

Abstract
Objective
Neurologic and psychiatric manifestations are severe complications of systemic lupus erythematosus (SLE). As commonly seen in patients, spontaneous development of lupus-like disease in MRL-lpr mice is accompanied by brain atrophy and behavioral dysfunction. We examined inflammatory and ultrastructural aspects of central nervous system (CNS) involvement using a nonselective cyclooxygenase-2 (COX-2) inhibitor and measuring effects on behavior, microglial activation, and neuronal morphology.
Methods
Ibuprofen (IBU) was provided in a rodent chow (375 ppm) for animals 5–19 weeks of age. Exploration of a novel environment and performance in the forced swim test assessed effects on behavior. Immunohistochemistry, fluoro-Jade B (FJB) staining, and flow cytometry were employed in neuropathological analysis. Transmission electron microscopy was used to examine ultrastructural morphology of cortical, hippocampal, hypothalamic, nigral, and cerebellar cells.
Results
Chronic IBU treatment failed to normalize immune status, behavior, and brain mass in lupusprone MRL-lpr mice. It also did not reduce density of CD3+ lymphocytes in the choroid plexus, or FJB+ neurons in the hypothalamus. Activated F4/80+ microglia increased with age, but IBU treatment was not effective in reducing their numbers. Although numerous dark cells were seen in functionally critical brain regions (e.g., paraventricular nucleus and subgranular zone), ultrastructural morphologies of classical apoptosis or necrosis were not detected.
Conclusion
The COX-dependent pathway does not seem to be critical in the etiology of CNS disease in this model of neuropsychiatric lupus. Reduced brain mass, increased microglial activation, and condensation of cytoplasm point to a metabolic perturbation (e.g., excitotoxic damage) that compromises function and survival of central neurons during lupus-like disease.
Key Indexing Terms: AUTOIMMUNITY, INFLAMMATION, IBUPROFEN, MICROGLIA, LUPUS, EXCITOTOXICITY, APOPTOSIS, ONCOSIS, SUBGRANULAR, ZONE
Although development of systemic autoimmunity and inflammation clearly account for peripheral manifestations (e.g., dermatitis, vasculitis, glomerulonephritis), mechanisms underlying central nervous system (CNS) damage in up to 50% of patients with systemic lupus erythematosus (SLE) remain largely unknown. Brain atrophy1,2 and biochemical signs of neuronal/astrocytic damage are reported in a significant proportion of patients with clinically verified CNS involvement3. To determine the relationship between SLElike disease and aberrant behavior, we study brain pathology and behavioral dysfunction in the MRL/MpJ-Faslpr (MRL-lpr) murine substrain. MRL-lpr mice lack a functional Fas receptor and develop lupus-like manifestations earlier in life than congenic MRL/MpJ (MRL+/+) controls4. Along with spontaneous onset of inflammation and autoimmunity, MRL-lpr mice show retarded brain growth, infiltration of leukocytes into the choroid plexus, neuronal atrophy, and deterioration in behavioral performance5–7. The constellation of behavioral deficits that distinguish 2 MRL substrains has been operationally defined as “autoimmunity-associated behavioral syndrome” 8, and has been proposed to model neuropsychiatric lupus (NP-SLE)9.
As reported in patients with NP-SLE10,11, the blood-brain barrier in MRL-lpr mice becomes more permeable with the progress of lupus-like disease12,13. Since T and B cells infiltrate brain14, their soluble mediators may drain into the cerebrospinal fluid (CSF), diffuse into neighboring interstitial tissue, and have detrimental effects on neuronal cell functioning15. Although recent evidence emphasizes the role of autoantibody-mediated neuronal damage16–18, inflammatory mechanisms in the etiology of mental dysfunction (e.g., depressive behavior) remain largely unexplored19–21. Circulating proinflammatory cytokines facilitate infiltration of macrophages and other monocytes into the brains of lupusprone mice by upregulating expression of cell adhesion molecules22,23. Activated microglial cells/macrophages often migrate to sites of injury where they proliferate and undergo morphological and functional changes24. This includes expression of molecules that orchestrate the inflammatory cascade, such as MHC class II, tumor necrosis factor-α (TNF- α), interleukin 1 (IL-1), and IL-6. Expressed primarily on activated microglial cells25, MHC class II molecules interact with T cells, contributing to local synthesis of inflammatory factors and recruitment of other inflammatory cells. Indeed, excessive levels of MHC class II (as determined by Ia mRNA) were observed in the diencephalon of MRL-lpr mice26. Similarly, overexpressed IL-6 and interferon-γ mRNA in the hippocampus and cerebellum27,28 further supported the notion of a central inflammatory response in these animals.
The limbic system of diseased MRL-lpr mice shows increased density of TUNEL+ cells29, but merely ~7% of this population is accounted for by degenerating neurons30. Although this suggests that the majority of dying neurons do not undergo synchronized DNA fragmentation, the prevalent mode of neuronal death within the apoptosis-necrosis continuum remains unclear. Our study focused on the “inflammatory aspect” of brain involvement by attenuating activity of cyclooxygenase-2 (COX-2), an enzyme instrumental in the inception of inflammatory processes31. In addition, given that neuronal necrosis is often accompanied by an inflammatory response32 and sustained microglial activation may contribute to chronic neuropathology33, the microglial/macrophage population and ultrastructural morphology of cells in several brain regions were examined.
Nonsteroidal antiinflammatory drugs (NSAID) are effective inhibitors of COX-1/COX-2 pathways34 and the microglia/macrophage-driven neurotoxic cascade35,36. In vitro studies showed that the nonselective COX-2 inhibitor ibuprofen (IBU) can induce apoptosis of activated microglial cells37, reduce glutamate neurotoxicity38, and attenuate drug-induced damage of dopaminergic neurons39, which are a proposed target of autoimmune processes in the MRL model40–42. Moreover, supplementing rodent chow with IBU led to attenuation of amyloid plaque deposition and microglia-mediated brain inflammation in murine models of Alzheimer’s disease43,44. These beneficial effects and stress-free chronic administration of the drug led us to select the above treatment for behavioral/neuropathological study. The overall expectation was that mice fed with IBU would show normalized behavioral performance, immune status, and brain morphology.
MATERIALS AND METHODS
Animals, drug treatment, and tissue collection
Twenty MRL-lpr and 10 MRL +/+ males (5 mice/cage) were obtained from a local specific-pathogen-free colony and maintained under standard laboratory conditions (light phase 8 AM – 8 PM, food and water ad libitum; temperature 24–26°C). Ten agematched CD1 males (Charles River Canada, Saint-Constant, PQ, Canada) were housed in the same colony room and used as non-autoimmune controls. IBU was purchased from Sigma Chemical Co. (St. Louis, MO, USA) and subsequently formulated into color-coded AIN-76A rodent diet (Research Diets, New Brunswick, NJ, USA) at a final concentration of 375 ppm. This dose was selected on the evidence that it prevents CNS pathology and inflammation in a mouse model for Alzheimer’s disease43. From 5 to 19 weeks of age half of each group was fed ad libitum with either drug-supplemented chow or control chow. At the end of the study, mice were anesthetized with Somnotol (intraperitoneal, 60 mg/kg body weight; MTC Pharmaceuticals, Cambridge, ON, Canada) and after terminal bleeding from the vena cava, they were intracardially perfused with 20 ml phosphate buffered saline (PBS) and 20 ml fresh 4% paraformaldehyde. Extracted brains and spleens were weighed on an analytical balance (AB54-S; Mettler Toledo, Switzerland). Brains were used to examine CD3+ cell (T lymphocyte) infiltration into the choroid plexus and brain parenchyma.
Given dissimilar fixation protocols, a second cohort of 4-week-old mice (MRL-lpr, MRL +/+, and CD1 males; n = 18/strain; 3 mice/cage) was used to examine effects of the IBU treatment on neuropathological indices (presence of FJB+ neurons, expression of a F4/80 marker on microglia/macrophage cell line) and behavioral performance (MRL-lpr vs MRL +/+ only). As with the first cohort, half of each group was fed ad libitum with either drug-supplemented chow or control chow from 4 to 18 weeks of age (n = 9 mice/strain/treatment). The body weight and food consumption were monitored weekly. Averaged food consumption was ~5 g/day/animal, resulting in a final daily dose of ~62.5 mg/kg in the IBU-treated groups. Mice were sacrificed at 18 weeks with Somnotol. After terminal bleeding from the vena cava, they were perfused as described above.
To assess the time-course of microglial/macrophage activation, PBS-perfused brains from 5, 12, and 18-week-old MRL-lpr (8/age group) and MRL +/+ mice (5/age group) were pooled for the purpose of flow cytometry. Mice were maintained under conditions as outlined above. Afourth cohort of males was used for the purpose of electron microscopy. It included three 23-weekold MRL-lpr mice, an asymptomatic 8-week old MRL-lpr mouse, a 22-weekold MRL +/+ congenic control, and a healthy, age-matched CD1 mouse. After overdose with Somnotol, they were heparinized and perfused by gravity via the left ventricle with lactated Ringer’s solution, followed by Karnofsky’s fixative (phosphate buffered 4% glutaraldehyde). The brains were dissected and postfixed in Karnofsky’s fixative at 4°C for 2 days. A stainless steel coronal brain matrix (Stoelting, Wood Dale, IL, USA) was used to obtain 1 mm-thick sections. Using a scalpel blade, blocks of tissue (~2 mm2) were dissected from hypothalamus, cerebellar cortex, hippocampus, and substantia nigra. To prevent tissue drying, sections remained submerged in Karnofsky’s fixative throughout the micro-dissection. All experimental protocols were approved by a local animal care committee and in accord with the regulations of the Canadian Council of Animal Care.
