

Abstract
Background
Various psychiatric manifestations of unknown etiology are common in systemic autoimmune disease lupus erythematosus (SLE). Profound heterogeneity at clinical and neuropathological levels suggests distinct subpopulations of SLE patients and multiple mechanisms in the pathogenesis of aberrant behavior. Using inbred mice prone to SLE-like condition, we presently examine whether subpopulations of diseased mice can be identified on the basis of their behavioral performance.
Methods
Hierarchical cluster analysis was used to classify 105 MRL-lpr males into clusters. Multivariate analysis of variance (MANOVA) and discriminant function analysis were used to detect overall differences and identify discriminative variables.
Results
Cluster 1 was characterized by blunted responsiveness to palatable stimulation, as well as increased spleen mass and serum levels of interleukin-1. Cluster 2 comprised of animals with reduced ambulation speed and enlarged spleen. Mice from cluster 3 showed profound dilatation of brain ventricles, reduced brain mass, impaired nutrition and performance in task reflective of emotional reactivity.
Conclusions
Present results suggest that systemic autoimmunity compromises brain function via non-Mendelian mechanisms. Although neuroactive cytokines may impair reward systems, brain atrophy seems to underlie deficits in ingestive behavior and emotional reactivity. This study supports the hypothesis that multiple neuroimmunological pathways are involved in the etiology of aberrant behavior during SLE-like disease.
Keywords: Neuropsychiatric lupus, autoimmunity, brain atrophy, multivariate analysis, MRL mice, animal model
Given its notorious heterogeneity in presentation, systemic lupus erythematosus (SLE) has been colloquially called “the disease with a thousand faces” (Senecal 1991; Hildenbrand 1992). Inflammation of the skin, kidneys, and joints are typical manifestations which largely vary with respect to onset, distribution, and severity. Although linkage and association studies confirmed the importance of multiple susceptibility genes, it is clear that environmental and humoral factors play an equally important role in the etiology of SLE (Tsao 2003). Recent epidemiological studies revealed clinically-distinct subpopulations among SLE patients (Jacobsen et al 1998; Tapanes et al 2000), thus further supporting the hypothesis that multiple mechanisms underlie the pathogenesis of this complex disorder.
In addition to diverse signs of systemic autoimmunity and inflammation, up to 70% of SLE patients present with a variety of neurologic and psychiatric problems of unknown etiology (Perry and Miller 1992; Denburg et al 1993). Although seizures and psychosis were included among the American Rheumatism Association criteria for the diagnosis of SLE, causes of central nervous system (CNS) dysfunction have been a matter of controversy for many years (Bruyn 1995). Nevertheless, contemporary imaging studies corroborate the notion that neuropsychiatric lupus (NP-SLE) is an autoimmunity-induced brain disorder, characterized by impaired blood flow, metabolic alterations, and progressive neuronal loss (Griffey et al 1990; Sibbitt and Sibbitt 1993; Sibbitt et al 1989, 1994; Brooks et al 1997). Recent research suggests the importance of soluble immune messengers, hormones, and neurotoxins (Hanson et al 1992; Denburg and Behmann 1994; Tsai et al 1994; Toubi et al 1995; Tincani et al 1996; Vogelgesang et al 1996; Ramos et al 1996; Jara et al 1998), but it remains unclear whether single or multiple mechanisms underlie the etiology of NP-SLE, and whether immune activity at the periphery or within the brain is instrumental for disease prognosis and therapy. Given this etiological complexity and clinical heterogeneity, it is likely that the pathogenesis of NP-SLE could be better understood if clinically homogenous cohorts of patients are systematically examined.
