

Abstract
Lactose permease is an integral membrane protein that uses the cell membrane's proton gradient for import of lactose. Based on extensive biochemical data and a substrate-bound crystal structure, intermediates involved in lactose/H+ co-transport have been suggested. Yet, the transport mechanism, especially the coupling of protonation states of essential residues and protein conformational changes involved in the transport, is not understood. Here we report molecular-dynamics simulations of membrane-embedded lactose permease in different protonation states, both in the presence and in the absence of lactose. The results analyzed in terms of pore diameter, salt-bridge formation, and substrate motion, strongly implicate Glu269 as one of the main proton translocation sites, whose protonation state controls several key steps of the transport process. A critical ion pair (Glu269 and Arg144) was found to keep the cytoplasmic entrance open, but via a different mechanism than the currently accepted model. After protonation of Glu269, the salt bridge between Glu269 and Arg144 was found to break, and Arg144 to move away from Glu269, establishing a new salt bridge with Glu126; furthermore, neutralization of Glu269 and the displacement of Arg144 and consequently of water molecules from the interdomain region was seen to initiate the closing of the cytoplasmic half channel (2.6–4.0 Å reduction in diameter in the cytoplasmic constriction region in 10 ns) by allowing hydrophobic surfaces of the N- and C-domains to fuse. Charged Glu269 was found to strongly bind the lactose permeant, indicating that proton transfer from water or another residue to Glu269 is a prerequisite for unbinding of lactose from the binding pocket.
INTRODUCTION
Membrane transport proteins facilitate the passage of specific molecules across the otherwise impermeable phospholipid bilayers that surround all cells and organelles. One large group of membrane transport proteins, the major facilitator superfamily, can be found in membranes from all life forms (1) including bacteria (2,3), fungi (4,5), plants (6,7), and animals (8,9). As a prime member of the major facilitator superfamily (10), lactose permease (LacY) of Escherichia coli is encoded by the lacY gene (11), the second structural gene of the lac operon. LacY utilizes the free energy stored in the transmembrane electrochemical proton gradient (ΔμH+) to drive the accumulation of lactose and other galactosides against their concentration gradient (12–17). The stoichiometry between lactose and H+ translocation is 1/1 (lactose/H+ symport). In the absence of ΔμH+, lactose permease catalyzes the reverse reaction, using free energy released from downhill translocation of galactosides to drive translocation of H+ with generation of ΔμH+, the polarity of which depends on the direction of the substrate concentration gradient. Lactose permease does not translocate H+ in the absence of substrate. However, a substrate concentration gradient can generate ΔμH+. Therefore, the most likely trigger for turnover is binding and release of the sugar on either side of the membrane (18).
Since the first report of LacY in 1956 (19), a large number of biochemical, biophysical, and structural studies (18,20,21) have been conducted to elucidate the molecular basis of the transport process. These studies have resulted in a significant amount of information regarding the role of individual residues in the transport mechanism and suggest that only six side chains are irreplaceable for active transport: Glu126; Arg144; Glu269; Arg302; His322; and Glu325 (22). The residues are important for ligand binding, proton translocation, and coupling of these two processes (21). Mutation of Glu325 to a neutral residue blocks the lactose/proton symport; active transport and proton translocation are blocked, but exchange, counterflow, and binding of sugar still occur (23). Furthermore, substrate-induced changes in the side-chain reactivity probed through various chemical modification reagents, site-directed fluorescence, and spin-labeling suggest that significant protein conformational changes take place during the transport process (21).
Structurally, LacY is composed of 12 transmembrane helices and the intervening loops. The two halves of the protein, known respectively as the N-domain and the C-domain, are each composed of six helices. These two domains together form a hydrophilic cavity in the middle, where substrate binding and translocation are believed to take place. One of the main binding sites of the substrate along its permeation path, which is captured by the crystal structure (22), is right in the middle of the transmembrane region of the protein at the interdomain space. The access of the binding pocket to the two sides of the membrane appears to be controlled by protein conformational changes that open and close cytoplasmic and periplasmic vestibules of the protein in a coordinated manner.
The currently proposed model for transport of lactose in LacY is based on sugar binding and dissociating coupled to a sequence of proton transfer events, and accompanying protein conformational changes. It is suggested that large interdomain protein conformational changes induced by binding, translocation, and dissociation of substrate and H+ result in a transition between the so-called outward-open and inward-open conformations, allowing the sugar to enter the internal cavity from the periplasm, and to be released into the cytoplasmic space, or vice versa. The crystal structure (22) of LacY represents the inward-open state of the transporter, in which the substrate is accessible only from the cytoplasmic cavity; the periplasmic half is closed and the substrate is ready to diffuse into the cell through the cytoplasmic cavity. Among the most important unknowns in the mechanism of sugar transport in LacY are the sequence of translocation and conformational events and the nature of protein conformational changes that switch substrate accessibility from the cytoplasmic half-channel to the periplasmic one.
The large body of experimental data, for example in Kaback et al. (18) and Kaback (1), and the crystal structure (22), have set the stage for identifying the lactose/proton co-transport mechanism with computational methods. For this purpose, one starts tests with a consistent set of hypotheses that establish an entire transport cycle, inspecting them computationally.
Fig. 1 provides a schematic representation of a possible lactose/proton co-transport mechanism based on an earlier suggestion (18,22). Intermediates e and f correspond to the inward-open conformations of ligand-bound and Apo forms of LacY, respectively, as seen in the published crystal structures (22,24); intermediate c corresponds to the outward-open conformation that has not yet been resolved structurally by observation. Several glutamate residues (Glu126, Glu269, and Glu325) and an arginine (Arg144), whose roles have been investigated in this study, are included in the scheme. It has to be noted that other residues (His322, Trp151, and Arg302) also play important functional roles in the transport mechanism of LacY (21), but are not included in the scheme, since their roles were not studied here. An unidentified group X in the periplasmic half of the lumen, representing a protein side chain or a water molecule, is proposed to relay the proton from the periplasmic region to Glu269. The putative transport process shown in Fig. 1 is composed of six steps:
The ground state LacY, in which Glu269 (E1) is protonated, takes up a proton at site X.
Sugar penetrates LacY from the periplasmic side.
The periplasmic entrance closes, and the sugar binds to its binding pocket.
A proton is transferred from Glu269 to Glu325 (E3) (most likely through His322, located between the two glutamate side chains), and Arg144 (R+) in the N-domain and deprotonated Glu269 form a salt bridge, inducing the inward-open conformation e. Meanwhile, sugar moves toward the cytoplasmic half of the lumen.
Sugar and the proton on Glu325 are released to the cytoplasm.
X transfers its proton to Glu269, Arg144 moves away from Glu269 establishing another salt bridge with Glu126 (E2), and the cytoplasmic-half channel closes.
FIGURE 1.

Putative lactose/proton co-transport mechanism of LacY. Six intermediates (a–f) are included. Sugar is denoted by a circled S. E1, E2, E3, and R+ represent the key residues (Glu269, Glu126, Glu325, and Arg144, respectively) involved in the lactose/proton co-transport. X represents a protein side chain or a water molecule in the periplasmic half-channel of LacY that relays the proton to Glu269. The charged states of residues are denoted in superscripts. The labels LacY(269H/325−), LacY(269−/325H), and ApoLacY for d, e, and f, respectively, refer to the three systems simulated in this study (see Table 1). The transport cycle is discussed in the text.
The mechanism in Fig. 1 provides only a rough sketch for transitions involved in sugar/H+ co-transport, and is far from complete. Primarily, it lacks the details of the structural responses of the protein to specific events (e.g., ligand binding/release, proton transfer steps, salt-bridge formation, etc.). Ideally, the mechanism would be tested by further structural data verifying the involvement of the proposed steps. However, determination of structures of membrane proteins is notoriously difficult because of their hydrophobicity and unstable nature. In this article, we have taken advantage of molecular dynamics (MD) simulation as a method for identifying further intermediate structures, and to explore the steps in the suggested transport scheme. We would like to identify, in particular, conformational changes that are induced by changes of protonation states of certain residues and by binding and unbinding of the substrate.
