blood vessels. Furthermore, because Matrigel is derived from the extracellular matrix of a murine sarcoma originating from the basement membrane (46, 47), it more or less faithfully simulates the normal extracellular environment of the connective tissue (48). Additionally, the analysis of vascularization is convenient because normally fluid Matrigel, along with embedded cells, solidifies at 37°C and forms easily excisable pellets. In a typical experiment, parental or Myc-expressing Rat-1A cells were mixed with chilled Matrigel at 4°C and injected into C57BL/6 mice. After 10 days, the animals were sacrificed, and the Matrigel pellets were excised, examined visually, reliquified at 4°C, and further assayed for hemoglobin content as described in “Materials and Methods” or subjected to histological staining.
In the first experiment, we determined that Myc-overexpressing Rat-1A cells are positive in this assay and that 4-OHT, an antiestrogen (49), does not interfere with the assay by further promoting or suppressing angiogenesis. To this end, we implanted mice with “empty” or Rat-1A/ LMycSN-containing Matrigels and treated the animals with either a suspension of 4-OHT in olive oil or vehicle alone. The drug and the vehicle were introduced directly into the alimentary canals using gastric gavage, as described in “Materials and Methods.” We observed that “empty” Matrigels were avascular in either drug- or vehicle-treated mice (Fig. 5A, □), but Matrigels with Myc-transformed Rat-1A cells (Rat-1A/LMycSN) were undergoing robust neovascularization (Fig. 5A, ■). On average, they contained five times more hemoglobin than “empty” Matrigels or Matrigels containing parental Rat-1A cells (Fig. 5B; data not shown). Importantly, neovascularization was not affected by 4-OHT because the slight difference between −4-OHT and +4-OHT was neither reproducible nor statistically significant. Hence, any effect that 4-OHT might have on neovascularization of Matrigels containing Rat-1A/MycER cells would not be due to changes in the hormone levels of the host.
Fig. 5.

Neovascularization of Matrigel implants containing Myc-expressing Rat-1 cells. Cells tested were expressing either constitutively active Myc (A) or the MycER derivative (B–F). A–C, the hemoglobin contents of implants from vehicle (−OHT)- and OHT (+OHT)-treated animals. MG refers to empty Matrigels, and Rat and Myc refer to Matrigels containing parental and LMycSN-infected Rat-1A cells, respectively. MERmass and MER-9 refer, respectively, to Matrigels containing mass cultures and a single cell clone of BabePuroMycER-infected Rat1A-cells. Each experiment was repeated at least twice, yielding similar results. Each bar, except for that of Matrigel alone, represents data obtained from at least four animals (2 pellets/animal). In the case of Rat-1A/MycER-9 cells, a total of 12 animals were analyzed. The variability in the experiment with 4-OHT-treated LMycSN Matrigels was negligible, resulting in an invisible error bar. D, gross appearance of Matrigels containing uninduced (left) and 4-OHT-induced (right) Rat-1A/My-cER-9 cells. E and F, histological staining of the two Matrigels shown in D (left and right, respectively). Small white arrows point at blood vessels in the surrounding connective tissue. Large white arrows point at blood vessels traversing the Matrigel.
The Matrigel neovascularization assay was repeated on Rat-1A/MycER cells in animals treated with either vehicle alone or 4-OHT. To account for possible differences in vascular potentials of different clones, we performed this experiment with the pool of clones selected for resistance to puromycin and proven to express the MycER gene (data not shown). Importantly, this pool had never been exposed to 4-OHT, and the cells retained their normal phenotype at the beginning of each experiment. As a negative control, we used parental Rat-1A cells that were not expected to respond to 4-OHT. Indeed, Rat-1A-containing Matrigel pellets were consistently avascular in both vehicle- and 4-OHT-treated animals, resulting in low hemoglobin contents (Fig. 5B, two left bars). Rat-1A/MycER-expressing cells, however, were avascular in control animals, but the majority of pellets in 4-OHT-treated mice were vascularized rather heavily (Fig. 5B, two right bars). Consequently, although some pellets exhibited poor neovascularization (presumably due to insufficiently high levels of functional Myc), resulting in average hemoglobin levels slightly lower than those observed with LMycSN cells, the difference between vehicle- and 4-OHT-treated Matrigels was reproducible and statistically significant (P < 0.03). When individual Rat-1A/MycER clones were tested, consistent neovascularization was also observed in 4-OHT-treated animals but not in control animals. Hemoglobin content data for one of these clones (MER-9) are shown in Fig. 5C. Interestingly, gross examination of Matrigel pellets revealed that activation of Myc also results in increased neovascularization of surrounding fascia that develops a network of small vessels in 4-OHT-treated mice (Fig. 5D, right specimen) but not in control (left specimen) mice.