Immunohistochemistry for CD3+ cells
Immunohistochemistry of the CD3 T lymphocyte marker was used to examine the degree of leukocyte infiltration into the choroid plexus and brain parenchyma in the first cohort of mice. Twelve micrometer coronal sections (Bregma ~ –1.0) were fixed in acetone at −20°C for 3 min and air dried. Following two 5 min washes in PBS, slides were placed into avidin-blocking solution (Vector Laboratories, Burlingame, CA, USA) for 15 min, washed in PBS, and immersed in biotin-blocking solution (Vector Laboratories) for 5 min. After a 5 min wash, sections were incubated with 10% normal goat serum (Vector Laboratories) in PBS for 30 min at room temperature. After incubation with primary antibody (hamster antimouse CD3e diluted 1:30 with 5% goat serum/PBS; BD Pharmingen, catalog no. 550275) overnight at 4°C, 3 additional PBS washes (5 min each) were followed by incubation with secondary antibodies (biotinylated anti-hamster IgG diluted 1:250 with 5% goat serum/PBS; Vector Laboratories, BA-9100) for 1 h at room temperature. After a 5 min wash in PBS, slides were immersed into 0.03% H2O2 in PBS for 10 min at room temperature, and after an additional wash, incubated with streptavidin/HRP (Anti-Ig HRP detection kit, BD Pharmingen 551013) for 30 min at room temperature. After 3 additional PBS washes (5 min each), slides were incubated with DAB for several minutes, and washed in tap water. A brief counterstain with hematoxylin and dehydration through graded alcohol and xylene preceded three 5-min PBS washes before cover-slipping. Counting of CD3+ cells from digitized photographs (depicting choroid plexus of the third ventricle) was performed in 1 section/brain under 400× magnification by an observer blinded to the group origin.
Behavioral testing
In comparison to congenic MRL +/+ controls, diseased MRL-lpr mice reliably show impaired locomotor activity in a novel environment and excessive floating in forced swim test6,21. In our study, the second cohort of age-matched MRL-lpr and MRL +/+ males was tested between 18 and 19 weeks in computerized activity boxes (Digiscan-16; Omnitech Electronics, Columbus, OH, USA). Distance traveled and time spent in ambulation over two 30-min periods (6:00–7:00 PM) were measured by VersaDat software (Accuscan Instruments, Columbus, OH, USA). Floating in the swimming pool (diameter 1.83 m, water temperature 25°C) was defined by the absence of any paw and tail movements over a 6-min trial and recorded by an unbiased observer, as described21.
Fluoro-Jade B staining
Fluoro-Jade B is an anionic fluorescein derivative used for the localization of degenerating neurons in brain tissue sections. This dye has an affinity for the entire degenerating neuron including cell body, dendrites, axon, and axon terminals. However, the degenerating tissue components (biomolecules) to which the dye has an affinity are currently unknown. Ten micrometer coronal sections (Bregma ~ –1.0) were cut with a Jung Frigocut 2800E cryostat and sections were mounted onto Aptex-coated slides according to protocol40. The sections were examined using a Zeiss argon laser scanning confocal microscope (wavelength 488 nm; LSM 510, Carl Zeiss Inc.). Two confocal micrographs from the paraventricular hypothalamic region were obtained using a Fluar 20×/0.75 objective in combination with a 1024 × 1024 pixel resolution, and Fluoro-Jade B+ cells were manually counted by an observer blinded to group origin.
Immunohistochemistry for F4/80+ cells
F4/80, a 120–160 kDa glycoprotein, is highly and constitutively expressed on most resident tissue macrophages45, including microglia46. Resting microglia possess a characteristic ramified morphology that can be visualized with antibodies to the F4/80 antigen. However, immunohistochemical staining to F4/80 is proposed to become more intense after microglial activation47. We used the rat monoclonal antibody F4/80 (1:100 dilution in 5% goat serum/PBS; Serotec) and biotinylated anti-rat IgG secondary antibodies (1:200 dilution in 5% goat serum/PBS; Serotec) to morphologically characterize microglia on horizontal sections. Incubation times for the primary and secondary antibodies were overnight at 4°C and 1 h at room temperature, respectively. After incubation, brain sections were treated with Vectastain ABC reagent (Vector Laboratories) according to the manufacturer’s directions, and the resulting avidinbiotin-peroxidase complex was visualized with DAB (DAB substrate kit, Vector Laboratories). The stained brains were observed by confocal light microscopy under differential interference contrast. In addition to more intense staining, activated microglia were distinguished by a more rounded morphology than when in a resting state48. Three adjacent 0.5 mm2 areas from the paraventricular region of the diencephalon were selected from each brain, and F4/80+ cells were manually counted by an observer blinded to group origin at 200× magnification. The sparse presence of F4/80+ cells in the cerebellum and cortex was also noted.
Flow cytometry for CD69+ F4/80+ cells
A useful approach to assess activated microglia/macrophages in the brain is the combination of immunohistochemistry and flow cytometry49. Therefore, the third cohort of mice was used to confirm activation of microglial/macrophage cell lineage during the progress of lupus-like disease in MRL-lpr mice. CD69 antigen (also known as an activation-inducer molecule or a very early activation marker) is a member of the natural killer (NK) cell gene complex family of signal-transducing receptors. CD69 expression can be induced in vitro on cells of most hematopoietic lineages, including T and B lymphocytes, NK cells, murine macrophages, neutrophils, and eosinophils50. According to a pilot study (data not shown), 0.5–1.5 × 105 of mononuclear cells can be obtained from a single MRL-lpr brain. Since this yield was not optimal for flow cytometry (~ 5 × 105), 5–8 brains for each age point and substrain were pooled from 5, 12, or 18-week-old MRL-lpr and MRL +/+ mice. Freshly extracted brains were minced in a glass homogenizer containing cell culture media, Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Invitrogen, Grand Island, NY, USA). Tissues were transferred into 50 ml centrifuge tubes, and a single-cell suspension was obtained by vigorous pipetting. Percoll separation was used to isolate the infiltrating mononuclear cells as described51. Briefly, 9 parts of Percoll (Amersham Bioscience, Uppsala, Sweden) to one part 10× PBS was added to brain tissue suspension at a final concentration of 30% Percoll and centrifuged at 27,000 g for 30 min at 4°C. This resulted in top myelin/cellular debris fraction, middle glial fraction, and bottom mononuclear cell fraction. The latter was collected and washed with DMEM. The number of living cells collected was counted in a hemacytometer.
Fluorescence-labeled antibodies used for staining were R-phycoerythrin (R-PE)-conjugated rat anti-mouse CD69 monoclonal antibody (0.25 μg for 1 × 106 cells; BD Pharmingen) and RPE-Cy5-conjugated rat anti-mouse F4/80 monoclonal antibody (5 μl for 1 × 106 cells; Serotec, Oxford, UK) as described52. In particular, aliquots of mononuclear cells (0.5–1 × 106) were suspended in 0.1 ml PBS containing 0.1% sodium azide and incubated with predetermined optimal concentrations of R-PE-conjugated anti-mouse CD69 and RPE-Cy5-conjugated anti-F4/80 for 30 min in the dark at 4°C. Cells were washed 3 times with PBS, resuspended in 0.3 ml PBS (containing 0.1% sodium azide), and subjected to FACScan analysis. Mononuclear splenocytes without fluorescence-labeled antibodies added were used as a negative control. Background autofluorescence was determined by incubating mononuclear cells (from brains of 18-week-old MRL-lpr mice) with PBS. The analysis was performed using FACScan (Becton Dickinson, Mountain View, CA, USA) and regions of interest were set according to forward scatter (roughly proportional to the diameter of the cell) and side scatter characteristics of splenocytes (proportional to the granularity). Atotal of 10,000 cells were analyzed and the frequency of each cell-surface marker was determined using WinMDI 5.1 software (J. Trotter, Scripps Clinic, La Jolla, CA, USA). Data were represented as dot plots, where each cell was represented by a dot, positioned according to the fluorescence intensities. These 2-color dot plots were divided into 4 quadrants, comprising double-negative cells, green only, red only, and double-stained cells. Percentage of different cell populations in each quadrant was calculated for comparative purposes.
Light microscopy and transmission
In preparation for electron microscopy analysis, 1 × 2 × 1 mm blocks of brain tissue were isolated from the regions of interest (described above) and stored in Karnofsky’s fixative until processing. Samples were rinsed in 0.1 M sodium cacodylate buffer twice for 10 min each, and post-fixed in 1% osmium-tetroxide (in 0.1 M sodium cacodylate buffer) for 1 h at room temperature. Tissue was dehydrated in ascending concentrations of ethanol, followed by 2 changes with propylene-oxide (10 min each). Then samples were immersed in 1:1 and 1:3 propylene-oxide:Spurr’s resin (Marivac-Canemco, St. Laurent, PQ, Canada) for 1 h before being placed into 100% Spurr’s resin overnight. The next day samples were embedded in fresh Spurr’s resin and polymerized overnight at 60°C. Blocks were trimmed and semithin sections (0.5 μm) were cut and stained for light microscopy with toluidine blue, which delineates both perikarya and proximal dendrites53. In brief, 5 g of toluidine blue powder (Marivac-Canemco) and 5 g sodium borate (Caledon, Georgetown, ON, Canada) were combined in deionized water to make a 500 ml staining solution. The solution was heated to ~65°C and sections were transiently immersed for staining. Sections were washed 3 times in distilled water before mounting and cover-slipping for assessment. Finite neuroanatomical areas were identified from each section. From each slide, an area common to a particular region was demarcated for further processing. Ultrathin sections (90 nm) of each finite area were then cut and placed onto 200-mesh copper-palladium grids. The grids were poststained with saturated uranyl acetate in 50% ethanol and Reynolds lead citrate. Grid sections were examined and photographed using a JEOL 1200 EX (Tokyo, Japan) transmission electron microscope at an accelerating voltage of 80 kV. Some sections were also processed with standard H&E as described54.
Indices of systemic disease
Profound changes in the systemic cytokine network are one of the earliest manifestations of autoimmune disease in murine models of lupus4. We presently measured TNF-α, which is proposed to contribute to progressive microglia-induced cell damage in neurodegenerative diseases55, and is elevated in MRL-lpr mice56,57. Similarly, high concentrations of IL-1ß may have detrimental effects on cultured neurons58 and hippocampal tissue59. Therefore, serum levels of TNF-α and IL-1ß were analyzed with an ELISA kit (R&D Systems, Minneapolis, MN, USA) as described57.