METHODS
System preparation
To investigate lactose/proton co-transport, we simulated three model systems, all with the protein embedded in a fully hydrated POPE lipid bilayer. In the first model, denoted LacY(269−/325H), Glu269 was deprotonated, and Glu325 was protonated. In the second model, denoted LacY(269H/325−), Glu269 was protonated, and Glu325 was deprotonated. In the third model, denoted ApoLacY, the substrate was removed from the binding pocket, and neither Glu269 nor Glu325 was protonated. Table 1 summarizes the simulated systems.
TABLE 1.
Systems simulated
| System name | Sugar | E325 | E269 | Simulation time |
|---|---|---|---|---|
| LacY(269−/325H) | + | Protonated | Deprotonated | 10 ns |
| LacY(269H/325−) | + | Deprotonated | Protonated | 10 ns |
| ApoLacY | − | Deprotonated | Deprotonated | 10 ns |
The three systems correspond to intermediates e, d, and f in Fig. 1, respectively.
The coordinates of E. coli lactose permease were obtained from the Protein Data Bank (PDB code 1PV7, which is the only available crystal structure of LacY with a substrate bound) (22). Gly154 was back-mutated to cysteine. The structure of lactose was adopted from a lactose-liganded protein congerin I (PDB 1C1L) (25). The topology file for lactose was generated by modifying the available CHARMM27 carbohydrate topology for pyranose (26). Missing hydrogen atoms were added by the psfgen program in VMD (27).
In LacY(269−/325H) and LacY(269H/325−), lactose was positioned in the binding pocket by superimposing the heavy atoms of the prebuilt lactose molecule onto the lactose analog β-d-galactopyranosyl-1-thio-β-d-galatopyranoside (TDG) as present in the crystal structure of LacY (22). Subsequently, TDG was replaced by the lactose molecule. ApoLacY was constructed by removing TDG from the 1PV7 structure.
All histidine residues were modeled in their neutral form. Their respective tautomeric states were assigned based on the interactions of the imidazole ring with their nearby residues. The tautomeric state of His322 in the binding pocket was determined by the protonation state of Glu269, which forms a hydrogen bond to this histidine (22).
We embedded the protein in a POPE bilayer by aligning hydrophilic residues with the lipid headgroups, and the hydrophobic surface of the protein with the acyl chains of the lipid bilayer. Although an approximate model, POPE is a reasonable choice of a pure lipid bilayer for simulation studies of the inner membrane of E. coli, which is mostly composed of PE. POPE has been previously used in simulation studies involving E. coli membrane proteins (28–30). Lipid molecules overlapping the protein were removed resulting in 348 POPE molecules included overall. The lipid-protein complex was then hydrated with 28,052 water molecules. Water molecules inside the inward facing cavity were removed. To neutralize the systems overall, eight and seven chloride ions were added to liganded and to the Apo systems, respectively. The total system sizes were ∼135,000 atoms, with the dimensions of 120 Å by 120 Å by 90 Å. A side view of one of the simulated systems is shown in Fig. 2 a.
FIGURE 2.

(a) Snapshot of a representative simulated system. The protein is shown in ribbon representation, with the N-domain in blue and the C-domain in red. The sugar is in van der Waals (vdW) representation, colored gray. The P and N atoms of the POPE bilayer are drawn in CPK and the rest of the lipid bilayer in licorice. Water molecules are in light blue. (b) RMSD per residue of the Cα atoms. The x axis denotes the residue number (from 1 to 417). The pink curve is the RMSD converted from the B-factor of the crystal structure (PDB: 1PV7) and scaled by a factor of 10 for better visualization. The blue, green, and red curves corresponds to the RMSD over the last 8 ns for LacY(269−/ 325H), LacY(269H/325−), and ApoLacY, respectively. The light gray regions denote the 12 transmembrane helices.
Simulation and analysis
MD simulations were performed with NAMD (31) using the CHARMM27 parameter set (32,33) for protein, lipid, water, and ions along with the carbohydrate force field for lactose (26). Periodic boundary conditions were imposed. After inserting the protein into the lipid bilayer and adding water, all systems were initially energy-minimized and simulated with the protein (and sugar) constrained at constant temperature (T = 310 K) and pressure (P = 1 atm) until both the total volume and the total area of the system stabilized (this was achieved in ∼300–400 ps). In this phase we allow water and lipid molecules to pack close to the protein and fill the crevices on the surface of the protein, without perturbing the original structure of the protein.
The crystal structure of LacY (1PV7) is devoid of water. We opted to remove water molecules initially added to the lumen by our approximate solvation method (superimposing a water box onto the system and removing overlapping water molecules), to avoid addition of any artificial water molecules at sensitive locations in the lumen. This resulted in a completely dry lumen at the beginning of the simulations. However, water molecules quickly penetrated the cytoplasmic opening of the protein, and a satisfactory level of hydration of the lumen was achieved during the protein-fixed phase of the simulations.
After releasing the constraints on the protein, another energy minimization was performed and NPT (T = 310 K, P = 1 atm) ensemble simulations were subsequently conducted for 10 ns. The particle-mesh Ewald method (34) was used for computation of electrostatic forces.
Trajectories were analyzed using VMD (27), and the program HOLE was used to calculate channel radii (35). All molecular images were prepared with VMD (27).
RESULTS AND DISCUSSION
It is known that large protein conformational changes are involved in the process of sugar transport in LacY (21). In this section, we first examine the flexibility of the protein in different simulations, and compare the dynamics of different regions. We will particularly analyze the fluctuations of the N- and C-domains, and their relative movement, which is an important dynamical property controlling stepwise access of substrate and proton to extracellular and cytoplasmic regions. We will then describe the results of simulations in which transitions from the crystal structure (intermediate e in Fig. 1) to a number of other intermediates in the outlined mechanism of sugar transport (see Fig. 1) are investigated.
Protein flexibility
LacY contains many proline and glycine residues in its transmembrane helices, which are expected to introduce a high degree of internal mobility to the protein (20,36). This aspect of protein dynamics can be readily calculated from MD simulations. Fig. 2 b shows the root mean-square deviation (RMSD) of individual residues in LacY for the three simulated systems (see Table 1), as well as for the crystal structure. The data for the simulations are time-averaged values calculated over the last 8 ns of each trajectory simulation, relative to the crystal structure: 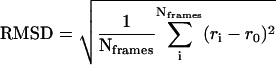 , where r0 and ri, are the initial (crystal structure) and actual positions of the Cα atom of the residue (Cα atoms of the whole LacY were used for the superposition of the simulated protein on the crystal structure). The values for the crystal structure were calculated from their Debye-Waller factor (β) by the relation
, where r0 and ri, are the initial (crystal structure) and actual positions of the Cα atom of the residue (Cα atoms of the whole LacY were used for the superposition of the simulated protein on the crystal structure). The values for the crystal structure were calculated from their Debye-Waller factor (β) by the relation 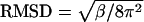 and scaled by a factor of 10 for better visualization in Fig. 2 b. The RMSD values for the simulations are higher than those for the crystal structure since the β-values for the crystal were measured at a lower temperature (100 K).
and scaled by a factor of 10 for better visualization in Fig. 2 b. The RMSD values for the simulations are higher than those for the crystal structure since the β-values for the crystal were measured at a lower temperature (100 K).
Both the calculated RMSD values from the simulations and the corresponding values from the crystal structure consistently show that the N-domain helices have a higher mobility than the C-domain ones. Furthermore, the cytoplasmic half of LacY generally exhibits a higher fluctuation than the periplasmic half.