The study of disease mechanisms has been facilitated by the development of inbred strains of autoimmune mice, which display serologic changes and organ damage similar to SLE (Andrews et al 1978; Theofilopoulos 1992). The MRL/MpJ-lpr (MRL-lpr) substrain spontaneously develops an accelerated lupus-like syndrome in comparison to the MRL/MpJ +/+ (MRL +/+) strain, which differs <.1% in genome and develops similar manifestations later in life (Theofilopoulos 1992). The availability of congenic MRL +/+ control and lack of inherited brain anomalies (Sherman et al 1987) helps to minimize confounding effects of dissimilar genetic makeup on behavior and neuromorphology, thus making the MRL-lpr model a suitable preparation for studying the effects of systemic autoimmunity on brain function (Alexander et al 1983; Sakic et al 1997b). Similar to SLE patients (van Dam et al 1994; Wekking 1993), adult MRL-lpr mice show disturbed emotional reactivity and affective behavior well before the function of peripheral organs is compromised (Sakic et al 1994b; Sakic et al 1994a). Moreover, breached blood-brain barrier, brain atrophy, and restorative effectiveness of immunosuppressive therapy in behaviorally-impaired autoimmune MRL-lpr mice (Vogelweid et al 1991; Sakic et al 2000a; Sakic et al 1998b; Saic et al 1995) show significant similarity to NP-SLE (Abbott et al 2003; Duprez et al 2001; Ramos et al 1996; Boumpas et al 1991).
A constellation ofbehavioral deficits (defined in respect to the behavioral profile of MRL +/+ mice) was operationally labeled “autoimmunity-induced behavioral syndrome,” or AABS (Sakic et al 1997b). Moreover, the brains from MRL-lpr mice show retarded growth, reduced branching of pyramidal neurons (Sakic et al 1998b), ventricular enlargement (Denenberg et al 1992), increased cell death in the periventricular region (Sakic et al 2000b), and infiltration of immune cells into the choroid plexus (Alexander et al 1983; Vogelweid et al 1991; Farrell et al 1997). The hypothesis that the onset of systemic autoimmune disease alters brain function has been supported by the contemporaneous emergence of serological and behavioral deficits (Sakic et al 1994a), as well as the effectiveness of immunosuppressive treatment in reducing behavioral differences between the MRL substrains, lymphocyte infiltration, and neuronal atrophy in brains of MRL-lpr mice (Sakic et al 1995; Farrell et al 1997; Sakic et al 2000a). However, the possibility that at least part of the behavioral dissimilarity between MRL-lpr and MRL +/+ mice is due to the subtle differences in their genome has never been discounted. Along the same line, despite the fact that inbred MRL-lpr mice are a genetically homogeneous population, we observed substantial within-group variability in the development of immunological, neuropathological, and behavioral manifestations. For example, age- and sex-matched MRL-lpr mice significantly differed in the number of infiltrated T-cells, the density of terminal deoxynucleotidyl transferase-meditated dUDP-biotin nick end labeling (TUNEL)-positive nuclei in the MRL-lpr brains (Sakic et al 2000b), and the pattern of behavioral abnormalities (Sakic et al 1994b).
Taken together, the accumulated data suggest that the progress of SLE-like disease produces several subpopulations that show dissimilar clinical features. The present study aims to identify these subsets and to further explore the relationships among autoimmunity/inflammation, early neuropathological changes, and behavioral deficits. To eliminate genetic dissimilarity between the two MRL substrains as a source of variability, a large cohort of inbred MRL-lpr males was used exclusively. Multivariate statistical procedures were used to identify subpopulations (i.e., clusters) on the basis of their performance in an established behavioral battery (Sakic et al 1994b; Sakic et al 1996) and to examine the relationships among serological markers of autoimmunity, gross neuropathology and behavioral deficits.
Methods and Materials
Animals
One hundred-five MRL/MpJ-lpr (MRL-lpr) mice were purchased from the Jackson Laboratory (Bar Harbour, Maine) when 6-weeks old. They were housed 5 per cage, kept under a 12-hour light cycle (8 am - 8 pm), with standard food (Purina 5001) and tap water provided ad lib. At 11 weeks, animals were single housed and tested in batches of 20-25 mice over 2 weeks. The behavioral, neuropathological, and immunopathological data (Table 1) were collected before skin lesions, generalized lymph-adenopathy, and other signs were apparent.
Table 1.