Considering that death of MRL-lpr mice is usually attributed to immune complex-mediated glomerulonephritis, and deposits of IgG and DNA immune complexes have been observed in the kidneys60, circulating immune complexes (CIC) and antibodies to double-stranded DNA (dsDNA) were measured as additional markers of disease severity. Blood samples were left to coagulate in 1.5 ml plastic vials and centrifuged for 10 min at 3000 rpm. Serum was separated from the clot and stored at –20°C until analysis. CIC and dsDNA concentrations were measured using qualitative sandwich ELISA kits (CIC: catalog no. 5900; anti-dsDNA: catalog no. 5100), according to the manufacturer’s instructions (Alpha Diagnostic International, San Antonio, TX, USA). In brief, serum samples were diluted 1:100 in diluent included in the kit, and applied to both experimental and control wells to assess the specificity of binding. The optical density of each well was determined using a microplate ELISA reader set to 450 nm. Given that splenomegaly is a well established marker of severe autoimmunity in the MRL model4, wet spleen was weighed immediately upon extraction.
Statistical analysis
Data were analyzed by analysis of variance with substrain (MRL-lpr vs MRL +/+; CD1) and treatment (IBU-rich vs control food) as between-group factors. Student’s t test was used in the post-hoc analysis because of the 2 × 2 design in most comparisons. Pearson’s correlation was used to measure association between variables. Significance level was set at p ≤ 0.05 and graphs show means ± SEM. All computations were performed using the SPSS 13 statistical package.
RESULTS
Dermatitis, alopecia, and necrosis of ear tips appeared earlier and were more common among the IBU-treated MRL-lpr mice in comparison to 3 other groups (~30% vs ~10%). This suggested that chronic ingestion of IBU-laced food exacerbated lupus-like disease in some animals. However, this observation needs to be repeated in a separate cohort by formulating an a priori hypothesis and by systematically measuring peripheral manifestations.
Infiltration of T cells and neuropathology
As shown previously29, T cells were dense in the choroid plexus and sparsely scattered throughout the brain parenchyma of MRL-lpr mice. Although the difference in the number of T lymphocytes in the choroid plexus and the third ventricle was not statistically significant, there was a trend for more T cells in brains from the IBU-treated MRL-lpr group (83 ± 23 vs 53 ± 13 in control MRL-lpr mice; t18 = 1.197, p > 0.1). Regardless of the treatment, T lymphocytes were not observed in brains of congenic or allogenic controls.
As expected, reduced brain weight (F(1,32) = 4.704, p < 0.05) was accompanied by increased Fluoro-Jade B and F4/80 positivity in brains of diseased MRL-lpr mice. Hypothalamic, cortical, and cerebellar regions were frequently populated with brightly stained fluoro-Jade B-positive neurons or Purkinje cells (Figure 1). In contrast to a scattered distribution in the hypothalamus (Figure 2A), clusters of intensely stained F4/80+ cells were observed in the parieto-temporal cortex of MRL-lpr brains (Figure 2C). The size and morphology of some F4/80+ cells were reminiscent of macrophage-like cells (Figure 2C inset). While F4/80+ cells were rarely seen in the cerebellum of MRL-lpr mice, discretely stained cells were frequently noted in proximity to the Purkinje layer (Figure 2E). These observations were more profound in comparison to brains of congenic MRL +/+ controls (Figures 2B, 2D, 2F) and healthy CD1 mice (data not shown). The treatment with IBU did not change established group differences, as evidenced by undiminished counts in the hippocampal region (Figure 3). However, similarly to enhanced T cell infiltration, there was a trend for more Fluoro-Jade B-positive neurons in the IBU-treated MRL-lpr mice than in MRL-lpr mice fed with the control diet (102 ± 8 vs 81 ± 7; t16 = 0.081; Figure 3A).
Figure 1.
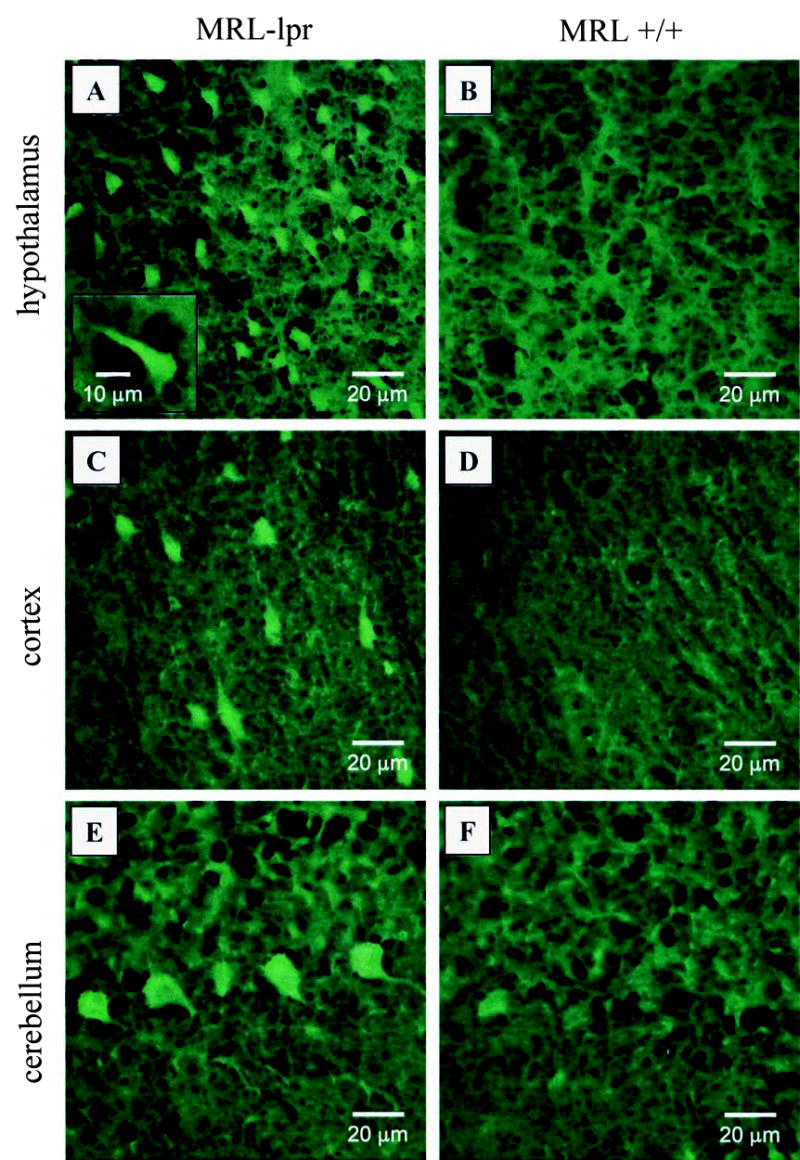
Representative fields of Fluoro-Jade B (FJB) staining in MRL brains. More numerous, brightly stained FJB+ cells suggest a neurodegenerative process in the diencephalon of MRL-lpr mice (A) in comparison to age and sex-matched MRL +/+ controls (B). Similarly, scattered FJB+ cells were commonly seen in the cortical parenchyma (C) in comparison to comparable areas from control brains (D). Some contiguous Purkinje cells were distinctly stained in MRL-lpr mice (E), but were not seen in congenic MRL +/+ controls (D).
Figure 2.
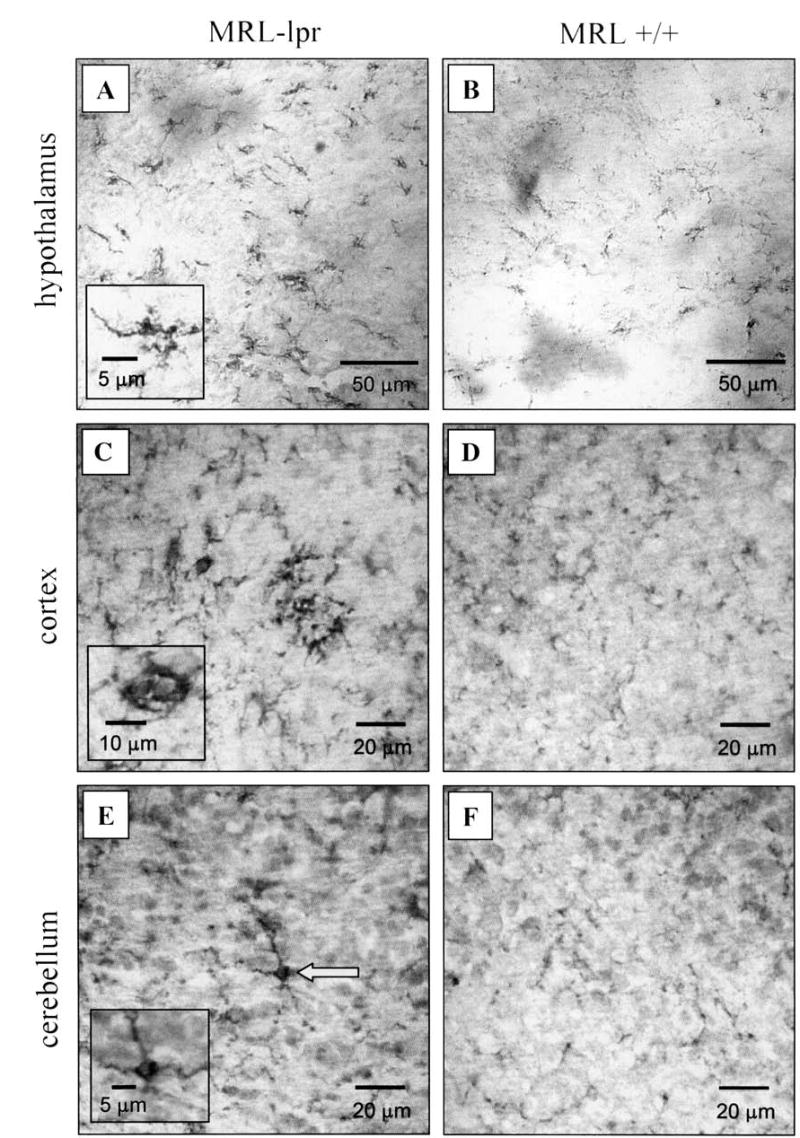
Representative fields of F4/80 staining in MRL brains. Intensely stained F4/80+ cells were commonly observed in hypothalmic regions from diseased lupus-prone mice (A), suggesting increased microglial activation. While also seen in control brains, F4/80+ cells appeared less dense (B). Patches of cortical microglia cells were frequently observed in lupus brains (C) in comparison to age-matched controls (D). Although sparse F4/80 staining was seen in the Purkinje layer of the MRL-lpr brain (E), this was not observed in age and sex-matched MRL+/+ controls (F).