In the N-domain, the cytoplasmic half of helix IV (in which Glu126 is located), helix V (in which Arg144 is located), and the short loop connecting them exhibit high RMSD values (Fig. 2 b), indicating a highly mobile region. The kink at Pro123 in helix IV and several glycine residues (e.g., Gly121, Gly141, and Gly147) in this region might account for the observed flexibility (37). As will be discussed in detail later, after protonation of Glu269, this region underwent the largest displacement among all the helices involved in the closing of the cytoplasmic cavity. This is in line with the results of a site-directed sulfhydryl labeling study suggesting large ligand-induced conformational change in this region (21,38,39). The cytoplasmic halves of the N-domain helices I, II, III, and the loop II/III are also quite flexible and moved in our simulation toward the C-domain upon protonation of Glu269. The loop II/III is conserved among a large group of membrane transport proteins that are evolutionarily related (40,41) and has been suggested to facilitate the conformational changes essential to the function of LacY (42).
The C-domain helices are generally less flexible. Nevertheless, helix VIII in which Glu269 is located and the conserved loop VIII/IX (40–42) exhibit relatively large RMSD values (Fig. 2 b). The hydrophobic face of the cytoplasmic half of helix VIII was observed to turn toward helix V in the N-domain after protonation of Glu269. A ligand-induced conformational change of the face of helix VIII has been reported earlier (39). The flexibility of loop VIII/IX might facilitate the conformational changes in LacY as also implied by mutagenesis studies (43).
The long loop VI/VII connecting the N- and C-domains exhibits the highest RMSD (Fig. 2 b) in all three simulated systems. In LacY(269H/325−) this loop moved closer to the cytoplasmic entrance (see Fig. 4 b), an event that was not observed in the other two simulated systems.
FIGURE 4.

Coupling of proton translocation and cytoplasmic closure. (a–f) Comparison of the initial and final structure of LacY(269−/325H) (left) and LacY(269H/325−) (right). In panels a and b, cyan and pink colors represent the N- and C-domains of the initial structure; blue and red represent the N- and C-domain of the final structure (t = 10 ns). (c–f) Front and back views of the interdomain region lined by helices V and VIII in the final structure of LacY as obtained from the simulation. White, green, blue, and red represent hydrophobic, hydrophilic, positive, and negative residues, respectively. Water molecules in the interface are shown in vdW representation only in front views (c,e). Back views (d,f) are obtained through a 180° rotation around the z axis. Glu269 and Arg144 are shown in licorice representation. (g) The interaction energy of the salt-bridges Glu126-Arg144 and Glu269-Arg144, defined as the sum of the electrostatic and vdW energies. (h) The length of the two salt-bridges Glu126-Arg144 and Glu269-Arg144, defined as the distance between Glu:Cδ and Arg:Cζ.
The periplasmic halves of all helices are more stable than their respective cytoplasmic halves. However, the helices in the periplasmic half were observed to be less tightly packed in LacY(269H/325−) than in the x-ray structure and in the other two systems.
Simulating transitions between intermediates
We report the results of three simulations in this article, as summarized in Table 1. The first simulation represents the inward-open conformation of LacY in the crystal structure (22) that corresponds to intermediate e in the mechanism outlined in Fig. 1. In this intermediate Glu325 is assumed to be protonated, Glu269 is deprotonated and forms a salt bridge with Arg144, and the substrate is present in the binding site. To model other intermediates in the cycle (Fig. 1), starting from intermediate e, we have changed the protonation states of certain residues in a second simulation, and removed the sugar from the binding pocket in a third one. These two simulations will investigate step 4 in Fig. 1, i.e., the transition from intermediate e to intermediate d and intermediate f in which sugar has been released from the protein. Comparison of the dynamics of the protein in these putative intermediate states allows us to deduce some of the protein conformational changes that are involved in the transport cycle. After giving an overview over the main findings of the three simulations, we will discuss the results and their relationship to the proposed mechanism in detail.
LacY(269−/325H)
This system (Fig. 3 a) represents the intermediate captured in the crystal structure (intermediate e). Here, Glu269 is deprotonated and expected to strongly bind lactose in the binding pocket; Glu325 is assumed protonated, and lactose is present in the binding cavity. Despite observed fluctuations, the protein's overall conformation, particularly in the interdomain space, remained close to the x-ray structure during the entire simulation. A salt bridge between Arg144 and Glu269 (observed in the crystal structure) was found to be stable, and lactose was strongly bound by Glu269 and Glu126, but showed an initial displacement toward the cytoplasm.
FIGURE 3.

Snapshots of the two simulated lactose-bound systems. The N- and C-domains are drawn in blue and purple, respectively. Lactose and several irreplaceable residues in the binding pocket are shown in licorice. (a) LacY(269−/325H); (b) LacY(269H/325−).
LacY(269H/325−)
Starting from the protein conformation in the crystal structure, we switched the protonation states of Glu325 and Glu269, as this transition has been suggested to be part of the transport cycle (22). Glu325 becomes deprotonated and Glu269 protonated. The sugar was kept in the binding pocket. Upon protonation of Glu269, the salt bridge between Glu269 and Arg144 broke immediately (Fig. 3 b). This in turn released the long side chain of Arg144, allowing it to move away from the binding pocket toward the cytoplasmic surface of the protein. Removing the two charges (departure of Arg144 and neutralization of Glu269) from the interdomain cleft was accompanied by water molecules spontaneously vacating the region. LacY underwent significant conformational changes, resulting in partial closure of its cytoplasmic entrance, and the substrate exhibited a 4 Å shift toward the periplasm.
ApoLacY
In this system, neither Glu269 nor Glu325 was protonated, and lactose was removed from the binding cavity. With regard to the overall structure of the protein, the simulated ApoLacY behaved very similar to LacY(269−/325H) i.e., no large protein conformational changes were observed and protein retained its inward open conformation during the simulation. However, the residues forming the binding pocket underwent major side-chain conformational changes after removing the substrate. Specifically, Arg144 moved very close to Glu126 and formed a stronger salt bridge with it at the cost of losing its salt bridge with Glu269 (see Fig. S1 in Supplementary Material). A very similar rearrangement of these amino acids has been reported in recently solved crystal structures of ApoLacY (24).
Protonation-induced closure of the cytoplasmic vestibule
As described in the previous section, in all simulated systems, the N-domain (helices I–VI) exhibits a higher internal mobility than the C-domain (helices VII–XII). The RMSD for Cα atoms of the N-domain helices (calculated from the last nanosecond of the simulation using only the N-domain Cα atoms for fitting) exceeds 2.0 Å, whereas the corresponding value for the C-domain helices (using only the C-domain Cα atoms for fitting) is <1.3 Å (see Fig. S2 in Supplementary Material). Therefore, we opted for using the less fluctuating C-domain as a reference to superimpose the resulting structures of the simulations on the crystal structure, which allows us to readily capture even small conformational changes between the two domains of LacY. Such overlays of the final (t = 10 ns) structures of the simulations onto the x-ray structure for LacY(269−/325H) and LacY(269H/325−) are shown in Fig. 4, a and b, respectively.
No appreciable interdomain conformational changes were observed in LacY(269−/ 325H) during the simulations, indicating that the inward-open conformation of the crystal structure is stable with the assigned protonation states of Glu325 and Glu269. In Fig. 4 a, only a slight deviation of the final structure of LacY(269−/325H) from the crystal structure, which can be attributed to the natural flexibility of the protein at the simulated temperature, is discernible.
A shift in the protonation states of Glu269 and Glu325 is suggested to be one of the key steps in the transport mechanism (22). As these residues are too far apart for a direct proton transfer, such a shift is most likely mediated by the side chain of His322, an irreplaceable residue in LacY function, which is positioned between the two glutamate side chains. Simulation of the actual proton transfer event through His322 is beyond the scope of this study, and we have only investigated the two states resulting from such proton transfer, i.e., LacY(269−/325H) and LacY(269H/325−). In contrast to LacY(269−/325H), protonation of Glu269 (LacY(269H/325−)), resulted in a large interdomain conformational change in the cytoplasmic half of the protein was observed over 10 ns. As shown in Fig. 4 b, in the final structure of this simulation the cytoplasmic half of the N-domain underwent a significant motion toward the C-domain, thus resulting in a partial closure of the cytoplasmic vestibule. Principal component analysis of the trajectory (results not shown) confirmed that the closure of the cytoplasmic half is the most dominant conformational change of the protein in LacY(269H/325−). Although a complete closure of the cytoplasmic entrance was not achieved in our simulations due to the limited timescale, the extent of the induced conformational change is particularly large for helices IV and V and the intervening short loop (loop IV/V) connecting them (residues 136–139). This loop is only comprised of four residues, which is unusually short for intervening loops in helical membrane proteins. The short length of this loop ensures close conformational coupling between helices IV and V, and facilitates their concerted inward and outward motions during the transport cycle. Evidently, the simulated proton transfer from Glu325 to Glu269 has triggered closure of the cytoplasmic vestibule of LacY.