Three Sets of Dependent Variables Measured Throughout the Study
| Description | |
|---|---|
| Behavior | |
| Food intake | Food mass (g) consumed over five days |
| Water intake | Water volume (ml) consumed over five days |
| Sucrose intake | Volume of 4% solution (ml) consumed in three 1-hour choice tests |
| Step-down latency | Time (s) elapsed to descend from an elevated platform within 5 min |
| Novel object exploration | Time (s) spent in contact with an object during the 10-min test |
| Floating time | Immobility time (s) in the 10-min forced swim test |
| Speed | Average movement speed in an activity monitor from 6–8 pm |
| Immunology | |
| Spleen mass | Mass (mg) of wet spleen after sacrifice |
| ANA | Optical density (OD) of serum anti-nuclear antibody levels (by ELISA) |
| AClA | OD of serum anti-cardiolipin antibody levels (by ELISA) |
| IL-1beta | Serum levels (pg/ml) of interleukin-1beta (by ELISA) |
| IFN-gamma | Serum levels (pg/ml) of interferon-gamma (by ELISA) |
| TNF-alpha | Serum levels (pg/ml) of tumor-necrosis factor-alpha (by ELISA) |
| Neuropathology | |
| Brain mass | Mass of the brain fixed upon sacrifice |
| Ventricle size | Total area of third and lateral ventricles/area of coronal sections |
| CD4+ cells | Total number of CD4+ cells in four coronal sections |
| CD8+ cells | Total number of CD8+ cells in four coronal sections |
ANA, anti-nuclear antibodies; AClA, anti-cardiolipin autoantibodies; IL, interleukin; IFN, interferon; TNF, tumor necrosis factor; ELISA, enzyme-linked immunosorbent assay.
Behavioral Testing
Food and Water Intake
Daily food intake was measured over 5 consecutive days by placing food pellets (about 13–14g) on the cage floor and re-weighing them 24 hours later. Daily water intake was assessed as the difference in mass of nonleaky 30-ml syringes filled with tap water.
Sucrose Preference Test
The brief sucrose preference test is proposed to measure “hedonic” response to a palatable stimulus (Muscat and Willner 1992; Monleon et al 1995). A plastic 10-ml syringe was added to a cage lid and 3 ml of 4 % aqueous sucrose solution was available daily over 3 days (the training phase). During the testing phase 7 ml of the sucrose solution was available between 9:30 – 10:30 pm. The volume ingested over three trials was used as an index of responsiveness to palatable stimulation (Sakic et al 1996; Sakic et al 1997a).
Step-Down Test
This task was designed to assess an anxiety-like response to elevated spaces (Egan and Royce 1973). A mouse was placed on wire mesh placed on a rectangular glass box(15 × 9 × 9 cm), and latency to step down on a black table was recorded in a 5-min test. The platform and surroundings were subsequently cleaned with a window cleaner.
Novel Object Test
The exploratory drive was assessed by allowing a mouse to explore a small arena in a Plexiglas cylinder for 5 min before a white, cone-like plastic funnel (diameter = 7 cm, height = 11.5 cm) was introduced. The exploration of the object was videotaped for 10 min and the contact time was assessed using the Observer Basic 3 software (Noldus Information Technology, Wageningen, The Netherlands). Contact was defined when mouse was sniffing, licking, biting or touching the funnel. The test environment was cleaned between the trials.
Forced Swim Test
Increased immobility of rodents in a no-escape situation was proposed to reflect a state of lower “mood,” which can be reduced by antidepressants and electro-convulsive shock (Porsolt et al 1977). Mice were gently lowered into a circular pool (diameter 1.83 m) filled with 25°C water and allowed to swim for 10 min (Sakic et al 1994b). The time spent in floating was recorded by an experimenter with a stopwatch.
Spontaneous Locomotor Activity
The spontaneous nocturnal activity was assessed as the speed of ambulation from 6 pm–8 am in computerized activity boxes (Omnitech Electronics Inc., Columbus, Ohio), as described before (Sakic et al 1992).
Sacrifice and Tissue Sampling
Mice were anesthetised with Somnotol (intraperitoneal [IP] 65 mg/kg, about .2 ml). The thoracic cavity was exposed to cut inferior vena cava and aspirate about 1.5 ml of blood with standard blood collecting tube. The animals were perfused intracardially with 20 ml of phosphate buffered saline (PBS), followed by 20 ml of a 4 % aqueous solution of paraformaldehyde (PFA). Brains were extracted and fixed in 4% PFA for 24 hours. After fixation, they were weighed, transferred to a 30% sucrose solution, and stored at 4°C until neuropathological analysis.