Figure 3.

Quantification of degenerating neurons and microglial cells, as revealed by fluoro-Jade B (FJB) and F4/80 staining. Compared to age-matched controls, the density of FJB+ counts was significantly higher in the hypothalamus of 19-week-old MRL-lpr mice than in other groups (A). Similarly, the number of intensely stained F4/80+ cells was increased in MRL-lpr mice in comparison to controls (B). Chronic treatment with IBU did not affect these measures.
A novel contribution to the neuropathology described previously5,7,29,30,54 was the observation that (regardless of treatment) intensely stained “activated microglia” were more common in the hypothalamus of diseased MRL-lpr mice (substrain: F(2,25) = 17.667, p < 0.001; Figure 3B). This notion of enhanced microglial activation was supported by the FACS analysis, which revealed a ~20-fold increase in the number of CD69+ F4/80+ cells from mice 5 to 18 weeks of age (Figure 4). Although resident CD69+ F4/80+ cells were less abundant than other leukocyte populations60a, their percentage increased with age and was higher in brains pooled from 12 and 18-week-old MRL-lpr mice than in age-matched MRL +/+ controls.
Figure 4.
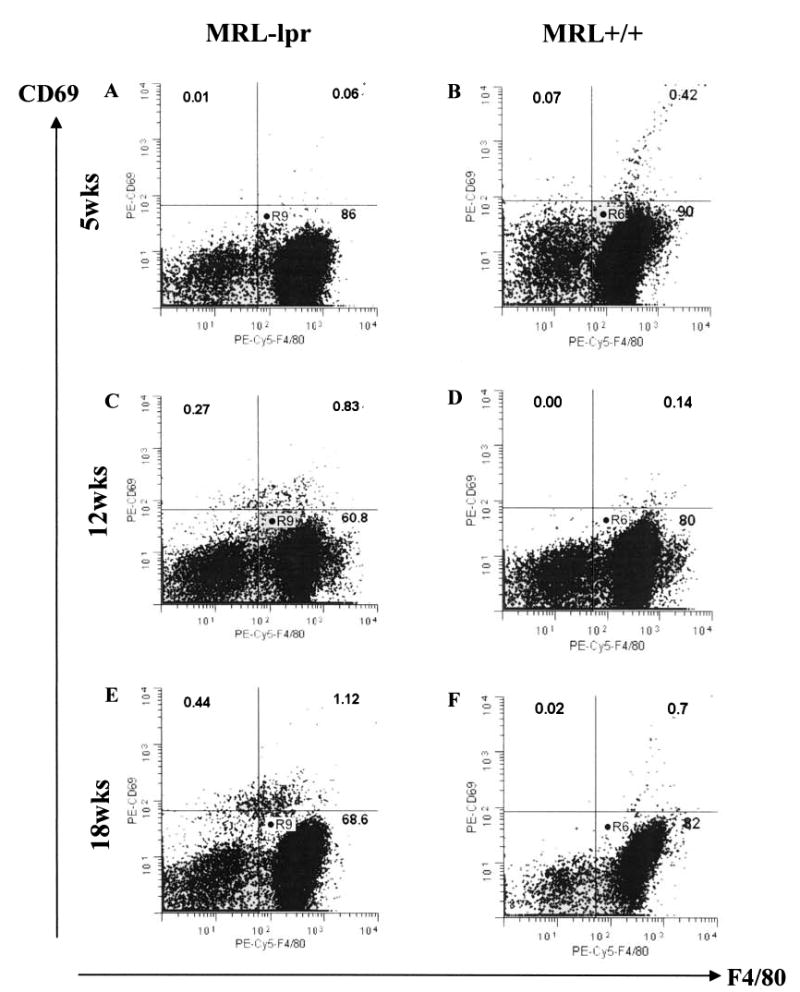
Dot-plot scatter analysis of CD69+ F4/80+ cell infiltration in MRL-lpr and MRL +/+ brains pooled at 5, 12, and 18 weeks of age. The percentage of CD69+ F4/80+ cells increased from 0.06% to 1.12% in the MRL-lpr groups (A, C, E), while the same population was less consistent (B, F) and less abundant (D, F) in age-matched MRL +/+ controls.
Behavior
MRL-lpr mice consumed less food and water compared to congenic controls, and the IBU-rich diet did not affect these measures (for food, substrain: F(1,32) = 6.639, p < 0.02; for water, substrain: F(1,32) = 7.062, p < 0.02). As expected, MRL-lpr males moved less and traveled shorter distances when exposed to a novel environment (activity box), and this deficit was not ameliorated by the IBU treatment (for movement time, substrain: F(1,32) = 9.153, p < 0.01; for distance, substrain: F(1,32) = 12.909, p < 0.001). Chronic IBU treatment also failed to reduce floating time in MRL-lpr mice (substrain: F(1,32) = 13.352, p < 0.01). Since these observations have been reported21,57,61, they are not shown here. The CD1 mice showed food/water consumption and behavioral performance similar to MRL +/+ mice (data not shown).
Indices of autoimmune disease
MRL-lpr mice had significantly larger spleen weights compared to the other 2 control groups (substrain: F(2,46) = 33.778, p < 0.001), but IBU did not attenuate this difference. Splenomegaly correlated with reduced brain weights within the MRL-lpr mice (r16 = –0.486, p < 0.05) and serum dsDNA levels (r16 = –0.573, p < 0.05), suggesting a relationship between systemic autoimmunity and brain damage in this substrain. Serum levels of TNF-α and IL- 1ß were elevated only in MRL-lpr mice, and the treatment with IBU did not alter these measures (for TNF-α substrain: F(2,47) = 8.058, p < 0.001; for IL-1ß substrain: F(2,47) = 4.719, p < 0.02). Similarly, increased levels of CIC and anti-dsDNA antibodies in sera of MRL-lpr mice were not attenuated by the antiinflammatory treatment (for CIC substrain: F(2,47) = 30.016, p < 0.001; for anti-dsDNA antibodies substrain: F(2,39) = 21.407, p < 0.001; data not shown). The MRL+/+ mice fed control chow and both groups of CD1 mice were negative for CIC and anti-dsDNA antibodies.
Microscopic observations
As expected, spleen weights of 3 MRL-lpr mice employed in the electron microscopy analysis were greatest in the 23-week-old MRL-lpr animals (0.45 ± 0.03 g) relative to controls (0.11 ± 0.02), confirming their autoimmune status (data not shown). Increased presence of “dark” cells in 2 MRL-lpr brains was observed at the level of light microscopy by enhanced toluidine blue staining (Figure 5). Interestingly, densely packed cells were prominent in the subgranular zone, one of few brain areas that is populated with neuronal and glial precursors62. This observation is consistent with our previous reports in which fluoro-Jade B-positive cells were detected in the dentate gyrus of MRL-lpr mice30 and a patient with NP-SLE54. Many scattered “dark” cells were seen in the substantia nigra, but less frequently in the periventricular hypothalamus and in the Purkinje and granular cell layers of the cerebellar vermis (Figure 5) and cerebral cortex (data not shown). Similarly to toluidine blue staining, H&E revealed accumulations of basophilic cells in the subgranular zone of the dentate gyrus (Figure 6). Although a degenerative process is anticipated30,54, it is generally considered that a clear distinction between cells in prophase and cells undergoing pycnosis or apoptosis cannot be made with H&E staining only63.
Figure 5.
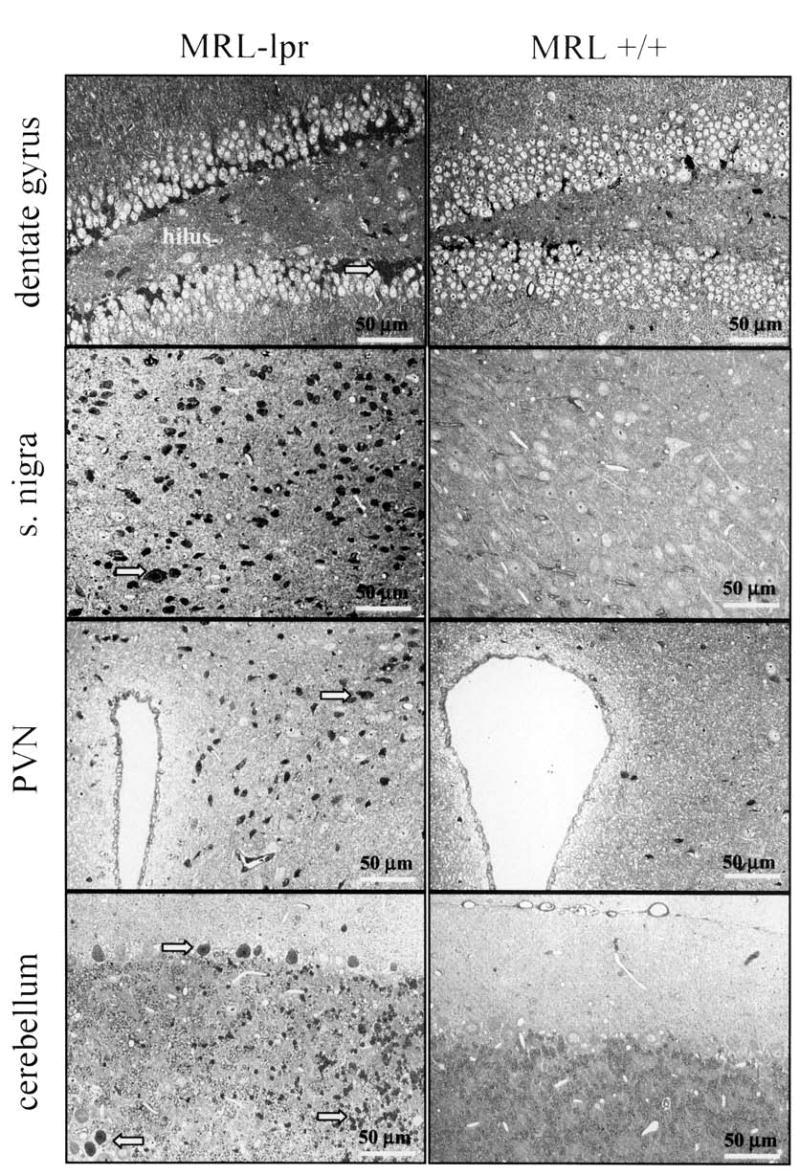
Toluidine blue staining of various brain regions inspected by light microscopy. The CA2/CA3 region (not shown) and subgranular zone of the dentate gyrus of 2 diseased MRL-lpr mice were frequently populated with densely packed, elongated dark cells. Although round, dark cells were clustered in the substantia nigra, they were scattered in the paraventricular nucleus (PVN), and Purkinje and granule cell layers of the cerebellum.