To confirm that MycER-expressing cells promote the growth of bona fide blood vessels, MER-9 pellets were also embedded in paraffin blocks and subjected to standard H&E histological staining. In vehicle-treated animals, we observed a small number of blood vessels in the connective tissue surrounding the Matrigel (white arrow); however, no blood vessels have traversed the Matrigel pellets, as evidenced by the lack of RBCs (Fig. 5E). In contrast, in the Matrigels from 4-OHT-treated mice (Fig. 5F), rather large vessels that contained numerous RBCs were readily detectable. Thus, transient activation of Myc in rodent fibroblasts can stimulate neovascularization in the absence of an in vivo selection for angiogenic clones and independently of tumor development.
Discussion
According to the paradigm, the transforming potential of the Myc oncoprotein is based essentially on its ability to promote unwarranted proliferation of quiescent cells, which are either deprived of stimulatory growth factors or exposed to inhibitory growth factors. Consistent with this idea, the majority of its putative target genes control entry into and progression through the cell cycle (1–3), with cdc25 phosphatase being the prime example (50). All those genes are up-regulated by Myc through interaction with Max (51) and subsequent binding to the so-called E-box DNA element (52–54). Myc is also known to act as a transcriptional repressor (55), and some of those targets also relate to cell cycle (56). To complicate the matter, there is a host of “orphan” targets whose relationship to neoplastic transformation by Myc remains obscure (57). Our initial identification of Tsp-1, a known inhibitor of angiogenesis, as a Myc-repressed gene afforded new insights into the role of Myc in neoplastic transformation (32). Specifically, it implicated Myc overexpression as a possible reason for the acquisition of the angiogenic phenotype by the target cell. In this study, we demonstrate that this indeed is the case and that activation of the myc oncogene in untransformed cells is sufficient to render them angiogenic.
This result argues that the oncogenic potential of the Myc protein has a dual basis. Not only does Myc trigger unwarranted proliferation of the preneoplastic cells, but it also, at least in fibroblasts, allows them to undergo neovascularization, which is known to promote survival and metastatic spread of neoplastic cells. Down-regulation of Tsp-1 turns out to be sufficient to tilt the balance of secreted growth factors toward inducers of angiogenesis, which are now able to realize their potential. Indeed, in rodent fibroblast, we observed abundant expression of VEGF, one of the major proangiogenic molecules, on which activation of Myc had no considerable effect. This suggests that high levels of Tsp-1 expression in parental Rat-1A cells are required to counteract the angiogenic potential of VEGF. Although we cannot rule out the possibility that some other yet-to-be-identified proangiogenic factors are positively influenced by Myc in Rat-1A fibroblasts, in our in vitro assays, neutralizing anti-Tsp-1 antibodies exert exactly the same effect as activation of Myc. We are thus inclined to believe that down-regulation of Tsp-1 by Myc, just like its down-regulation via loss of p53 (16), is sufficient to explain the angiogenic switch during neoplastic transformation. This effect of Myc would be of particular importance in cells retaining functional p53, such as Rat-1A cells (37). Admittedly, during normal development the outcome of tsp-1 loss can be less dramatic because the tsp-1 knockout mice do not exhibit exaggerated angiogenesis (58). Thus, regulation by Myc of other gene products might contribute to angiogenesis. One interesting possibility is that Myc affects expression of the closely related Tsp-2 gene, whose disruption in the germ line can provoke exaggerated vascularization (19). In any event, it appears likely that the incipient Myc-overexpressing tumor would not need to undergo selection for angiogenic clones.
Myc therefore has the potential to act as a “complete” oncogene capable of inducing tumors rapidly and without relying on propitious genetic events, e.g., loss of p53. This idea is supported by a recent finding that activation of MycER chimera in keratinocytes leads not only to uncontrolled proliferation but also to appreciable angiogenesis (59). It remains to be seen whether similar events occur in other cell types and whether they involve Tsp. The tsp-1 mRNA is not expressed in either parental or Myc-transformed B lymphocytes,5 implicating other angiogenic factors in Myc-induced tumors of B lineage. Furthermore, overexpression of N-Myc in human neuroblastoma cells has been reported to down-modulate several inhibitors of endothelial cell proliferation, none of which has the biochemical properties of Tsp-1 (60). However, in colon cancers, in which Myc is often over-expressed due to the loss of the APC tumor suppressor (61), inactivation of Tsp-1 is thought to play an important role in the angiogenic switch (62). Although the patterns of regulation of the tsp-1 gene in primary and Myc-overexpressing colonocytes has not as yet been established, the ability of Myc to function as an angiogenic trigger is likely to be exploited in these cells and in other neoplasias.