Brain Pathology
Reduced brain mass (Sakic et al 1998b), ventricular enlargement (Denenberg et al 1992), and infiltration of T-lymphocytes into the brain tissue (Sakic et al 2000b) are common in diseased MRL-lpr mice and they were presently used as markers of brain pathology.
Preparation of Brain Tissue
A stainless steel matrix for mouse brain (Stoelting Co., Wood Dale, Illinois) was used to facilitate standardized access to regions of interest. The central slice (about 5 mm away from the rostral end of olfactory bulb) was placed in a foil cup with optimal cutting temperature compound (OCT) medium. The tissue was frozen in slurry of 2-methylbutane and dry ice, and stored at −20°C. Eight-micron sections were cut using a Leitz 1720 cryostat. Four sections from each brain were placed on microscope slides coated with 2-aminopropyl-triethoxysilane (APTEX), air dried, and refrigerated until immunohistochemical analysis.
Lymphocyte Infiltration
Slides were brought to room temperature (RT), and washed in PBS for 15 min. The primary monoclonal antibodies to mouse CD4 (Sigma-Aldrich Canada Ltd, Oakville, Ontario, Canada) or CD8 (Pharmingen, San Diego, California) antigens were applied. The antibodies were diluted 1:100 (CD4) and 1:200 (CD8) in diluting buffer (Dako, Mississauga, Ontario, Canada), with 1% goat serum as a blocking agent. The slides were incubated overnight at RT. Subsequently, they were rinsed and then washed in PBS for 15 min. A secondary fluorescein isothicyanate (FITC)-conjugated goat anti-rat IgG was diluted 1:50 in diluting buffer and mixed with 1% goat serum, then applied for 4 hours at RT. After secondary incubation, the slides were rinsed with PBS and cover slipped using Fluorescence Mounting Medium (Dako, Mississauga, ON). Fluorescence was visualized using a Zeiss Diastar fluorescent microscope, with an FITC filter. Stained cells showed round, with no visible processes or elongation, and nucleus usually visible. The number of cells in four entire sections was recorded by an unbiased observer.
Brain Planimetry
Brains were extracted, fixed, and cut at the level of lateral and third ventricles. However, sections from 18 brains appeared either wrinkled or damaged under the microscope, thus precluding a reliable planimetric analysis and reducing the sample size to n = 87. Slides with 8 μm-thick sections were stained with hematoxylin and eosin according to standard histological procedures. A SC505 camera (VSP Inc., Michigan) was used to acquire digital images of each tissue section, which were illuminated using a light box (Imaging Research Inc., St. Catharines, Ontario, Canada). They were analyzed using the corresponding MCID system by tracing the outline of ventricles and a brain section. The system was calibrated for mm2 outputs, and the ventricular area versus total area ratio was calculated.
Indices of Systemic Autoimmunity
Together with enlargement of spleen mass, high levels of pro-inflammatory cytokines and autoantibodies are typical manifestations of systemic lupus-like disease in the MRL-lpr substrain (Theofilopoulos 1992). The wet spleen weight was determined on an analytical scale upon extraction. Blood was allowed to clot in 1.5 ml vials at RT. The serum was separated after 10-min centrifugation at 3000 rpm and stored at −20°C.
Serum Cytokine Levels
Commercial enzyme-linked immunosorbant assay (ELISA) kits (R&D Systems Inc., Minneapolis, Minnesota) were used to assess the serum levels of IL-1 beta, IFN-gamma and TNF-alpha. In brief, 50 μl of thawed serum was added to the plate and incubated at RT for 2 hours. The plate was washed five times with wash buffer, dried, and 100 μl of secondary antibody conjugated to horseradish peroxidase added to each well. After 2 hours of incubation the plate was washed and the chromogenic substrate added to each well. The reaction was stopped after 30 min of incubation with HCl solution. Optical density was measured at 450 nm, and internal standards were used to calculate cytokine concentration (pg/ml).