Figure 6.

H&E staining of dentate gyrus inspected by light microscopy. The subgranular zone of diseased MRL-lpr mice was populated with densely packed basophilic cells in both hemispheres (contralateral hemisphere not shown). Although H&E cannot reliably distinguish degenerating cells from cells in the prophase, the notion of proliferating, immature neurons “at risk” is consistent with our reports of hippocampal damage in MRL-lpr mice30,54. Inset: enlarged area showing neighboring cells with dark, basophilic nucleoli.
Using electron microscopy, an abundance of “electron-dense” cells was confirmed in the hypothalamus, hippocampus, cerebellum (Figure 7), and the substantia nigra. Nuclear and cytoplasmic condensation and clumping/condensation of chromatin were ubiquitous across different brain regions. However, “dark” cells neither contained apoptotic bodies nor showed evidence of budding, which are classical signs of apoptosis64. Although intracellular organelles showed good ultrastructural preservation and membrane integrity, their swollen shape suggested an initial stage of a metabolic insult. Cells adjacent to the “dark Purkinje layer” were often of normal appearance despite increased amounts of lipofuscin-like particles. Occasionally, the cellular plasma membrane was ruffled, giving a scalloped appearance to these cells (Figure 7C). Electron-dense cells were confirmed in the subgranular zone and the CA2/CA3 region, with mitochondria preserving double-membrane appearance. While cristae appeared condensed, they were relatively intact within the condensed cytoplasm of dark neurons. Blebbing of cell membranes or profound internal changes in organelles was generally absent. The dysmorphic cells in MRL-lpr brains resembled reversible ultrastructural changes documented by Auer and colleagues, who proposed nonlethal alterations to the neurons after hypoglycemic brain damage65. In contrast to the above observations, cells in control brains had evenly dispersed nuclear chromatin and rounded/oval shaped nuclei with prominent nucleoli (data not shown).
Figure 7.
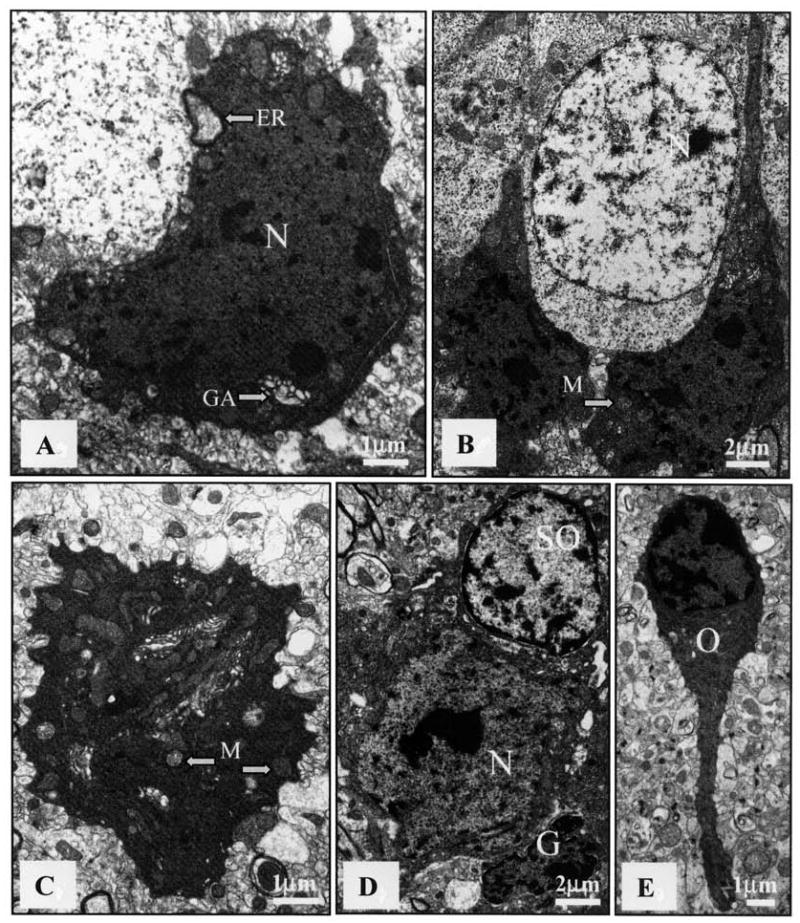
Electron microscopy (EM) revealing ultrastructural features of dark cells in brain of an MRL-lpr mouse. The increased electron density was unaccompanied by blebbing of cell membranes or internal changes in organelles, other than occasional swelling. A. Hypothalamic neuron (N) with densely compacted karyoplasm and cytoplasm, as well as an enlarged Golgi apparatus (GA) and endoplasmic reticulum (ER) (bar = 1 μm). B. Densely packed dark cells between healthy neurons (N) in the subgranular zone (bar = 2 μm). C. Cerebellar neuron with condensed cytoplasm, swollen mitochondria (M), and ruffled outer membrane (bar = 1 μm). D. Dark neuron (N) surrounded by healthy-looking satellite oligodendrocyte (SO) and shrunken glial cell (G) (bar = 2 μm). E. Hippocampal oligodendrocyte (O) with electron-dense cytoplasm and karyoplasm. No evidence of apoptotic bodies, nuclear fragmentation, or ruptured membranes could be observed in any preparation (bar = 1 μm).
Overall, these dark cells failed to exhibit classical signs of apoptosis, such as formation of apoptotic bodies secondary to nuclear and cytoplasmic fragmentation, or blebbing of the cytoplasmic membrane. Moreover, there was no evidence of large discrete masses (crescentic caps) of chromatin aggregation around the perimeter of nuclear membranes66. These negative findings would argue against a classical form of apoptosis within brain cells of Fas-deficient autoimmune mice. Similarly, there were no signs of classical oncosis or necrosis, characterized by vacuolization, nuclear and plasma membrane breaks, and spilling of cell contents. Despite such an “intermediate form” of cellular pathology67, a large proportion of cells in 2 out of 3 brains extracted postmortem from aged MRL-lpr mice appeared to have undergone profound metabolic perturbations.
DISCUSSION
Immunosuppression with cyclophosphamide attenuates infiltration of leukocytes5, reduces neuronal atrophy68, lowers serum and CSF levels of TNF-α, and abolishes in vitro CSF cytotoxicity in lupus-prone MRL-lpr mice40. Since cyclophosphamide is a cytotoxic drug that affects broad populations of cells, it was of interest to understand whether inflammatory pathways associated with prostaglandin production play a role in the etiology of brain damage. We used a nonsteroid antiinflammatory drug, ibuprofen, that is commonly used in treatment of SLE. The experimental dose was comparable to therapeutic doses used in other models of CNS inflammation43,44,69. In this model of NP-SLE, however, the IBU-rich diet did not attenuate behavioral dysfunction, serologic markers of autoimmune disease, T lymphocyte infiltration, or microglia/macrophage activation. This lack of beneficial effects was also observed in prospective clinical trials in patients with Alzheimer’s disease70, and when kidney pathology was examined in lupus-prone mice71. As for infiltrated CD3+ lymphocytes, chronic IBU treatment showed a trend for increased severity of leukocytosis in the third brain ventricle. The commonly reported adverse CNS reactions to IBU in patients with SLE72–75 are consistent with this observation. In addition, our negative finding on the importance of the inflammatory pathway is in agreement with a classic description of brain pathology in NP-SLE, generally negative for vasculitis76. Although this reasoning indirectly points to the importance of autoimmune and/or COX-independent mechanisms, one may assume the experimental treatment regimen failed due to limited penetration of IBU into the brain77, or because systemic inflammation in MRL-lpr mice is more aggressive than in models with Alzheimer’s-like brain pathology43,44. However, given that the blood-brain barrier is breached in diseased MRL-lpr mice12,13, the former possibility is unlikely. Using light and electron microscopy, numerous dark cells were observed throughout brains from diseased MRL-lpr mice. Condensed nuclear and cytoplasmic material, swollen mitochondria, and ruffled cell membranes suggest events that may precede excitotoxic/oncotic cell death. Transient dark neurons, however, are common in ischemic brain65, and also in normal CNS78 as a consequence of improper fixation or handling of brain tissue64. Although our sample size was not large enough to estimate the severity of the phenomenon, profound differences between MRL-lpr and control brains suggest that dark cells are not procedural artifacts. Rather, they point to profound metabolic perturbations (in both neurons and accessory cells) during the development of systemic autoimmune disease.
The Fas antigen (Fas/Apo-1/CD95) is a cell-surface receptor that is critical in mediating apoptosis in the CNS79. This receptor is not expressed in brains of MRL-lpr mice80, which is generally consistent with the lack of typical apoptotic morphology in this study. However, an apoptotic mode of neuronal demise can be mediated by mechanisms such as TNF-α or granzyme B receptors. If so, there are at least 2 lines of supportive evidence. For example, Alexander and colleagues combined immunohistochemistry of the neurofilament, TUNEL staining, DNA laddering, and caspase-3 activity to infer increased neuronal apoptosis in MRL-lpr brains81. Additionally, caspase-3-mediated apoptosis in animal preparations treated with neurotoxic CSF16 points to a causal relationship between NR2 receptor-reactive autoantibodies and brain damage in patients with NP-SLE82. However, the inability of the TUNEL method to discriminate apoptosis from necrosis in solid tissues calls into question the specificity of this assay83–85. Similarly, the evidence that caspase-3 activation also occurs in neuronal necrosis86 is in accord with the general consensus that transmission electron microscopy is the most reliable identification method87. This dilemma on whether apoptotic or necrotic mechanisms prevail in autoimmune brain is further complicated by markers of excitotoxic and/or pro-oncotic signaling. In particular, significant increases in glutamine, glutamate, and lactate concentrations have recently been reported in MRL-lpr brains88. In addition to free oxygen species, intracellular glutamine accumulation and release of glutamate are known to result in both apoptotic and necrotic neuronal demise89. However, acute excitotoxic damage is considered physiologically closer to the necrotic end of the apoptosis-necrosis continuum67,90.