Materials and Methods
Generation and Propagation of Myc-expressing Clones
Rat-1A cells and Rat-1A-derived clones were maintained in either DMEM supplemented with 10% FCS (Life Technologies, Inc., Gaithersburg, MD) or in fibroblast growth medium (FGM-2; Clonetics). The Rat-1A/LMycSN cells have been described previously (32). The pBabePuro vector (63) expressing the MycER™ protein (36) was introduced into the Rat-1A cells via electroporation, which was performed using the Bio-Rad apparatus and the following conditions: 320 V and 960 μF. Selection of Rat-1A clones producing MycER was performed in puromycin-containing media (1 mg/ ml; Sigma, St. Louis, MO). 4-OHT was purchased from Research Biochemicals International (Natick, MA) or Sigma (catalogue number H6278). Cells to be used as a source of CM were grown in complete medium on 10-cm dishes to approximately 90% confluence. They were then washed twice with Ca2+-, Mg2+-free PBS and placed in serum-free FGM-2 medium for 24–36 h. CM were collected and chilled on ice. The protease inhibitor phenylmethylsulfonyl fluoride (Sigma) was added to the final concentration of 0.1 mM. CM were then spun in Ultrafree concentrators (Millipore, Bedford, MA) with a cutoff size of 5 kDa for approximately 2 h, until the volume of the retentate decreased to 0.25 ml. The retentate was then diluted with 15 ml of Ca2+-,Mg2+-free PBS, and the centrifugation was repeated. The final protein concentrations in CM were in the range of 1–1.5 mg/ml.
Tumor Growth
Rat-1A or Rat-1A/LMycSN cells (1 × 106) harvested from semiconfluent plates were injected s.c. into the flanks of immunodeficient Rag-1−/− mice. Implanted cell masses were excised 10 days later, weighed, photographed, fixed in formalin, and subjected to standard histological staining with H&E.
TUNEL Apoptosis Assay
Cells were plated in DMEM supplemented with 10% FCS at ~60% density (6.5 × 105 cells/35-mm plate) and allowed to attach overnight. They were then washed twice with Ca2+ -, Mg2+ -free PBS and refed with either DMEM + 10% FCS, serum-free DMEM, or FGM-2 (Clonetics). Twenty-four h later, both adherent and floating cells were collected, counted, washed, and resuspended in cold 1% BSA in PBS at a concentration of 107 cells/ml. Aliquots (100 ml) were used in the TUNEL assay with the kit and the protocol from Boehringer Mannheim. DNA breaks characteristic of apoptosis were labeled with FITC-dUTP, followed by detection of labeled cells using a flow cytometer (Becton Dickinson).
Corneal Neovascularization Assay
Polyvinyl sponges preirradiated with 2000 Gy of γ-irradiation (cesium source) were cut into 0.4 × 0.4 × 0.2 mm pieces, and test material (0.2–0.5 μl) was introduced into each sponge. The loaded sponges were dried, covered with a layer of 12% Hydron, and then dried under vacuum. Sponges were then introduced into a surgically created micropocket created in the avascular cornea of adult mice as described previously (64). For positive control, sponges were loaded with 100 ng of bFGF. Mouse eyes were examined for neovascularization daily, using an ophthalmic microscope. On day 7 after implantation, 300 μl of FITC-conjugated high molecular weight dextran (Mr 1,000,000–2,000,000) were injected into the tail vein, and the animal was sacrificed after 5 min. The eyeball was enucleated from the orbital cavity and fixed for 5 min in 4% paraformaldehyde. The cornea with the adjacent limbus was dissected, rinsed in PBS, and mounted on a glass slide in 10% glycerol. Phase-contrast microscopy and fluorescence microscopy were used to visualize the general layout of the cornea and the presence of perfused blood vessels, respectively.
Endothelial Cell Chemotaxis Assay
The bovine adrenal capillary endothelial cells were maintained in DMEM supplemented with 10% donor calf serum, 2 mM glutamine, and 100 μg/ml endothelial cell mitogen. For the endothelial cell chemotaxis assay, serum-starved cells were plated in serum-free medium supplemented with 0.1% BSA on the bottom side of gelatinized microporous membrane (pore size, 5 μm; Nucleopore Corp.) in the inverted modified Boyden chamber (Neuroprobe Science). The cells were allowed to attach for 1–1.5 h, and then the chambers were reinverted, and test substances in media (20 mg/ml) were added to the top part of the wells and incubated for 3–4 h. The chambers were then disassembled, the membranes were fixed and stained, and the number of cells that migrated to the top part of the filter was determined in 10 high-power fields.