A very similar behavior to LacY(269−/325H) was also observed for ApoLacY, i.e., no major conformational changes in the overall structure of the protein were observed, and the inward-open conformation was found to be stable during the timescale of the simulation. However, removal of the substrate from the binding pocket resulted in a marked rearrangement of side chains in this region. The side chain of Arg144 moves away from Glu269 and forms a closer, and likely stronger, salt bridge with Glu126 (see Fig. S1 in Supplementary Material). The observed rearrangement is in close agreement with the recently solved crystal structure of ApoLacY (24), suggesting that the binding pocket formation is to some extent induced by the substrate. Although the short timescale of simulations might not allow one to draw definitive conclusions, comparison of the behavior of LacY(269H/325−) and ApoLacY in 10 ns suggests that the protonation state of Glu269 is the main factor in determining whether the cytoplasmic cavity stays open or starts to close, and that the titration state of Glu325 is not critical in gating of the cytoplasmic entrance, since Glu325 has the same titration state in the two simulations. The coupling of the protonation state of Glu269 and the observed large protein conformational change is of great importance in the transport cycle and we will provide a more detailed mechanism for it below.
In Fig. 4, c–f, closer views of the interdomain region, specifically of helices V (in the N-domain) and VIII (in the C-domain), which form a major part of this region, are shown. The interaction of Arg144 with side chains of Glu269 and Glu126 in LacY(269−/325H) and LacY(269H/325−) were also monitored during the 10-ns duration of the two simulations in terms of interaction energies (Fig. 4 g) and distances between these residues (Fig. 4 h). As will be described later, these salt bridges were found to be very important in controlling the protein conformational changes and substrate binding.
The interdomain faces of helices V and VIII are mainly composed of hydrophobic residues. The cytoplasmic halves of the helices are far enough from each other in LacY(269−/325H) to allow for a wide open cytoplasmic vestibule, which was filled with water molecules, but retained its shape during the simulation. In the middle of the interdomain region, close to the lactose binding site, a salt bridge between Arg144 and Glu269, which further attracts water molecules into the cytoplasmic cleft in LacY(269−/ 325H), was found stable over the entire simulation. The distance between Glu269:Cδ and Arg144:Cζ in LacY(269−/325H) (Fig. 4 h) is consistently <5 Å, and the electrostatic interaction energy between the two residues is ∼−70 kcal/mol (Fig. 4 g).
In LacY(269H/325−), on the other hand, protonation of Glu269 results in an immediate disruption of the Arg144-Glu269 salt bridge. The distance between Glu269:Cδ and Arg144:Cζ in LacY(269H/325−) (Fig. 4 h) increases during the simulation to >10 Å, and the electrostatic interaction energy between these residues drops significantly (Fig. 4 g). In LacY(269H/325−), the hydrophobic faces of helices V and VIII are in much closer contact at the end of the simulation. Helices V and VIII are almost parallel to each other and in close contact in the cytoplasmic half of LacY(269H/325−), whereas in LacY(269−/325H) the two helices still are in the V-like conformation seen in the x-ray structure. As depicted in Fig. 4 e, the fusion of the hydrophobic faces of helices V and VIII in LacY(269H/325−) is accompanied by a decrease in water occupancy of the interdomain region.
Altogether the observed dramatic changes in LacY(269H/325−) suggest that there is a link between the breakage of the Arg144-Glu269 salt bridge, and water occupancy and protein conformational changes at the interdomain, interfacial region. Visual examination of the simulation provides a clear sequence for these events. The breaking of the salt bridge between Arg144 and Glu269, after protonation of the latter, frees the long side chain of the arginine, allowing it to undergo a large displacement and establish a stronger connection to Glu126, which is positioned closer to the cytoplasmic entrance. Translocation of Arg144 to a location closer to the surface not only results in a vacant space in the middle of the protein, it also removes a charge from a relatively hydrophobic region. Together with neutralization of Glu269 this results in a significant increase of hydrophobicity and a simultaneous drop of water occupancy of the interdomain interface, which in turn facilitates the movement of helices V and VIII toward each other, thus closing the cytoplasmic vestibule.
It is important to note that Arg144 and Glu269 are located in the N-domain and the C-domain of the protein, respectively. Glu269 is approximately in the middle of the transmembrane region of the protein, while Arg144 occupies a position in the cytoplasmic half. One would expect that formation of a salt bridge between these residues would bring the two domains together in the cytoplasmic half, i.e., favors an inward-closed conformation. This is in sharp contrast with the mechanism discovered by our simulations indicating that the breaking of this salt bridge is in fact a prerequisite for closing of the cytoplasmic half. We note that the results of our simulations are in close agreement with experimental data, which could not be satisfactorily explained even after solving the protein's structure. The concurrent disruption of this salt bridge and closing of the cytoplasmic entrance have been presented as one of the key steps in a previously proposed transport cycle (22). The proposed scheme is somewhat misleading in regard to the location of the involved amino acids, in that both residues are drawn schematically in the periplasmic half of the protein, which is clearly not in line with their location in the crystal structure (22).
It has been suggested that after protonation of Glu269, Arg144 forms a salt bridge with another acidic residue, Glu126 (22). Indeed, we observed the formation of a stronger salt bridge between Glu126 and Arg144 in LacY(269H/325−) right after energy minimization (Fig. 4, g and h). Interestingly, even in LacY(269−/325H) where the charged Glu269 is the main salt-bridge partner for Arg144, a somewhat weaker salt bridge is present between Arg144 and Glu126 (Fig. 4, g and h). From examination of the trajectories in LacY(269−/325H) it appears that Arg144 is coordinating the positions of the side chains of both Glu126 and Glu269 for an optimal interaction with the substrate. Any change in protonation state of either glutamate, as simulated for example in LacY(269H/325−), will result in a shift in the balance of the two salt bridges making one stronger at the expense of weakening or disrupting the other. The supporting role of the two salt bridges for lactose binding and recognition is consistent with the results of previous biochemical studies (21,24,44).
As described earlier, vacation of the interdomain region by Arg144 and water molecules, which is induced by the protonation of Glu269 in LacY(269H/325−), results in inward motions of helices V and VIII toward each other. This motion is then propagated to the entire N- and C-domains (Fig. 4 b), through vast interhelical contacts between the helices in each domain that keep the helices together. In the N-domain, the conformational coupling of helices V and IV (where Glu126 is located) is probably the strongest, as in addition to their proximal positions, these two helices are interconnected by a very short loop. Furthermore, Pro123 and Gly111, respectively located in the cytoplasmic and periplasmic halves of helix IV, render the helix highly flexible, allowing it to closely follow helix V. In the C-domain, helix VIII (where Glu269 is located) is more flexible than the others, as indicated by their RMSD values (Fig. 2 b). The overall flexibilities of these helices are prerequisites for the large conformational changes that control cytoplasmic and periplasmic access in LacY, e.g., as induced by the proton transfer in LacY(269H/325−). Similar salt-bridge mediated large-scale domain motions have been reported for other systems, e.g., for the maltose-binding protein (45).
In none of our simulations did a noticeable opening of the periplasmic half-channel of LacY occur. Apparently, the inward-open state of the crystal structure that was used as a starting point in our simulations is more prone to conformational changes in the cytoplasmic half. This is consistent with the overall higher structural flexibility of the cytoplasmic half of the protein as reported in the previous section (Fig. 2 b). A model for the outward-open conformation might be derived from the extensive thiol cross-linking studies (46). However, these experiments usually tend to underestimate distances between the linked residues. Furthermore, technical difficulties in arresting the protein in a single state during such experiments makes it hard to use the data directly in a model. Much longer simulations, in which sugar translocation and proton transfer steps are modeled, would be required to address whether an outward-open intermediate is part of the transport cycle.