Numerous factors can induce excitotoxic damage, and in injured or immunologically challenged brain, cytokine-producing microglia play an important role91,92. Together with class II MHC upregulation26, deposition of complement proteins C3 and C981, and increased mRNA expression for proinflammatory cytokines in brains of MRL-lpr mice27,28, our results are consistent with the notion of microglia-induced neuronal excitotoxicity. Observation of densely packed dark cells in the subgranular zone is potentially important in light of the evidence that CSF from lupus-prone mice is cytotoxic to proliferating brain cells93.
In addition to immune-mediated insults, altered production of steroid hormones is likely to be another important factor in the pathogenic circuitry during systemic autoimmune disease. Plasma corticosterone is chronically elevated in MRL-lpr mice94, and in contrast to its suppressive effect on peripheral inflammation, it may exacerbate excitotoxicity by glutamate accumulation and non-apoptotic death of central neurons95,96. Despite the relative colocalization of Fluoro-Jade B-positive and F4/80+ cells, our results do not clarify whether age-dependent microglial activation causes neuronal demise, or reflects a “scavenging response” to necrosis, or alternatively, a reparative process in the CNS97,98. In the same way, as commonly seen in neurodegenerative disorders associated with glutamate receptor-mediated excitotoxicity67, we could not provide conclusive evidence that documents typical modes of cell death in brains of autoimmune mice. The presence of clustered and scattered dark cells may not necessarily indicate the same pathogenic mechanism in major divisions of the brain because peripheral inflammation, permeable blood-brain barrier, disturbed ionic transport, and altered glucose metabolism may individually and/or synergistically compromise survival of mature and immature neurons in different brain regions12,13,15,88,93.
Defining characteristics of typical apoptotic or necrotic cell death were not found in MRL-lpr brains, despite relatively broad electron microscopic screening. However, condensation of nuclear and cytoplasmic material, swollen mitochondria, and ruffled appearance of cell membranes suggest profound metabolic perturbations that may precede excitotoxic/oncotic cell death. This possibility requires further exploration in a larger sample, throughout the whole brain, and using a combination of methodological approaches. A better understanding of neuronal death in the MRL model of NP-SLE may provide a basis for novel, non-antiinflammatory drugs in treating this poorly understood neuroimmunological condition.
Acknowledgments
We thank Marnie Timleck and Ernie Spitzer for their technical assistance. We are also grateful to Dr. Monalisa Sur for her expertise in interpreting electron microscopy results.
Footnotes
D.A. Ballok, PhD; X. Ma, MSc, Department of Psychiatry and Behavioral Neurosciences; J.A. Denburg, MD, FRCP, Department of Medicine; L. Arsenault, PhD, Department of Pathology and Molecular Medicine; B. Sakic, PhD, Department of Psychiatry and Behavioral Neurosciences, The Brain-Body Institute (St. Joseph’s Healthcare).
Supported by doctoral research award grant MOP 38065 from the Canadian Institutes of Health Research to D. Ballok, and grant 1R21 AR49163-01 from the National Institutes of Health to B. Sakic. Mr. Sakic is a recipient of the Father Sean O’Sullivan Research Centre career development award.
References
- 1.Steens SC, Admiraal-Behloul F, Bosma GP, et al. Selective gray matter damage in neuropsychiatric lupus. Arthritis Rheum. 2004;50:2877–81. doi: 10.1002/art.20654. [DOI] [PubMed] [Google Scholar]
- 2.Bosma GP, Middelkoop HA, Rood MJ, Bollen EL, Huizinga TW, van Buchem MA. Association of global brain damage and clinical functioning in neuropsychiatric systemic lupus erythematosus. Arthritis Rheum. 2002;46:2665–72. doi: 10.1002/art.10574. [DOI] [PubMed] [Google Scholar]
- 3.Trysberg E, Nylen K, Rosengren LE, Tarkowski A. Neuronal and astrocytic damage in systemic lupus erythematosus patients with central nervous system involvement. Arthritis Rheum. 2003;48:2881–7. doi: 10.1002/art.11279. [DOI] [PubMed] [Google Scholar]
- 4.Theofilopoulos AN. Murine models of lupus. In: Lahita RG, editor. Systemic lupus erythematosus. 2nd ed. New York: Churchill Livingstone; 1992. pp. 121–94. [Google Scholar]
- 5.Farrell M, Sakic B, Szechtman H, Denburg JA. Effect of cyclophosphamide on leucocytic infiltration in the brain of MRL/lpr mice. Lupus. 1997;6:268–74. doi: 10.1177/096120339700600310. [DOI] [PubMed] [Google Scholar]
- 6.Sakic B, Szechtman H, Keffer M, Talangbayan H, Stead R, Denburg JA. A behavioral profile of autoimmune lupus-prone MRL mice. Brain Behav Immun. 1992;6:265–85. doi: 10.1016/0889-1591(92)90048-s. [DOI] [PubMed] [Google Scholar]
- 7.Sakic B, Szechtman H, Denburg JA, Gorny G, Kolb B, Whishaw IQ. Progressive atrophy of pyramidal neuron dendrites in autoimmune MRL-lpr mice. J Neuroimmunol. 1998;87:162–70. doi: 10.1016/s0165-5728(98)00085-x. [DOI] [PubMed] [Google Scholar]
- 8.Sakic B, Szechtman H, Denburg JA. Neurobehavioral alteration in autoimmune mice. Neurosci Biobehav Rev. 1997;21:327–40. doi: 10.1016/s0149-7634(96)00018-8. [DOI] [PubMed] [Google Scholar]
- 9.Szechtman H, Sakic B, Denburg JA. Behaviour of MRL mice: an animal model of disturbed behaviour in systemic autoimmune disease. Lupus. 1997;6:223–9. doi: 10.1177/096120339700600302. [DOI] [PubMed] [Google Scholar]
- 10.Abbott NJ, Mendonca LL, Dolman DE. The blood-brain barrier in systemic lupus erythematosus. Lupus. 2003;12:908–15. doi: 10.1191/0961203303lu501oa. [DOI] [PubMed] [Google Scholar]
- 11.Duprez T, Nzeusseu A, Peeters A, Houssiau FA. Selective involvement of the choroid plexus on cerebral magnetic resonance images: a new radiological sign in patients with systemic lupus erythematosus with neurological symptoms. J Rheumatol. 2001;28:387–91. [PubMed] [Google Scholar]
- 12.Vogelweid CM, Johnson GC, Besch-Williford CL, Basler J, Walker SE. Inflammatory central nervous system disease in lupus-prone MRL/lpr mice: comparative histologic and immunohistochemical findings. J Neuroimmunol. 1991;35:89–99. doi: 10.1016/0165-5728(91)90164-3. [DOI] [PubMed] [Google Scholar]
- 13.Sidor MM, Sakic B, Malinowski PM, Ballok DA, Oleschuk CJ, Macri J. Elevated immunoglobulin levels in the cerebrospinal fluid from lupus-prone mice. J Neuroimmunol. 2005;165:104–13. doi: 10.1016/j.jneuroim.2005.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zameer A, Hoffman SA. B and T cells in the brains of autoimmune mice. J Neuroimmunol. 2004;146:133–9. doi: 10.1016/j.jneuroim.2003.10.052. [DOI] [PubMed] [Google Scholar]
- 15.Maric D, Millward JM, Ballok DA, et al. Neurotoxic properties of cerebrospinal fluid from behaviorally impaired autoimmune mice. Brain Res. 2001;920:183–93. doi: 10.1016/s0006-8993(01)03060-8. [DOI] [PubMed] [Google Scholar]
- 16.DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med. 2001;7:1189–93. doi: 10.1038/nm1101-1189. [DOI] [PubMed] [Google Scholar]
- 17.Kowal C, DeGiorgio LA, Nakaoka T, et al. Cognition and immunity; antibody impairs memory. Immunity. 2004;21:179–88. doi: 10.1016/j.immuni.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 18.Huerta PT, Kowal C, DeGiorgio LA, Volpe BT, Diamond B. Immunity and behavior: Antibodies alter emotion. Proc Natl Acad Sci USA. 2006;103:678–83. doi: 10.1073/pnas.0510055103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27:24–31. doi: 10.1016/j.it.2005.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakic B, Denburg JA, Denburg SD, Szechtman H. Blunted sensitivity to sucrose in autoimmune MRL-lpr mice: a curve-shift study. Brain Res Bull. 1996;41:305–11. doi: 10.1016/s0361-9230(96)00190-6. [DOI] [PubMed] [Google Scholar]
- 21.Sakic B, Szechtman H, Talangbayan H, Denburg SD, Carbotte RM, Denburg JA. Disturbed emotionality in autoimmune MRL-lpr mice. Physiol Behav. 1994;56:609–17. doi: 10.1016/0031-9384(94)90309-3. [DOI] [PubMed] [Google Scholar]
- 22.Marshall D, Dangerfield JP, Bhatia VK, Larbi KY, Nourshargh S, Haskard DO. MRL/lpr lupus-prone mice show exaggerated ICAM-1-dependent leucocyte adhesion and transendothelial migration in response to TNF-alpha. Rheumatology Oxford. 2003;42:929–34. doi: 10.1093/rheumatology/keg251. [DOI] [PubMed] [Google Scholar]