VEGF Assay
Rat-1A or Rat-1A/LMycSN cells (5 × 105 cells) were seeded 24 h before the experiment in duplicate in 35-mm plates. Cells were refed with regular growth medium or FGM-2 and incubated for an additional 24 h. CM (500 μl) were harvested to determine VEGF concentrations. The mouse VEGF ELISA kit from R&D was used according to the manufacturer’s recommendations. Optical densities were measured using a plate reader (Fisher).
Radioimmunoprecipitation
Approximately 5 × 106 cells grown in either DMEM/10% FCS or FGM-2 were labeled for 4 h with 50 μCi of [35S]methionine (DuPont New England Nuclear, Boston, MA) in DMEM/5% dialyzed FCS. Cells were lysed in the Ab buffer (65). The A4.1 anti-Tsp-1 monoclonal antibody (2.5 μl; Life Technologies, Inc.) was added to cell lysates and incubated overnight at 4°C. Secondary goat antimouse IgM antiserum (2.5 μl; TAGO Inc., Camarillo, CA) was added, along with 50 μl of Gamma-Bind G-Sepharose (Pharmacia Biotech, Piscataway, NJ), and incubation continued at room temperature for another 90 min. Standard collection and washing techniques were applied, and the immunoreactive proteins were run on 7.5% denaturing PAGE.
Matrigel Angiogenesis Assay
Logarithmically growing Rat1A, Rat1A/LMycSN, and Rat1A/MycER cells were harvested by treatment with 1 mM EDTA in Ca2+ -, Mg2+ -free PBS. Cell pellets were resuspended in cold Ca2+ -, Mg2+ -free PBS at a concentration of 108 cells/ml. Five × 106 cells in 50 μl were combined with 0.5 ml of thawed Matrigel (Collaborative Biomedical Products, Bedford, MA) and placed briefly on ice. Using a 27-gauge needle, cells were then injected s.c. between the dermis and the peritoneum of 5–7-week-old C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME). Aliquots (50 mg) of 4-OHT powder (Sigma) were resuspended in 2.5 ml of pharmacy-grade olive oil using a Model 50 Sonic Dismembrator (Fisher Scientific, Hampton, NH). 4-OHT was delivered p.o. (5 mg in 250 μl of olive oil) every 2 days using a 20-gauge intubation needle (Popper and Sons, Inc., New Hyde Park, NY). Animals in the control groups received the same dose of pure oil. Matrigel pellets were excised after 10 days and reliquefied for 24 h at 4°C in 0.3 ml of either PBS or the Matrisperse solution (Collaborative Biomedical Products). When a significant fraction of Matrigel-embedded cells remained in the solid phase (due to vigorous cell proliferation), pellets were further disrupted using an ultrasonic dismembrator. Hemoglobin content was determined using the Drabkin reagent (Sigma). Alternatively, solid Matrigel pellets, along with the surrounding connective tissue, were photographed and fixed in 10% phosphate-buffered formalin, embedded in paraffin blocks, cut into thin sections, and stained with H&E.
Acknowledgments
We thank Drs. William M. F. Lee (University of Pennsylvania, Philadelphia, PA), Maxine Linial (Fred Hutchinson Cancer Research Center, Seattle, WA), and Noel Bouck (Northwestern University, Chicago, IL) for advice and many helpful discussions. We thank Dr. Michael Goldschmidt (University of Pennsylvania) for help with histological analyses. The plasmid pBabePuroMycER was a kind gift of Martin Eilers (European Molecular Biology Laboratory, Heidelberg, Germany). The bovine adrenal capillary endothelial cells were a kind gift of Dr. J. Folkman (Harvard Medical School, Boston, MA).
Footnotes
Supported by National Cancer Institute Grant R29 CA 71881 and a grant from the University of Pennsylvania Research Foundation (to A. T-T.). During the preliminary phase of this work, A. T-T. was a Special Fellow of the Leukemia Society of America.
The abbreviations used are: Tsp, thrombospondin; VEGF, vascular endothelial growth factor; bFGF, basic fibroblast growth factor; 4-OHT, 4-hydroxytamoxifen; CM, conditioned media; TUNEL, terminal deoxynucleotidyl transferase-mediated nick end labeling.