Structure, dynamics, and hydration of the lumen
Here we describe the structure and dynamics, and the pattern of hydration of LacY's lumen formed at the interface between the N- and C-domains. The lumen can be divided into the periplasmic and cytoplasmic half channels, with the sugar binding pocket almost in the middle of the transmembrane segment of the protein. Half-channels are connected to the two sides of the membrane through the periplasmic and cytoplasmic entrances, respectively. In the crystal structure the lumen of the protein is wide open to the cytoplasm but, effectively closed to the periplasm. Monitoring variations in the pore size in LacY during the simulations is an effective means of identifying partial closure or opening of the two half-channels. We will also analyze the hydration of the lumen and present how it varies along the channel axis. The hydration of the lumen is an important aspect in regard to proton transfer events during the transport cycle, and has to be closely controlled by the protein to prevent undesired proton leaks.
Pore diameter
In Fig. 5 a we present a snapshot (t = 10 ns) of water molecules within the lumen of LacY(269H/325−). The protein is drawn to scale with the pore diameter and water occupancy profiles in Fig. 5. An effective pore diameter d of the lumen was computed for the x-ray structure and for the three simulated systems (calculated from the last nanosecond of the simulations) using the program HOLE (35). The diameter profiles indicate that the lumen of LacY has two narrow constriction regions: one above (cytoplasmic constriction region) and one below (periplasmic constriction region) the binding pocket centered at ∼z = 0. The cytoplasmic constriction region is the wider of the two, and the pore is widest around the central binding cavity.
FIGURE 5.

Pore size and hydration. (a) Snapshot showing water in the lumen of the final conformation of LacY(269−/325H). N- and C-domains of the protein are shown in blue and red colors with helices V and VIII hidden for clarity. Lactose is shown in gray vdW representation. Water molecules inside the pore are shown in vdW representation. Bulk water molecules on the two sides of the membrane are shown in light blue line representation. Putative residues functioning as gates are shown in licorice representation. The protein is drawn to scale with pore diameter (b) and water occupancy profiles (c). (b) Pore diameter profile. (c) Water occupancy measured in molecules per 1 Å slabs along the channel axis. The orange band in panels b and c highlights the cytoplasmic constriction region (also see Fig. S3 in Supplementary Material).
Relaxation of the side chains from their starting configuration in the crystal structure resulted in an overall change of the diameter profile in all simulations. In the periplasmic half-channel, the diameter profile shows slight deviations from the crystal structure, but in general the periplasmic half-channel remains essentially closed in all simulated systems. In the cytoplasmic half-channel, on the other hand, all the simulated systems exhibit a tendency to narrow. However, the effect is much more pronounced in LacY(269H/325−) and extends all the way toward the cytoplasmic entrance of the lumen. The significant decrease of the cytoplasmic pore diameter in LacY(269H/325−), 2.6–4.0 Å in the cytoplasmic constriction region (−13 Å < z < −5 Å, see Fig. 5), is a result of the partial closure of the cytoplasmic half channel in response to the simulated proton transfer from Glu325 to Glu269.
The conformational changes localized around the binding pocket are probably due to the relaxation of the binding pocket and penetration of water molecules during the simulations, and not directly due to the presence of lactose, as ApoLacY also exhibits very similar behaviors in this region. Although removal of the substrate from the binding pocket does not significantly alter the maximal diameter and its position along the pore axis, the unbinding event is accompanied by a significant conformational shift of Arg144 toward Glu126, in close agreement with the crystal structure of ApoLacY (24).
Pore hydration
The hydration of LacY's lumen was quantified by calculating the average number of water molecules per 1 Å slabs of the lumen during the simulations. Comparison of the time evolution of water occupancy of the cytoplasmic constriction region (−13 Å < z < −5 Å) in LacY(269−/325H) and LacY(269H/325−) (see Fig. S3 in Supplementary Material) permits one to further characterize the protein's conformational changes in this region. Both LacY(269−/325H) and LacY(269H/325−) show a sudden drop in pore hydration in the beginning of the simulations, which is probably the initial response of the protein to free dynamics. However, in line with larger cytoplasmic closure observed for LacY(269H/325−), a larger dehydration is achieved and maintained for the rest of the simulation in LacY(269H/325−). The number of water molecules in the cytoplasmic constriction region averaged over the last 5 ns of the simulation is 29.5 and 17.7 for LacY(269−/325H) and LacY(269H/325−), respectively, supporting a larger closure of the cytoplasmic half in the latter. In both LacY(269−/325H) and LacY(269H/325−), the cytoplasmic half-channel becomes easily and quickly hydrated during the first nanosecond of the simulations. The periplasmic half-channel, on the other hand, is a narrower region, and is found to be less accessible to water in all simulated systems.
At the end of the simulations (t = 10 ns), the highest water occupancy (5–6 H2O/Å) is observed around the central binding cavity (z ≅ 0) in both systems. The locations of the hydration peaks in the two simulations, however, are different; in LacY(269−/325H) hydration is maximal at z ≅ −2 Å, while in LacY(269H/325−) the maximum is found at z ≅ 2 Å. Since the pore diameters of the two systems are identical at this position (d ≅ 9 Å, Fig. 5 b), the observed differences are most likely due to variations in the substrate's position and its orientation within the binding cavity. As will be discussed in detail in the next section, the final position of lactose in LacY(269H/325−) is ∼4 Å closer to the cytoplasm than in LacY(269−/325H), a displacement that closely corresponds to and accounts for the shift of the hydration peak toward the periplasmic half-channel in LacY(269H/325−).
According to the hydration patterns, the least hydrated region of LacY in all simulated systems is found to be the periplasmic constriction region, which is lined by side chains of Ile40 and Asn245. The narrow size of the pore in this region together with a relatively low polarity of the lining residues effectively act as a hydrophobic gate resulting in very little or no hydration in this region. A similar gating region might form at the cytoplasmic constriction region by side chains of Phe334 and Tyr350 during the closure of the cytoplasmic half-channel. As proposed in the previous section, the charged side chain of Arg144 and its salt bridge(s) with two glutamates seem to be one of the factors that controls the opening of the cytoplasmic half-channel through hydration modulation of this region. The two hydrophobic gating regions play an important role in coordinating proton transfer events, and in preventing undesired proton flow across the LacY lumen. These gates coordinate the opening and closure of the two half-channels, and thus ensure that no free pathway for uncontrolled proton transfer is formed at any time during the transport cycle.
SUBSTRATE BINDING AND DYNAMICS
Below, we first discuss sugar binding in the LacY binding cavity; subsequently, dynamics of the sugar molecule and its hydration are addressed.
Sugar binding
Lactose exhibits different binding modes in intermediates d and e (Fig. 1); not only do different side chains interact with lactose in the two intermediates, but the strength of the interactions is also significantly different in the two simulations. In Table 2 we summarize all residues that interact with the sugar at any time during the simulations along with their average interaction energy with the sugar (sum of electrostatic and van der Waals (vdW) energies). Any residue coming closer than 3 Å of the sugar was selected for this analysis. Based on the strength of their interaction with the sugar and their role in the proposed transport cycle (22), we have selected seven residues from Table 2 and presented the time evolution of their interaction with lactose in Fig. 2. To present a more detailed picture, these energies will also be discussed in terms of interactions of individual glucose (GLC) and galactose (GAL) rings of the lactose with the protein.
TABLE 2.