- 23.Zameer A, Hoffman SA. Increased ICAM-1 and VCAM-1 expression in the brains of autoimmune mice. J Neuroimmunol. 2003;142:67–74. doi: 10.1016/s0165-5728(03)00262-5. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez-Scarano F, Baltuch G. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci. 1999;22:219–40. doi: 10.1146/annurev.neuro.22.1.219. [DOI] [PubMed] [Google Scholar]
- 25.Nakanishi H. Microglial functions and proteases. Mol Neurobiol. 2003;27:163–76. doi: 10.1385/MN:27:2:163. [DOI] [PubMed] [Google Scholar]
- 26.McIntyre KR, Ayer-LeLievre C, Persson H. Class II major histocompatibility complex (MHC) gene expression in the mouse brain is elevated in the autoimmune MRL/Mp-lpr/lpr strain. J Neuroimmunol. 1990;28:39–52. doi: 10.1016/0165-5728(90)90039-p. [DOI] [PubMed] [Google Scholar]
- 27.Tomita M, Holman BJ, Williams LS, Pang KC, Santoro TJ. Cerebellar dysfunction is associated with overexpression of proinflammatory cytokine genes in lupus. J Neurosci Res. 2001;64:26–33. doi: 10.1002/jnr.1050. [DOI] [PubMed] [Google Scholar]
- 28.Tomita M, Holman BJ, Santoro TJ. Aberrant cytokine gene expression in the hippocampus in murine systemic lupus erythematosus. Neurosci Lett. 2001;302:129–32. doi: 10.1016/s0304-3940(01)01679-2. [DOI] [PubMed] [Google Scholar]
- 29.Sakic B, Maric I, Koeberle PD, et al. Increased TUNEL-staining in brains of autoimmune Fas-deficient mice. J Neuroimmunol. 2000;104:147–54. doi: 10.1016/s0165-5728(99)00277-5. [DOI] [PubMed] [Google Scholar]
- 30.Ballok DA, Millward JM, Sakic B. Neurodegeneration in autoimmune MRL-lpr mice as revealed by Fluoro Jade B staining. Brain Res. 2003;964:200–10. doi: 10.1016/s0006-8993(02)03980-x. [DOI] [PubMed] [Google Scholar]
- 31.Dubois RN, Abramson SB, Crofford L, et al. Cyclooxygenase in biology and disease. FASEB J. 1998;12:1063–73. [PubMed] [Google Scholar]
- 32.Kagan T, Zakeri Z. Detection of apoptotic cells in the nervous system. In: Harry J, Tilson HA, editors. Neurodegeneration methods and protocols. Totowa, NJ: Humana Press; 1999. pp. 105–23. [DOI] [PubMed] [Google Scholar]
- 33.Hanisch UK. Microglia as a source and target of cytokines. Glia. 2002;40:140–55. doi: 10.1002/glia.10161. [DOI] [PubMed] [Google Scholar]
- 34.Garavito RM, Mulichak AM. The structure of mammalian cyclooxygenases. Annu Rev Biophys Biomol Struct. 2003;32:183–206. doi: 10.1146/annurev.biophys.32.110601.141906. [DOI] [PubMed] [Google Scholar]
- 35.Klegeris A, McGeer PL. Cyclooxygenase and 5-lipoxygenase inhibitors protect against mononuclear phagocyte neurotoxicity. Neurobiol Aging. 2002;23:787–94. doi: 10.1016/s0197-4580(02)00021-0. [DOI] [PubMed] [Google Scholar]
- 36.Shibata H, Katsuki H, Nishiwaki M, Kume T, Kaneko S, Akaike A. Lipopolysaccharide-induced dopaminergic cell death in rat midbrain slice cultures: role of inducible nitric oxide synthase and protection by indomethacin. J Neurochem. 2003;86:1201–12. doi: 10.1046/j.1471-4159.2003.01929.x. [DOI] [PubMed] [Google Scholar]
- 37.Elsisi NS, Darling-Reed S, Lee EY, Oriaku ET, Soliman KF. Ibuprofen and apigenin induce apoptosis and cell cycle arrest in activated microglia. Neurosci Lett. 2005;375:91–6. doi: 10.1016/j.neulet.2004.10.087. [DOI] [PubMed] [Google Scholar]
- 38.Iwata R, Kitagawa K, Zhang NY, Wu B, Inagaki C. Non-steroidal anti-inflammatory drugs protect amyloid beta protein-induced increase in the intracellular Cl- concentration in cultured rat hippocampal neurons. Neurosci Lett. 2004;367:156–9. doi: 10.1016/j.neulet.2004.05.103. [DOI] [PubMed] [Google Scholar]
- 39.Carrasco E, Casper D, Werner P. Dopaminergic neurotoxicity by 6-OHDA and MPP(+): Differential requirement for neuronal cyclooxygenase activity. J Neurosci Res. 2005;81:121–31. doi: 10.1002/jnr.20541. [DOI] [PubMed] [Google Scholar]
- 40.Ballok DA, Earls AM, Krasnik C, Hoffman SA, Sakic B. Autoimmune-induced damage of the midbrain dopaminergic system in lupus-prone mice. J Neuroimmunol. 2004;152:83–97. doi: 10.1016/j.jneuroim.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 41.Sakic B, Lacosta S, Denburg J, Szechtman H. Altered neurotransmission in brains of autoimmune mice: pharmacological and neurochemical evidence. J Neuroimmunol. 2002;129:84–96. doi: 10.1016/s0165-5728(02)00171-6. [DOI] [PubMed] [Google Scholar]
- 42.Anderson KK, Ballok DA, Prasad N, Szechtman H, Sakic B. Impaired response to amphetamine and neuronal degeneration in the nucleus accumbens of autoimmune MRL-lpr mice. Behav Brain Res. 2006;166:32–8. doi: 10.1016/j.bbr.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lim GP, Yang F, Chu T, et al. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer’s disease. J Neurosci. 2000;20:5709–14. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan Q, Zhang J, Liu H, et al. Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer’s disease. J Neurosci. 2003;23:7504–9. doi: 10.1523/JNEUROSCI.23-20-07504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morris L, Graham CF, Gordon S. Macrophages in haemopoietic and other tissues of the developing mouse detected by the monoclonal antibody F4/80. Development. 1991;112:517–26. doi: 10.1242/dev.112.2.517. [DOI] [PubMed] [Google Scholar]
- 46.Perry VH, Hume DA, Gordon S. Immunohistochemical localization of macrophages and microglia in the adult and developing mouse brain. Neuroscience. 1985;15:313–26. doi: 10.1016/0306-4522(85)90215-5. [DOI] [PubMed] [Google Scholar]
- 47.Chen Z, Duan RS, Quezada HC, et al. Increased microglial activation and astrogliosis after intranasal administration of kainic acid in C57BL/6 mice. J Neurobiol. 2005;62:207–18. doi: 10.1002/neu.20099. [DOI] [PubMed] [Google Scholar]
- 48.Perry VH, Gordon S. Microglia and macrophages. In: Keane RW, Hickey WF, editors. Immunology of the nervous system. New York: Oxford University Press; 1997. pp. 155–72. [Google Scholar]
- 49.Merrill JE, Kono DH, Clayton J, Ando DG, Hinton DR, Hofman FM. Inflammatory leukocytes and cytokines in the peptide-induced disease of experimental allergic encephalomyelitis in SJL and B10.PL mice. Proc Natl Acad Sci USA. 1992;89:574–8. doi: 10.1073/pnas.89.2.574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marzio R, Mauel J, Betz-Corradin S. CD69 and regulation of the immune function. Immunopharmacol Immunotoxicol. 1999;21:565–82. doi: 10.3109/08923979909007126. [DOI] [PubMed] [Google Scholar]
- 51.Lindsley MD, Rodriguez M. Characterization of the inflammatory response in the central nervous system of mice susceptible or resistant to demyelination by Theiler’s virus. J Immunol. 1989;142:2677–82. [PubMed] [Google Scholar]
- 52.Luzina IG, Knitzer RH, Atamas SP, et al. Vasculitis in the Palmerston North mouse model of lupus: phenotype and cytokine production profile of infiltrating cells. Arthritis Rheum. 1999;42:561–8. doi: 10.1002/1529-0131(199904)42:3<561::AID-ANR22>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 53.Trump BF, Smuckler EA, Benditt EP. A method for staining epoxy sections for light microscopy. J Ultrastruct Res. 1961;5:343–8. doi: 10.1016/s0022-5320(61)80011-7. [DOI] [PubMed] [Google Scholar]
- 54.Ballok DA, Woulfe J, Sur M, Cyr M, Sakic B. Hippocampal damage in mouse and human forms of systemic autoimmune disease. Hippocampus. 2004;14:649–61. doi: 10.1002/hipo.10205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ahmad I, Tang L, Pham H. Identification of neural progenitors in the adult mammalian eye. Biochem Biophys Res Commun. 2000;270:517–21. doi: 10.1006/bbrc.2000.2473. [DOI] [PubMed] [Google Scholar]
- 56.Tsai CY, Wu TH, Huang SF, et al. Abnormal splenic and thymic IL-4 and TNF-alpha expression in MRL-lpr/lpr mice. Scand J Immunol. 1995;41:157–63. doi: 10.1111/j.1365-3083.1995.tb03548.x. [DOI] [PubMed] [Google Scholar]
- 57.Ballok DA, Szechtman H, Sakic B. Taste responsiveness and diet preference in autoimmune MRL mice. Behav Brain Res. 2003;140:119–30. doi: 10.1016/s0166-4328(02)00276-0. [DOI] [PubMed] [Google Scholar]
- 58.Strijbos PJ, Rothwell NJ. Interleukin-1 beta attenuates excitatory amino acid-induced neurodegeneration in vitro: involvement of nerve growth factor. J Neurosci. 1995;15:3468–74. doi: 10.1523/JNEUROSCI.15-05-03468.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Depino A, Ferrari C, Pott Godoy MC, Tarelli R, Pitossi FJ. Differential effects of interleukin-1 beta on neurotoxicity, cytokine induction and glial reaction in specific brain regions. J Neuroimmunol. 2005;168:96–110. doi: 10.1016/j.jneuroim.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 60.Theofilopoulos AN, Dixon FJ. Murine models of systemic lupus erythematosus. Adv Immunol. 1985;37:269–390. doi: 10.1016/s0065-2776(08)60342-9. [DOI] [PubMed] [Google Scholar]