Serum Anti-Nuclear And Anti-Cardiolipin Antibody Levels
To confirm the autoimmune status and test for a role of circulating anti-cardiolipin autoantibodies (AClA) in brain damage (Martinez-Cordero et al 1997; Schwartz et al 1998; Hanly et al 1999), serum levels of anti-nuclear antibodies (ANA) and AClA and were measured by ELISA kits (Alpha Diagnostic International, San Antonio, Texas)(Hess et al 1993; Sakic et al 2000a). Sera were diluted 1:100 with sample diluent, and 100 μl was added to coated and noncoated (control) wells in the plate. The plate was incubated for 30 min at RT, washed four times with wash buffer, and 100 μl of goat anti-mouse IgG (conjugated to horseradish peroxidase) was added to each well. After 30 min at RT, the chromogenic substrate was added and the reaction stopped with HCl after 15 min. Optical density was determined at 450 nm, and the data expressed as relative optical densities.
Due to significant volume demands for cytokine ELISAs, there was not enough serum for the assessment of AClA in ten cases. Therefore, the sample size for the MANOVA analysis was ninety-five.
Data Analysis
All calculations were performed using SPSS (v. 11, SPSS Inc, Chicago, Illinois). Bar graphs show means ± standard error, and significant pair-wise differences of p < .05, p < .01 and p < .001 are indicated by *, **, and *** respectively. Table 1 provides the summary of variables collected.
Cluster Analysis
This statistical method allows the classification of subjects on the basis of their trait similarities. In the present study behavioral performance was used as the criterion to identify subpopulations (clusters) within the cohort of diseased, age- matched MRL-lpr males. The overall expectation was that clusters that differ in behavioral performance will also show a specific immunological status and/or brain morphology. Based on the assumption that the subgroups have common traits important in control of behavioral performance (genetic background, sex, age, proclivity to systemic disease) hierarchical cluster analysis was used to examine the level of similarity among the subjects.
Previous multivariate analysis of the behavioral profile in MRL mice suggested that measures taken in a specific task are highly inter-correlated and that performance in different tasks reflects different domains of behavior (Sakic et al 1994b). This implies that constructs, such as anxiety-related behavior, spontaneous locomotion, exploratory drive, and depressive-like behavior can be depicted by a rather parsimonious list of variables. To avoid high inter-correlations among variables measured in the same task and to improve the subject-to-variable ratio, the spontaneous activity was measured by ambulatory speed, instead of distance traversed and ambulatory time. Similarly, latency to approach a novel object was not used due to the significant negative correlation with the exploratory time. Given that the presently selected behavioral variables differed significantly on the scales in which they were measured (e.g., speed from 7 – 15 cm/s; step-down latency 40 – 300 s), the raw data were converted to z-scores to obtain a standardized contribution in the cluster analysis. The proximity matrix was calculated using squared Euclidean distance as the measure of similarity between pairs of mice. The clustering algorithm applied to this distance matrix was Ward’s minimum variance method. This is an agglomerative method, which examines all possible mergers of two clusters at each iteration. Subsequently, it selects the merger that results in the smallest increase in the variance of the cluster, as calculated by the sum of the squares of the differences between each object in the cluster and the cluster mean (Romesburg 2003). This method is regarded as very efficient, although it tends to create clusters of rather small size. A tree diagram (dendogram), illustrating the hierarchical relationships among individual mice, was generated by the SPSS graphical module. Two, three, and four putative clusters were identified from the dendogram, and their validity in detecting distinct subpopulations was tested by 3 separate multivariate analyses of variance (MANOVA), where cluster membership was used as an independent variable, and the behavioral, autoimmunity, and neuropathology variables were used as dependent variables, respectively. Discriminant functions analysis (DFA) was also conducted, using the behavioral variables as predictors of cluster membership. This analysis was used to quantify rates of correct classification based on the behavioral variables, and also to explore the patterns of behavioral variables that differentiate mice in different clusters.
Results
The results of cluster analysis are shown in the dendogram (Figure 1), from which the relationships between each case can be seen. Although two-, three- and four-cluster solutions appeared viable, the three-cluster solution solely distinguished the subpopulations on all behavioral measures (Figure 2), as confirmed by MANOVA and ANOVA tests (Table 2). Moreover, such classification distinguished the clusters on the basis of two neuropathological and two immunological measures. Dissimilar brain weight and ventricle size rendered the three-cluster solution consistent with a report on “mild,” “moderate,” and “severe” ventricular enlargement in diseased MRL-lpr mice (Denenberg et al 1992). At the same time, this solution revealed that spleen size and levels of circulating pro-inflammatory cytokine IL-1 beta differ across the groups. Body weight and leukocyte infiltration did not differ among the clusters, despite high within-group variability. However, more CD4+ cells than CD8+ were seen overall (256 ± 20 vs. 116 ± 13, respectively). The majority of infiltrates were detected in the choroid plexus of the third ventricle, in the lateral ventricles and the interventricular foramina. Cells were also occasionally seen in the meninges, in the longitudinal fissure on the dorsal surface of the brain, hippocampus, and in the parenchyma.