Residues of N- and C-domains of LacY interacting with lactose during the simulation of intermediates e and d and their corresponding interaction energies (E) in kcal/mol
| Intermediate (LacY(269−/325H))
|
Intermediate (LacY(269H/325−))
|
||||||
|---|---|---|---|---|---|---|---|
| N-dom. | E | C-dom. | E | N-dom. | E | C-dom. | E |
| Tyr19 | −3.1 ± 2.7 | Tyr236 | −0.1 ± 0.5 | ||||
| Phe20 | −1.5 ± 0.6 | Asp237 | −2.5 ± 4.4 | Phe20 | −0.3 ± 0.3 | Asp237 | −10.0 ± 3.1 |
| Met23 | −2.4 ± 0.8 | Asp240 | −3.5 ± 9.4 | Met23 | −1.0 ± 0.6 | Asp240 | −9.8 ± 8.4 |
| Tyr26 | −0.3 ± 0.6 | Gln241 | −0.0 ± 0.5 | Tyr26 | −0.9 ± 0.9 | Gln241 | −2.6 ± 2.8 |
| Phe27 | −2.2 ± 1.2 | Thr265 | 0.3 ± 0.5 | Phe27 | −4.6 ± 0.9 | Thr265 | 0.4 ± 0.4 |
| Gly268 | −1.0 ± 1.4 | Phe30 | −0.8 ± 0.3 | ||||
| Phe49 | −0.2 ± 0.4 | Glu269 | −27.4 ± 14.7 | Glu269 | −5.4 ± 2.7 | ||
| Asn272 | 0.6 ± 1.5 | Ile52 | −0.1 ± 0.2 | Asn272 | −1.7 ± 1.1 | ||
| Phe118 | −2.0 ± 2.1 | Lys319 | −11.2 ± 8.7 | Phe118 | −0.5 ± 0.4 | Lys319 | −25.2 ± 5.7 |
| Asn119 | −0.5 ± 2.4 | Hse322 | −4.4 ± 3.9 | Asn119 | −1.0 ± 1.8 | Hsd322 | −6.2 ± 2.2 |
| Ala122 | −0.6 ± 1.7 | Met323 | −1.0 ± 1.0 | Met323 | −2.4 ± 0.8 | ||
| Pro123 | −0.2 ± 0.6 | Val326 | −0.7 ± 0.7 | Val326 | −0.9 ± 0.6 | ||
| Glu126 | −22.2 ± 11.5 | Leu330 | −0.2 ± 0.3 | Leu330 | 0.2 ± 0.2 | ||
| Arg144 | −0.5 ± 5.2 | Lys358 | 0.7 ± 3.3 | Arg144 | 2.7 ± 2.3 | Lys358 | 4.0 ± 1.3 |
| Cys148 | −0.3 ± 0.3 | Gln359 | −0.1 ± 0.3 | ||||
| Trp151 | −1.8 ± 1.1 | Met362 | −0.0 ± 0.2 | Trp151 | −2.9 ± 1.1 | Met362 | 0.1 ± 0.1 |
Residues in contact with sugar were identified as residues having any atom within a distance of 3 Å of any sugar atom during the simulation. Boldface entries denote amino acids showing strongest interaction with lactose.
In both intermediates d and e, the strongest interaction between sugar and the binding site stems from hydrogen bonds between protein and GAL ring. In intermediate e, the largest affinity arises from the electrostatic attraction of Glu126 in the N-domain, and deprotonated Glu269 in the C-domain, which is expected due to direct hydrogen bonds between the sugar and these (negatively charged) residues. The carboxylate groups of Glu126 and Glu269 form two bifurcated hydrogen bonds with four hydroxyl groups of the GAL ring (Fig. 2 a), keeping this ring very strongly bound. As shown in Fig. 2 c, the interactions of sugar with Glu126 and Glu269 are anticorrelated. Glu269 binds the sugar strongly from the beginning of the simulation, although their interaction starts to fluctuate significantly toward the end. Glu126, on the other hand, has only weak interaction with the sugar at the beginning of the simulation, but establishes a steady and strong interaction during the last 5 ns of the simulation. The position of Glu126 is closer to the cytoplasmic half-channel than Glu269. The observed anticorrelation is indicative of the sugar's tendency to move toward the cytoplasm in intermediate e relative to intermediate d.
In intermediate d, the largest contribution to the binding energy also results from electrostatic interaction with charged residues of the protein, but not with Glu126 or Glu269. For this intermediate, Lys319 (helix X), and Asp240 (helix VII), both in the C-domain, exhibit the strongest interactions with the sugar. The loss of affinity to Glu269 after its protonation in intermediate d, leads to displacement of the sugar toward the periplasm and its detachment from Glu126, which despite being charged can no longer interact strongly with the sugar, due to the large distance between the two.
Surprisingly, direct interaction between the sugar and Arg144 was not observed during the simulations in either intermediate d or e, a finding that appears to contradict the experimentally suggested important role of this residue in binding (21,22,47). This apparent contradiction can be explained through an indirect interaction of Arg144 with the sugar that was observed in the simulations. In LacY(269−/325H), Arg144 directly, and strongly, interacts with both Glu126 and Glu269, acting as a bridge between the two negative residues. This bridging interaction not only coordinates the positions of these two glutamates for an optimal interaction with the sugar, it also plays an important role in “handing” the sugar from one glutamate to the other during the transport cycle. It is important to note that, due to the charged states of these two glutamates in LacY(269−/325H), they would repel each other and adopt very different conformations in the absence of Arg144, possibly affecting the binding affinity of lactose.
The interactions of the protein and the GLC ring are weaker than those with the GAL ring, probably due to a relatively higher mobility of GAL, as will be discussed later. In LacY(269−/325H) the charge pair of Lys319 and Asp240, which is located closer to the periplasmic half-channel than Glu126 and Glu269, interact with the GLC ring at the beginning of the simulation. This interaction weakens toward the end of the simulation (Fig. 2, a and c) and is replaced by a new contact between Asp237 and the sugar, established after rotation of the GLC ring away from Lys319 and Asp240.
In LacY(269H/325−) (intermediate d), on the other hand, the charge pair of Lys319 and Asp240 interacts with the sugar strongly and stably (Fig. 2, b and d). Asp240 forms a bifurcated hydrogen bond with both GAL and GLC rings, while Lys319 forms a hydrogen bond with the GAL ring. These interactions force the sugar to move to the periplasmic half-channel, resulting in loss of interaction between Glu126 and the sugar as stated above. In both intermediates, the aromatic rings of Trp151 (helix V) and Phe27 (helix I) in the N-domain help orient the sugar rings through hydrophobic interactions, although the interaction energies are rather small (Fig. 2, c and d).
It is known that alkylation of Cys148 inactivates LacY by blocking sugar binding, and that substrate binding in turn protects this residue from the alkylating agent (18). These observations have been attributed to the close proximity of Cys148 and the sugar binding pocket, implying that a bound sugar and an alkylated Cys148 cannot tolerate each other due to steric effects (22). In both intermediates d and e, however, Cys148 is ∼7–8 Å away from the sugar, arguing against the significance of direct steric effects in the observed results. Our simulations suggest that alkylation of Cys148 might have an indirect effect on substrate binding; Cys148 is positioned almost between Arg144 and Glu269, and its alkylation will most likely disrupt the salt bridge between Arg144 and Glu126 and Glu269, which was found to play an important role in the binding of substrates.
Similarly, alkylation of the Ala122→Cys mutant or its replacement by phenylalanine or tyrosine abolishes binding and transport (48), suggesting that this position is in the vicinity of the substrate binding site (38). Our simulations support the suggested mechanism, since the sugar in intermediate e is constantly in close proximity of Ala122. It is noteworthy that after the protonation of Glu269 in intermediate d, the sugar exhibits a large translocation in the binding pocket resulting in a substantial increase of the distance between Ala122 and the sugar.
Sugar dynamics and hydration
For both intermediates d and e, the GAL ring is more strongly bound to the binding cavity than the GLC ring. This can give rise to different dynamics of the two sugar rings. To compare the dynamics of the two rings, an overlay of several simulation snapshots of intermediate e is shown in Fig. 6 a, and the RMSD values for the GLC and GAL rings are presented in Fig. 6 b. As seen in both intermediates d and e, GLC exhibits a significantly larger RMSD than does GAL. Moreover, lactose is significantly more confined in intermediate d than in intermediate e, probably due to the displacement of lactose toward the more open cytoplasmic half-channel in intermediate e. As the cytoplasmic half-channel is in a more open state in intermediate e, one would expect that the space between helices V and VIII should permit more extensive sugar movement in intermediate e than in intermediate d, which is also suggested by comparison of Fig. 4, c and e.