- 60a.Ma X, Foster J, Sakic B. Distribution and prevalence of leukocyte phenotypes in brains of lupus-prone mice. J Neuroimmunol. 2006 Aug 8; doi: 10.1016/j.jneuroim.2006.06.023. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 61.Sakic B, Szechtman H, Braciak TA, Richards CD, Gauldie J, Denburg JA. Reduced preference for sucrose in autoimmune mice: a possible role of interleukin-6. Brain Res Bull. 1997;44:155–65. doi: 10.1016/s0361-9230(97)00107-x. [DOI] [PubMed] [Google Scholar]
- 62.Alvarez-Buylla A, Lim DA. For the long run: maintaining germinal niches in the adult brain. Neuron. 2004;41:683–6. doi: 10.1016/s0896-6273(04)00111-4. [DOI] [PubMed] [Google Scholar]
- 63.Brenner RM, Slayden OD, Rodgers WH, et al. Immunocytochemical assessment of mitotic activity with an antibody to phosphorylated histone H3 in the macaque and human endometrium. Hum Reprod. 2003;18:1185–93. doi: 10.1093/humrep/deg255. [DOI] [PubMed] [Google Scholar]
- 64.Schmechel D. Assessment of ultrastructural changes associated with apoptosis. In: Hannun YA, Boustany R, editors. Apoptosis in neurobiology. Boca Raton: CRC Press; 1999. pp. 153–81. [Google Scholar]
- 65.Auer RN, Kalimo H, Olsson Y, Siesjo BK. The temporal evolution of hypoglycemic brain damage. I. Light- and electron-microscopic findings in the rat cerebral cortex. Acta Neuropathol Berl. 1985;67:13–24. doi: 10.1007/BF00688120. [DOI] [PubMed] [Google Scholar]
- 66.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C. Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: A perspective on the contributions of apoptosis and necrosis. Brain Res Bull. 1998;46:281–309. doi: 10.1016/s0361-9230(98)00024-0. [DOI] [PubMed] [Google Scholar]
- 68.Sakic B, Kolb B, Whishaw IQ, Gorny G, Szechtman H, Denburg JA. Immunosuppression prevents neuronal atrophy in lupus-prone mice: evidence for brain damage induced by autoimmune disease? J Neuroimmunol. 2000;111:93–101. doi: 10.1016/s0165-5728(00)00364-7. [DOI] [PubMed] [Google Scholar]
- 69.Teismann P, Tieu K, Choi DK, et al. Cyclooxygenase-2 is instrumental in Parkinson’s disease neurodegeneration. Proc Natl Acad Sci USA. 2003;100:5473–8. doi: 10.1073/pnas.0837397100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147 (Suppl 1):S232–S240. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kelley VE, Sneve S, Musinski S. Increased renal thromboxane production in murine lupus nephritis. J Clin Invest. 1986;77:252–9. doi: 10.1172/JCI112284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Samuelson CO, Jr, Williams HJ. Ibuprofen-associated aseptic meningitis in systemic lupus erythematosus. West J Med. 1979;131:57–9. [PMC free article] [PubMed] [Google Scholar]
- 73.Sonnenblick M, Abraham AS. Ibuprofen hypersensitivity in systemic lupus erythematosus. BMJ. 1978;1:619–20. doi: 10.1136/bmj.1.6113.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hoppmann RA, Peden JG, Ober SK. Central nervous system side effects of nonsteroidal anti-inflammatory drugs. Aseptic meningitis, psychosis, and cognitive dysfunction. Arch Intern Med. 1991;151:1309–13. [PubMed] [Google Scholar]
- 75.Mou SS, Punaro L, Anton J, Luckett PM. Severe systemic hypersensitivity reaction to ibuprofen: a presentation of systemic lupus erythematosus. J Rheumatol. 2006;33:171–2. [PubMed] [Google Scholar]
- 76.Johnson RT, Richardson EP. The neurological manifestations of systemic lupus erythematosus. Medicine. 1968;47:337–69. doi: 10.1097/00005792-196807000-00002. [DOI] [PubMed] [Google Scholar]
- 77.Mannila A, Rautio J, Lehtonen M, Jarvinen T, Savolainen J. Inefficient central nervous system delivery limits the use of ibuprofen in neurodegenerative diseases. Eur J Pharm Sci. 2005;24:101–5. doi: 10.1016/j.ejps.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 78.Cohen EB, Pappas GD. Dark profiles in the apparently-normal central nervous system. A problem in the electron microscopic identification of early anterograde axonal degeneration. J Comp Neurol. 1969;136:375–96. doi: 10.1002/cne.901360403. [DOI] [PubMed] [Google Scholar]
- 79.Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270:1189–92. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- 80.Park C, Sakamaki K, Tachibana O, Yamashima T, Yamashita J, Yonehara S. Expression of Fas antigen in the normal mouse brain. Biochem Biophys Res Commun. 1998;252:623–8. doi: 10.1006/bbrc.1998.9572. [DOI] [PubMed] [Google Scholar]
- 81.Alexander JJ, Jacob A, Bao L, Macdonald RL, Quigg RJ. Complement-dependent apoptosis and inflammatory gene changes in murine lupus cerebritis. J Immunol. 2005;175:8312–9. doi: 10.4049/jimmunol.175.12.8312. [DOI] [PubMed] [Google Scholar]
- 82.Omdal R, Brokstad K, Waterloo K, Koldingsnes W, Jonsson R, Mellgren SI. Neuropsychiatric disturbances in SLE are associated with antibodies against NMDA receptors. Eur J Neurol. 2005;12:392–8. doi: 10.1111/j.1468-1331.2004.00976.x. [DOI] [PubMed] [Google Scholar]
- 83.Yasuda M, Umemura S, Osamura RY, Kenjo T, Tsutsumi Y. Apoptotic cells in the human endometrium and placental villi: pitfalls in applying the TUNEL method. Arch Histol Cytol. 1995;58:185–90. doi: 10.1679/aohc.58.185. [DOI] [PubMed] [Google Scholar]
- 84.Charriaut-Marlangue C, Ben-Ari Y. A cautionary note on the use of the TUNEL stain to determine apoptosis. Neuroreport. 1995;7:61–4. [PubMed] [Google Scholar]
- 85.Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995;21:1465–8. doi: 10.1002/hep.1840210534. [DOI] [PubMed] [Google Scholar]
- 86.Niquet J, Allen SG, Baldwin RA, Wasterlain CG. Evidence of caspase-3 activation in hyposmotic stress-induced necrosis. Neurosci Lett. 2004;356:225–7. doi: 10.1016/j.neulet.2003.11.063. [DOI] [PubMed] [Google Scholar]
- 87.Otsuki Y, Li Z, Shibata MA. Apoptotic detection methods — from morphology to gene. Prog Histochem Cytochem. 2003;38:275–339. doi: 10.1016/s0079-6336(03)80002-5. [DOI] [PubMed] [Google Scholar]
- 88.Alexander JJ, Zwingmann C, Quigg R. MRL/lpr mice have alterations in brain metabolism as shown with [(1)H-(13)C] NMR spectroscopy. Neurochem Int. 2005;47:143–51. doi: 10.1016/j.neuint.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 89.Brown GC, Bal-Price A. Inflammatory neurodegeneration mediated by nitric oxide, glutamate, and mitochondria. Mol Neurobiol. 2003;27:325–55. doi: 10.1385/MN:27:3:325. [DOI] [PubMed] [Google Scholar]
- 90.Gwag BJ, Koh JY, Demaro JA, Ying HS, Jacquin M, Choi DW. Slowly triggered excitotoxicity occurs by necrosis in cortical cultures. Neuroscience. 1997;77:393–401. doi: 10.1016/s0306-4522(96)00473-3. [DOI] [PubMed] [Google Scholar]
- 91.Piani D, Spranger M, Frei K, Schaffner A, Fontana A. Macrophage-induced cytotoxicity of N-methyl-D-aspartate receptor positive neurons involves excitatory amino acids rather than reactive oxygen intermediates and cytokines. Eur J Immunol. 1992;22:2429–36. doi: 10.1002/eji.1830220936. [DOI] [PubMed] [Google Scholar]
- 92.Allan SM, Rothwell NJ. Cytokines and acute neurodegeneration. Nat Rev Neurosci. 2001;2:734–44. doi: 10.1038/35094583. [DOI] [PubMed] [Google Scholar]
- 93.Sakic B, Kirkham DL, Ballok DA, et al. Proliferating brain cells are a target of neurotoxic CSF in systemic autoimmune disease. J Neuroimmunol. 2005;169:68–85. doi: 10.1016/j.jneuroim.2005.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lechner O, Dietrich H, Oliveira dos SA, et al. Altered circadian rhythms of the stress hormone and melatonin response in lupus-prone MRL/MP-fas (Ipr) mice. J Autoimmun. 2000;14:325–33. doi: 10.1006/jaut.2000.0375. [DOI] [PubMed] [Google Scholar]
- 95.Roy M, Sapolsky RM. The exacerbation of hippocampal excitotoxicity by glucocorticoids is not mediated by apoptosis. Neuroendocrinology. 2003;77:24–31. doi: 10.1159/000068337. [DOI] [PubMed] [Google Scholar]
- 96.Dinkel K, Ogle WO, Sapolsky RM. Glucocorticoids and central nervous system inflammation. J Neurovirol. 2002;8:513–28. doi: 10.1080/13550280290100914. [DOI] [PubMed] [Google Scholar]
- 97.Lazarov-Spiegler O, Solomon AS, Zeev-Brann AB, Hirschberg DL, Lavie V, Schwartz M. Transplantation of activated macrophages overcomes central nervous system regrowth failure. FASEB J. 1996;10:1296–302. doi: 10.1096/fasebj.10.11.8836043. [DOI] [PubMed] [Google Scholar]
- 98.Prewitt CM, Niesman IR, Kane CJ, Houle JD. Activated macrophage/microglial cells can promote the regeneration of sensory axons into the injured spinal cord. Exp Neurol. 1997;148:433–43. doi: 10.1006/exnr.1997.6694. [DOI] [PubMed] [Google Scholar]