A. Janz, C. Sevignani, K. Kenyon, C. V. Ngo, and A. Thomas-Tikhonenko. Activation of the Myc oncoprotein leads to increased turnover of the thrombospondin-1 mRNA, submitted for publication.
C. V. Ngo and A. Thomas-Tikhonenko, unpublished observations.
References
- 1.Bouchard C, Staller P, Eilers M. Control of cell proliferation by Myc. Trends Cell Biol. 1998;8:202–206. doi: 10.1016/s0962-8924(98)01251-3. [DOI] [PubMed] [Google Scholar]
- 2.Zornig M, Evan GI. Cell cycle: on target with Myc. Curr Biol. 1996;6:1553–1556. doi: 10.1016/s0960-9822(02)70769-0. [DOI] [PubMed] [Google Scholar]
- 3.Grandori C, Eisenman RN. Myc target genes. Trends Biochem Sci. 1997;22:177–181. doi: 10.1016/s0968-0004(97)01025-6. [DOI] [PubMed] [Google Scholar]
- 4.Campisi J, Gray HE, Pardee AB, Dean M, Sonenshein GE. Cell-cycle control of c-myc but not c-ras expression is lost following chemical transformation. Cell. 1984;36:241–247. doi: 10.1016/0092-8674(84)90217-4. [DOI] [PubMed] [Google Scholar]
- 5.Kelly K, Cochran BH, Stiles CD, Leder P. Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell. 1983;35:603–610. doi: 10.1016/0092-8674(83)90092-2. [DOI] [PubMed] [Google Scholar]
- 6.Roussel MF, Cleveland JL, Shurtleff SA, Sherr CJ. Myc rescue of a mutant CSF-1 receptor impaired in mitogenic signalling. Nature (Lond) 1991;353:361–363. doi: 10.1038/353361a0. [DOI] [PubMed] [Google Scholar]
- 7.Shibuya H, Yoneyama M, Ninomiya-Tsuji J, Matsumoto K, Taniguchi T. IL-2 and EGF receptors stimulate the hematopoietic cell cycle via different signaling pathways: demonstration of a novel role for c-myc. Cell. 1992;70:57–67. doi: 10.1016/0092-8674(92)90533-i. [DOI] [PubMed] [Google Scholar]
- 8.Eilers M, Picard D, Yamamoto KR, Bishop JM. Chimaeras of Myc oncoprotein and steroid receptors cause hormone-dependent transformation of cells. Nature (Lond) 1989;340:66–68. doi: 10.1038/340066a0. [DOI] [PubMed] [Google Scholar]
- 9.Biro S, Fu YM, Yu ZX, Epstein SE. Inhibitory effects of antisense oligodeoxynucleotides targeting c-myc mRNA on smooth muscle cell proliferation and migration. Proc Natl Acad Sci USA. 1993;90:654–658. doi: 10.1073/pnas.90.2.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heikkila R, Schwab G, Wickstrom E, Loke SL, Pluznik DH, Watt R, Neckers LM. A c-myc antisense oligodeoxynucleotide inhibits entry into S phase but not progress from G0 to G1. Nature (Lond) 1987;328:445–450. doi: 10.1038/328445a0. [DOI] [PubMed] [Google Scholar]
- 11.Kinzler KW, Vogelstein B. Life (and death) in a malignant tumour. Nature (Lond) 1996;379:19–20. doi: 10.1038/379019a0. [DOI] [PubMed] [Google Scholar]
- 12.Fidler IJ, Ellis LM. The implications of angiogenesis for the biology and therapy of cancer metastasis. Cell. 1994;79:185–188. doi: 10.1016/0092-8674(94)90187-2. [DOI] [PubMed] [Google Scholar]
- 13.Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 14.Bouck N, Stellmach V, Hsu SC. How tumors become angiogenic. Adv Cancer Res. 1996;69:135–174. doi: 10.1016/s0065-230x(08)60862-3. [DOI] [PubMed] [Google Scholar]
- 15.Good DJ, Polverini PJ, Rastinejad F, Le Beau MM, Lemons RS, Frazier WA, Bouck NP. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci USA. 1990;87:6624–6628. doi: 10.1073/pnas.87.17.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science (Washington DC) 1994;265:1582–1584. doi: 10.1126/science.7521539. [DOI] [PubMed] [Google Scholar]
- 17.Stellmach V, Volpert OV, Crawford SE, Lawler J, Hynes RO, Bouck N. Tumour suppressor genes and angiogenesis: the role of TP53 in fibroblasts. Eur J Cancer. 1996;32A:2394–2400. doi: 10.1016/s0959-8049(96)00385-1. [DOI] [PubMed] [Google Scholar]
- 18.Volpert OV, Tolsma SS, Pellerin S, Feige JJ, Chen H, Mosher DF, Bouck N. Inhibition of angiogenesis by thrombospondin-2. Biochem Biophys Res Commun. 1995;217:326–332. doi: 10.1006/bbrc.1995.2780. [DOI] [PubMed] [Google Scholar]