FIGURE 6.

Sugar dynamics, hydration, and ring-based interactions with the protein's binding cavity. (a) Snapshot from the simulation of LacY325H. The N-domain is shown in surface representation. The connecting loop between C- and N-domains (residues 200–220 connecting helices VI and VII) is displayed as an orange tube. Helices VII and VIII of the C-domain are rendered as silver ribbons. Twenty snapshots of the galactose (GAL) and glucose (GLC) rings, depicted in CPK representation in green and purple, respectively, illustrate their mobility within the binding pocket. The side chains of residues E126, R144, and E269 are shown for reference. (b) RMSD fluctuations of the GAL and GLC ring. Before the RMSD calculation, lactose was aligned based on the initial positions of Cα atoms in the N-domain. (c) Hydration of lactose. The hydration number was taken as the number of water molecules within 3 Å of any hydroxyl group of lactose. (d) Protein-lactose electrostatic (El) and vdW interaction energy as a function of time calculated for GAL and GLC individually. For color coding see e. (e) Averaged protein-lactose interaction energies (in kcal/mol).
Differences in confinement also relate to the significantly higher hydration of lactose in the binding cavity of intermediate e than intermediate d (Fig. 6 c). We determined sugar hydration numbers as the number of water molecules within 3 Å of any sugar's hydroxyl group. From Fig. 6 c we find average (2–10 ns) hydration numbers of 15 ± 3 and 10 ± 2 for intermediates e and c, respectively. This is in line with a larger water content of the central binding cavity in intermediate e than in intermediate d (Fig. 5 c).
Interestingly, differences in the affinity of LacY for the two sugar rings is most prominent in intermediate e. This is quantified in Fig. 6, d and e. The average (2–10 ns) LacY-GAL electrostatic interaction (Eel) energy is Eel = −45.5 ± 16.1 kcal/mol (Fig. 6 e). The corresponding LacY-GLC electrostatic interaction is ∼10 kcal/mol weaker; Eel = −36.1 ± 9.2 kcal/mol. In contrast, average vdW interaction energies (EvdW) between the two sugar rings and LacY are nearly identical, namely, −5 ± 3.2 kcal/mol (LacY-GAL) and −7 ± 2.9 kcal/mol (LacY-GLC). For intermediate d, the average LacY-GAL interaction energies are Eel = −29.7 ± 6.9 kcal/mol and EvdW = −10.7 ± 2.5 kcal/mol, respectively. The corresponding energies for interactions with the GLC ring are Eel = −27.0 ± 6.4 kcal/mol and EvdW = −9.4 ± 2 kcal/mol, respectively. Overall this reiterates that lactose is more weakly bound in intermediate e than in intermediate e, suggesting that sugar translocation in and out of the binding cavity is closely coupled to changes in protonation states of key binding residues such as Glu126, Glu269, and Glu325. Furthermore, the higher mobility of the GLC ring and its weaker interaction with the protein (when compared with GAL) suggest a mechanism of permeation.
CONCLUSIONS
Several intermediates of the transport cycle in lactose permease (LacY) have been simulated in this study. Starting from the crystal structure, transitions between the intermediates were induced either by removing the substrate, lactose, from the binding pocket, or through changing the protonation states of key residues. Conformational analysis of the simulated systems indicates that the N-domain exhibits a larger mobility than does the C-domain, suggesting that the N-domain is more actively engaged in the transport cycle. Despite the limited timescale, the simulations were able to capture a significant closure of the cytoplasmic half-channel in response to the induced proton transfer event. The partial closure of the cytoplasmic half-channel is triggered by the loss of a salt bridge between Arg144 and Glu269, after protonation of the latter. The observed closure is facilitated by migration of water molecules from the domain interfacial region, allowing the hydrophobic surfaces of the two domains to fuse. The protonation state of Glu269 also plays an essential role in binding of the substrate. Furthermore, the two rings of lactose have very different interactions with the protein. A very strong interaction between negatively charged Glu269 and the GAL ring of lactose suggests that unbinding of the substrate can only happen after protonation of Glu269.
The sequence of proton transfer events, stepwise substrate translocation events, and the intervening protein conformational changes involved in the function of LacY is almost completely unknown. Unraveling the detailed mechanism of proton/lactose symport in LacY presents a challenge to both experimental and theoretical investigations. The results of the present study are based on a small number of simulations, and need to be statistically improved by further calculations. However, they provide a dynamic picture for a number of proposed intermediates and plausible mechanisms for transitions between them. A more extensive description of the transport cycle will require a larger set of calculations in which sugar permeation through the channel will be investigated by nonequilibrium calculations. Combining the methodology with variations in protonation states of key residues, similar to that presented in this study, will enable us to shed light onto mechanistic details and structural elements that directly contribute to the function of LacY, and improve the current transport cycle model of this prototype membrane transporter.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Supplementary Material
Acknowledgments
The authors thank H. R. Kaback and his group for stimulating and insightful discussion. All molecular images are made with VMD (27).
The work was supported by grants from the National Institutes of Health (No. P41-RR05969 and No. R01-GM067887). The authors gladly acknowledge supercomputer time provided through the National Science Foundation Large Resource Allocation Committee (grant No. MCA93S028) at the National Center for Supercomputing Applications and the Pittsburgh Supercomputer Center.
Morton Ø. Jensen's present address is D. E. Shaw Research, New York, New York.
References
- 1.Kaback, H. R. 2005. Structure and mechanism of the lactose permease. C. R. Biol. 328:557–567. [DOI] [PubMed] [Google Scholar]
- 2.West, I. C. 1970. Lactose transport coupled proton movement in Escherichia coli. Biochem. Biophys. Res. Commun. 41:655–661. [DOI] [PubMed] [Google Scholar]
- 3.Wilson, T. H., and D. M. Wilson. 1983. Sugar cation co-transport systems in bacteria. In Cell Membranes: Methods and Reviews, Vol. 1. E. Elson, W. Frazier, and L. Galaser, editors. Plenum Press, NY. 1–39.
- 4.Eddy, A. A., and J. A. Nowachi. 1971. Stoichiometrical proton and potassium ion movements accompanying the absorption of amino acids by the yeast Saccharomyces carlsbergensis. Biochem. J. 122:701–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Slayman, C. L., and C. W. Slayman. 1974. Depolarization of the plasma membrane of Neurospora during active transport of glucose: evidence for a proton-dependent co-transport system. Proc. Natl. Acad. Sci. USA. 71:1935–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Komor, E. 1973. Proton-coupled hexose transport in Chlorella vulgaris. FEBS Lett. 38:16–18. [DOI] [PubMed] [Google Scholar]
- 7.Komor, E., and W. Tanner. 1974. The hexose-proton symport system of Chlorella vulgaris. Specificity, stoichiometry and energetics of sugar-induced proton uptake. Eur. J. Biochem. 44:219–223. [DOI] [PubMed] [Google Scholar]
- 8.Crane, R. K. 1960. Intestinal absorption of sugars. Physiol. Rev. 40:789–825. [DOI] [PubMed] [Google Scholar]
- 9.Crane, R. K. 1962. Hypothesis for mechanism of intestinal active transport of sugars. Fed. Proc. 21:891–895. [PubMed] [Google Scholar]
- 10.M. H. Saier, Jr. 2000. Families of transmembrane sugar transport proteins. Mol Microbiol. 35:699–710. [DOI] [PubMed] [Google Scholar]
- 11.Müller-Hill, B. 1996. The lac Operon. Walter de Gruyter, New York.
- 12.Mitchell, P. 1963. Molecule, group and electron transport through natural membranes. Biochem. Soc. Symp. 22:142–168. [Google Scholar]
- 13.Mitchell, P. 1968. Chemiosmotic Coupling and Energy Transduction. Glynn Research, Bodminster, UK.