- 19.Kyriakides TR, Zhu YH, Smith LT, Bain SD, Yang Z, Lin MT, Danielson KG, Iozzo RV, LaMarca M, McKinney CE, Ginns EI, Bornstein P. Mice that lack thrombospondin 2 display connective tissue abnormalities that are associated with disordered collagen fibrillogenesis, an increased vascular density, and a bleeding diathesis. J Cell Biol. 1998;140:419–430. doi: 10.1083/jcb.140.2.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kerbel RS, Viloria-Petit A, Okada F, Rak J. Establishing a link between oncogenes and tumor angiogenesis. Mol Med. 1998;4:286–295. [PMC free article] [PubMed] [Google Scholar]
- 21.Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T, Kerbel RS. Mutant ras oncogenes up-regulate VEGF/VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Res. 1995;55:4575–4580. [PubMed] [Google Scholar]
- 22.Larcher F, Robles AI, Duran H, Murillas R, Quintanilla M, Cano A, Conti CJ, Jorcano JL. Up-regulation of vascular endothelial growth factor/vascular permeability factor in mouse skin carcinogenesis correlates with malignant progression state and activated H-ras expression levels. Cancer Res. 1996;56:5391–5396. [PubMed] [Google Scholar]
- 23.Jiang BH, Agani F, Passaniti A, Semenza GL. V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: involvement of HIF-1 in tumor progression. Cancer Res. 1997;57:5328–5335. [PubMed] [Google Scholar]
- 24.Mukhopadhyay D, Tsiokas L, Zhou XM, Foster D, Brugge JS, Sukhatme VP. Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature (Lond) 1995;375:577–581. doi: 10.1038/375577a0. [DOI] [PubMed] [Google Scholar]
- 25.Slack JL, Bornstein P. Transformation by v-src causes transient induction followed by repression of mouse thrombospondin-1. Cell Growth Differ. 1994;5:1373–1380. [PubMed] [Google Scholar]
- 26.Mettouchi A, Cabon F, Montreau N, Vernier P, Mercier G, Blangy D, Tricoire H, Vigier P, Binetruy B. SPARC and thrombospondin genes are repressed by the c-jun oncogene in rat embryo fibroblasts. EMBO J. 1994;13:5668–5678. doi: 10.1002/j.1460-2075.1994.tb06905.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bein K, Ware JA, Simons M. Myb-dependent regulation of thrombospondin 2 expression. Role of mRNA stability. J Biol Chem. 1998;273:21423–21429. doi: 10.1074/jbc.273.33.21423. [DOI] [PubMed] [Google Scholar]
- 28.Zabrenetzky V, Harris CC, Steeg PS, Roberts DD. Expression of the extracellular matrix molecule thrombospondin inversely correlates with malignant progression in melanoma, lung and breast carcinoma cell lines. Int. J Cancer. 1994;59:191–195. doi: 10.1002/ijc.2910590209. [DOI] [PubMed] [Google Scholar]
- 29.Arbiser JL, Moses MA, Fernandez CA, Ghiso N, Cao Y, Klauber N, Frank D, Brownlee M, Flynn E, Parangi S, Byers HR, Folkman J. Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proc Natl Acad Sci USA. 1997;94:861–866. doi: 10.1073/pnas.94.3.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Petit AM, Rak J, Hung MC, Rockwell P, Goldstein N, Fendly B, Kerbel RS. Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: angiogenic implications for signal transduction therapy of solid tumors. Am J Pathol. 1997;151:1523–1530. [PMC free article] [PubMed] [Google Scholar]
- 31.Perrotte P, Matsumoto T, Inoue K, Kuniyasu H, Eve BY, Hicklin DJ, Radinsky R, Dinney CP. Anti-epidermal growth factor receptor antibody C225 inhibits angiogenesis in human transitional cell carcinoma growing orthotopically in nude mice. Clin Cancer Res. 1999;5:257–265. [PubMed] [Google Scholar]
- 32.Tikhonenko AT, Black DJ, Linial ML. Viral Myc oncoproteins in infected fibroblasts down-modulate thrombospondin-1, a possible tumor suppressor gene. J Biol Chem. 1996;271:30741–30747. doi: 10.1074/jbc.271.48.30741. [DOI] [PubMed] [Google Scholar]
- 33.Thompson TC, Southgate J, Kitchener G, Land H. Multistage carcinogenesis induced by ras and myc oncogenes in a reconstituted organ. Cell. 1989;56:917–930. doi: 10.1016/0092-8674(89)90625-9. [DOI] [PubMed] [Google Scholar]
- 34.Land H, Parada LF, Weinberg RA. Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature (Lond) 1983;304:596–602. doi: 10.1038/304596a0. [DOI] [PubMed] [Google Scholar]
- 35.Ribatti D, Vacca A, Nico B, Fanelli M, Roncali L, Dammacco F. Angiogenesis spectrum in the stroma of B-cell non-Hodgkin’s lymphomas: an immunohistochemical and ultrastructural study. Eur J Haematol. 1996;56:45–53. doi: 10.1111/j.1600-0609.1996.tb00293.x. [DOI] [PubMed] [Google Scholar]
- 36.Littlewood TD, Hancock DC, Danielian PS, Parker MG, Evan GI. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995;23:1686–1690. doi: 10.1093/nar/23.10.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dhanaraj SN, Marcus AM, Korah RM, Iwata K, Small MB. Characterization of c-myc-transformed rat fibroblasts resistant to apoptosis induced ny growth factor deprivation. Exp Cell Res. 1996;224:52–62. doi: 10.1006/excr.1996.0110. [DOI] [PubMed] [Google Scholar]
- 38.Small MB, Hay N, Schwab M, Bishop JM. Neoplastic transformation by the human gene N-myc. Mol Cell Biol. 1987;7:1638–1645. doi: 10.1128/mcb.7.5.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brown JM. The hypoxic cell: a target for selective cancer therapy. Cancer Res. 1999;59:5863–5870. [PubMed] [Google Scholar]
- 40.Evan GI, Wyllie AH, Gilbert CS, Littlewood TD, Land H, Brooks M, Waters CM, Penn LJZ, Hancock DC. Induction of apoptosis in fibroblasts by c-myc protein. Cell. 1992;69:119–128. doi: 10.1016/0092-8674(92)90123-t. [DOI] [PubMed] [Google Scholar]
- 41.Shi Y, Glynn JM, Guilbert LJ, Cotter TG, Bissonette RP, Green DR. Role for c-myc in activation-induced apoptotic cell death in T cell hybridomas. Science (Washington DC) 1992;257:212–214. doi: 10.1126/science.1378649. [DOI] [PubMed] [Google Scholar]
- 42.Polverini PJ, Bouck NP, Rastinejad F. Assay and purification of naturally occurring inhibitor of angiogenesis. Methods Enzymol. 1991;198:440–450. doi: 10.1016/0076-6879(91)98044-7. [DOI] [PubMed] [Google Scholar]
- 43.Conn G, Soderman DD, Schaeffer MT, Wile M, Hatcher VB, Thomas KA. Purification of a glycoprotein vascular endothelial cell mitogen from a rat glioma-derived cell line. Proc Natl Acad Sci USA. 1990;87:1323–1327. doi: 10.1073/pnas.87.4.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwenk F, Kuhn R, Angrand PO, Rajewsky K, Stewart AF. Temporally and spatially regulated somatic mutagenesis in mice. Nucleic Acids Res. 1998;26:1427–1432. doi: 10.1093/nar/26.6.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kibbey MC, Grant DS, Kleinman HK. Role of the SIKVAV site of laminin in promotion of angiogenesis and tumor growth: an in vivo Matrigel model. J Natl Cancer Inst. 1992;84:1633–1638. doi: 10.1093/jnci/84.21.1633. [DOI] [PubMed] [Google Scholar]
- 46.Orkin RW, Gehron P, McGoodwin EB, Martin GR, Valentine T, Swarm R. A murine tumor producing a matrix of basement membrane. J Exp Med. 1977;145:204–220. doi: 10.1084/jem.145.1.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Danielson KG, Martinez-Hernandez A, Hassell JR, Iozzo RV. Establishment of a cell line from the EHS tumor: biosynthesis of basement membrane constituents and characterization of a hybrid proteoglycan containing heparan and chondroitin sulfate chains. Matrix. 1992;12:22–35. doi: 10.1016/s0934-8832(11)80101-0. [DOI] [PubMed] [Google Scholar]
- 48.Yurchenco PD, Schittny JC. Molecular architecture of basement membranes. FASEB J. 1990;4:1577–1590. doi: 10.1096/fasebj.4.6.2180767. [DOI] [PubMed] [Google Scholar]