- 14.Kaback, H. R. 1986. Active transport in Escherichia coli: passage to permease. Annu. Rev. Biophys. Biophys. Chem. 15:279–319. [DOI] [PubMed] [Google Scholar]
- 15.Kaback, H. R. 1989. Molecular biology of active transport: from membranes to molecules to mechanism. Harvey Lect. 83:77–103. [PubMed] [Google Scholar]
- 16.Kaback, H. R. 1992. In and out and up and down with the lactose permease of Escherichia coli. Int. Rev. Cytol. 137:97–125. [DOI] [PubMed] [Google Scholar]
- 17.Kaback, H. R. 1976. Molecular biology and energetics of membrane transport. J. Cell. Phys. 89:575–593. [DOI] [PubMed] [Google Scholar]
- 18.Kaback, H. R., M. Sahin-Tóth, and A. B. Weinglass. 2001. The kamikaze approach to membrane transport. Nat. Rev. Mol. Cell Biol. 2:610–620. [DOI] [PubMed] [Google Scholar]
- 19.Rickenberg, H. V., G. N. Cohen, G. Buttin, and J. Monod. 1956. The galactoside permease of Escherichia coli. Ann. Inst. Pasteur (Paris). 91:829–857. [PubMed] [Google Scholar]
- 20.Abramson, J., S. Iwata, and H. R. Kaback. 2004. Lactose permease as a paradigm for membrane transport proteins. (Review.) Mol. Membr. Biol. 21:227–236. [DOI] [PubMed] [Google Scholar]
- 21.Guan, L., and H. Kaback. 2006. Lessons from lactose permease. Annu. Rev. Biophys. Biomol. Struct. 35:67–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abramson, J., I. Smirnova, V. Kasho, G. Verner, H. R. Kaback, and S. Iwata. 2003. Structure and mechanism of the lactose permease of Escherichia coli. Science. 301:610–615. [DOI] [PubMed] [Google Scholar]
- 23.Sahin-Tóth, M., A. Karlin, and H. R. Kaback. 2000. Unraveling the mechanism of the lactose permease of Escherichia coli. Proc. Natl. Acad. Sci. USA. 97:10729–10732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mirza, O., L. Guan, G. Verner, S. Iwata, and H. R. Kaback. 2006. Structural evidence for induced fit and a mechanism for sugar/H+ symport in LacY. EMBO J. 25:1177–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shirai, T., C. Mitsuyama, Y. Niwa, Y. Matsui, H. Hotta, T. Yamane, H. Kamiya, C. Ishii, and K. M. T. Ogawa. 1999. High-resolution structure of the conger eel galectin, congerin I, in lactose-liganded and ligand-free forms: emergence of a new structure class by accelerated evolution. Structure. 7:1223–1233. [DOI] [PubMed] [Google Scholar]
- 26.Kuttel, M., J. W. Brandy, and K. J. Naidoo. 2002. Carbohydrate solution simulations: producing a force field with experimentally consistent primary alcohol rotational frequencies and populations. J. Comput. Chem. 23:1236–1243. [DOI] [PubMed] [Google Scholar]
- 27.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD—visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 28.Tieleman, D. P., L. R. Forrest, M. S. P. Sansom, and H. J. C. Berendsen. 1998. Lipid properties and the orientation of aromatic residues in OmpF, Influenza M2, and alamethicin systems: molecular dynamics simulations. Biochemistry. 37:17554–17561. [DOI] [PubMed] [Google Scholar]
- 29.Tajkhorshid, E., P. Nollert, M. Ø. Jensen, L. J. W. Miercke, J. O'Connell, R. M. Stroud, and K. Schulten. 2002. Control of the selectivity of the aquaporin water channel family by global orientational tuning. Science. 296:525–530. [DOI] [PubMed] [Google Scholar]
- 30.Wang, Y., K. Schulten, and E. Tajkhorshid. 2005. What makes an aquaporin a glycerol channel: a comparative study of AqpZ and GlpF. Structure. 13:1107–1118. [DOI] [PubMed] [Google Scholar]
- 31.Phillips, J. C., R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kale, and K. Schulten. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schlenkrich, M., J. Brickmann, A. D. MacKerell, Jr., and M. Karplus. 1996. Empirical potential energy function for phospholipids: criteria for parameter optimization and applications. In Biological Membranes: A Molecular Perspective from Computation and Experiment. K. M. Merz and B. Roux, editors. Birkhauser, Boston, MA. 31–81.
- 33.Feller, S. E. 2000. Molecular dynamics simulations of lipid bilayers. Curr. Op. Coll. Interf. Sci. 5:217–223. [Google Scholar]
- 34.Darden, T., D. York, and L. Pedersen. 1993. Particle mesh Ewald. An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98:10089–10092. [Google Scholar]
- 35.Smart, O., J. Goodfellow, and B. Wallace. 1993. The pore dimensions of Gramicidin A. Biophys. J. 65:2455–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Locher, K. P., R. B. Bass, and D. C. Rees. 2003. Breaching the barrier. Science. 301:603–604. [DOI] [PubMed] [Google Scholar]
- 37.Weinglass, A. B., and H. R. Kaback. 1999. Conformational flexibility at the substrate binding site in the lactose permease of Escherichia coli. Proc. Natl. Acad. Sci. USA. 96:11178–11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwaw, I., K.-C. Zen, Y. Hu, and H. R. Kaback. 2001. Sited-directed sulfhydryl labeling of the lactose permease of Escherichia coli: helices IV and V that contain the major determinants for substrate binning. Biochemistry. 40:10491–10499. [DOI] [PubMed] [Google Scholar]
- 39.Frillingos, S., and H. R. Kaback. 1997. The role of helix VIII in the lactose permease of Escherichia coli: II. Site-directed sulfhydryl modification. Protein Sci. 6:438–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Henderson, P. J. F. 1990. Proton-linked sugar transport systems in bacteria. J. Bioenerg. Biomembr. 22:525–569. [DOI] [PubMed] [Google Scholar]
- 41.Griffith, J. K., M. E. Baker, D. A. Rouch, M. G. Page, R. A. Skurray, I. T. Paulsen, K. F. Chater, S. A. Baldwin, and P. J. Henderson. 1992. Membrane transport proteins: implications of sequence comparisons. Curr. Opin. Cell Biol. 4:684–695. [DOI] [PubMed] [Google Scholar]
- 42.Jessen-Marshall, A. E., N. J. Paul, and R. J. Brooker. 1995. The conserved motif, GXXX(D/E)(R/K)XG[X](R/K)(R/K), in hydrophilic loop 2/3 of the lactose permease. J. Biol. Chem. 270:16251–16257. [DOI] [PubMed] [Google Scholar]
- 43.Pazdernik, N. J., A. E. Jessen-Marshall, and R. J. Brooker. 1997. Role of conserved residues in hydrophilic loop 8–9 of the lactose permease. J. Bacteriol. 179:735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sahin-Toth, M., J. le Coutre, D. Kharabi, G. le Marie, J. C. Lee, and H. R. Kaback. 1999. Characterization of Glu126 and Arg144, two residues that are indispensable for substrate binding in the lactose permease of Escherichia coli. Biochemistry. 38:813–819. [DOI] [PubMed] [Google Scholar]
- 45.Stockner, T., H. J. Vogel, and D. P. Tieleman. 2005. A salt-bridge motif involved in ligand binding and large scale domain motions of the maltose-binding protein. Biophys. J. 89:3362–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sorgen, P. L., Y. Hu, L. Guan, H. R. Kaback, and M. E. Girvin. 2002. An approach to membrane protein structure without crystals. Proc. Natl. Acad. Sci. USA. 99:14037–14040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Venkatesan, P., and H. R. Kaback. 1998. The substrate binding site in the lactose permease of Escherichia coli. Proc. Natl. Acad. Sci. USA. 95:9802–9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guan, L., M. Sahin-Tóth, and H. R. Kaback. 2002. Changing the lactose permease of Escherichia coli into a galactose-specific symporter. Biochemistry. 99:6613–6618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.