- 49.Murphy CS, Langan-Fahey SM, McCague R, Jordan VC. Structure-function relationships of hydroxylated metabolites of tamoxifen that control the proliferation of estrogen-responsive T47D breast cancer cells in vitro. Mol Pharmacol. 1990;38:737–743. [PubMed] [Google Scholar]
- 50.Galaktionov K, Chen X, Beach D. Cdc25 cell-cycle phosphatase as a target of c-myc. Nature (Lond) 1996;382:511–517. doi: 10.1038/382511a0. [DOI] [PubMed] [Google Scholar]
- 51.Blackwood EM, Eisenman RN. Max: a helix-loop-helix-zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science (Washington DC) 1991;251:1211–1217. doi: 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- 52.Amati B, Dalton S, Brooks MW, Littlewood TD, Evan GI, Land H. Transcriptional activation by the human c-Myc oncoprotein in yeast requires interaction with Max. Nature (Lond) 1992;359:423–426. doi: 10.1038/359423a0. [DOI] [PubMed] [Google Scholar]
- 53.Kretzner L, Blackwood EM, Eisenman RN. Myc and Max proteins possess distinct transcriptional activities. Nature (Lond) 1992;359:426–429. doi: 10.1038/359426a0. [DOI] [PubMed] [Google Scholar]
- 54.Grandori C, Mac J, Siebelt F, Ayer DE, Eisenman RN. Myc-Max heterodimers activate a DEAD box gene and interact with multiple E box-related sites in vivo. EMBO J. 1996;15:4344–4357. [PMC free article] [PubMed] [Google Scholar]
- 55.Li L, Nerlov C, Prendergast GC, Ziff EB. c-Myc represses transcription in vivo by a novel mechanism dependent on the initiator element and Myc box II. EMBO J. 1994;13:4070–4079. doi: 10.1002/j.1460-2075.1994.tb06724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Marhin WW, Chen S, Facchini LM, Fornace AJ, Penn LZ. Myc represses the growth arrest gene gadd45. Oncogene. 1997;14:2825–2834. doi: 10.1038/sj.onc.1201138. [DOI] [PubMed] [Google Scholar]
- 57.Facchini LM, Penn LZ. The molecular role of Myc in growth and transformation: recent discoveries lead to new insights. FASEB J. 1998;12:633–651. [PubMed] [Google Scholar]
- 58.Lawler J, Sunday M, Thibert V, Duquette M, George EL, Rayburn H, Hynes RO. Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J Clin Investig. 1998;101:982–992. doi: 10.1172/JCI1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pelengaris S, Littlewood T, Khan M, Elia G, Evan GI. Reversible activation of c-Myc in skin: induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol Cell. 1999;3:565–577. doi: 10.1016/s1097-2765(00)80350-0. [DOI] [PubMed] [Google Scholar]
- 60.Fotsis T, Breit S, Lutz W, Rossler J, Hatzi E, Schwab M, Schweigerer L. Down-regulation of endothelial cell growth inhibitors by enhanced MYCN oncogene expression in human neuroblastoma cells. Eur J Biochem. 1999;263:757–764. doi: 10.1046/j.1432-1327.1999.00575.x. [DOI] [PubMed] [Google Scholar]
- 61.He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science (Washington DC) 1998;281:1509–1512. doi: 10.1126/science.281.5382.1509. [DOI] [PubMed] [Google Scholar]
- 62.Rak J, Filmus J, Kerbel RS. Reciprocal paracrine interactions between tumour cells and endothelial cells: the “angiogenesis progression” hypothesis. Eur J Cancer. 1996;32A:2438–2450. doi: 10.1016/s0959-8049(96)00396-6. [DOI] [PubMed] [Google Scholar]
- 63.Morgenstern JP, Land H. A series of mammalian expression vectors and characterisation of their expression of a reporter gene in stably and transiently transfected cells. Nucleic Acids Res. 1990;18:1068. doi: 10.1093/nar/18.4.1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muthukkaruppan V, Auerbach R. Angiogenesis in the mouse cornea. Science (Washington DC) 1979;205:1416–1418. doi: 10.1126/science.472760. [DOI] [PubMed] [Google Scholar]
- 65.Tikhonenko AT, Hartman AR, Linial M. Overproduction of v-Myc in the nucleusits excess over Max are not required for avian fibroblast transformation. Mol Cell Biol. 1993;13:3623–3631. doi: 10.1128/mcb.13.6.3623. [DOI] [PMC free article] [PubMed] [Google Scholar]