Figure 1.
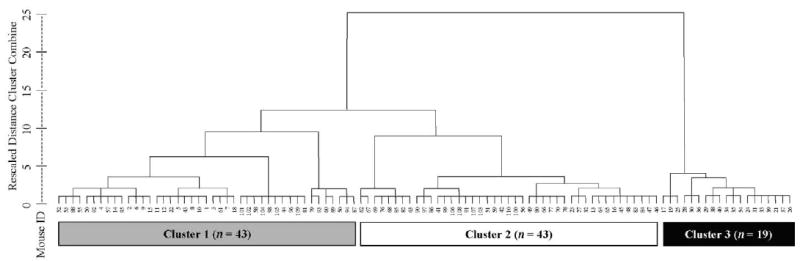
Dendogram output from the hierarchical cluster analysis of performance in the battery of behavioral tasks. The figure can be read from bottom to top as a summary of the clustering process. At each step, starting with each mouse as its own cluster, the most similar clusters are amalgamated at each stage. Cases are listed along the x-axis, while the y-axis indicates a rescaled measure of multivariate similarity in behavioral performance. When clusters are less similar to their nearest neighbors, the vertical distance between the formation of the cluster and its amalgamation will be larger. Small numbers are code numbers for each MRL-lpr male. The 3-cluster solution is emphasized by the shaded boxes at the bottom of the figure, with the numbers in each box indicating sample size of each cluster.
Figure 2.

Statistical differences in behavioral, neuropathological, and immune measures among three clusters. It can be seen that most of behavioral deficits are associated with markers of brain atrophy in Cluster 3.
Table 2.
The Differences Between Clusters (as Detected by MANOVA) in Case of the Three-Cluster Solution
| df | F | p | |
|---|---|---|---|
| Behavioral Measures | |||
| Hotelling’s Trace | 14, 190 | 4.067 | <.001 |
| Food intake | 2, 102 | 72.125 | <.001 |
| Water intake | 2, 102 | 44.464 | <.001 |
| Sucrose intake | 2, 102 | 6.315 | <.005 |
| Step-down latency | 2, 102 | 48.055 | <.001 |
| Novel object exploration | 2, 102 | 24.929 | <.001 |
| Floating time | 2, 102 | 3.505 | <.05 |
| Speed | 2, 102 | 15.958 | <.001 |
| Indices of Autoimmunity | |||
| Hotelling’s Trace | 12, 172 | 2.651 | .003 |
| Relative spleen mass | 2, 92 | 9.675 | <.001 |
| Anti-nuclear antibodies | 2, 92 | 1.245 | .293 |
| Anti-cardiolipin antibodies | 2, 92 | 1.134 | .326 |
| IL-1beta | 2, 92 | 4.148 | <.02 |
| IFN-gamma | 2, 92 | .537 | .587 |
| TNF-alpha | 2, 92 | 2.252 | .111 |
| Neuropathological Measures | |||
| Hotelling’s Trace | 8, 160 | 10.672 | <.001 |
| Brain mass | 2, 84 | 43.719 | <.001 |
| Ventricle | 2, 84 | 8.052 | <.001 |
| Section area | |||
| CD4+ cells | 2, 84 | .884 | .417 |
| CD8+ cells | 2, 84 | 2.389 | .098 |
MANOVA, multivariate analysis of variance; IL, interleukin, IFN, interferon; TNF, tumor necrosis factor.
The behavioral variables were also used as predictors of the cluster memberships in DFA. Jack-knifed classification accuracy was calculated using observed cluster frequencies as prior probabilities. Two discriminant functions, accounting for 68% and 32% of the between-cluster variance respectively, classified the mice into the correct clusters with 92.4% accuracy (Table 3). Unstandardized values of discriminant functions evaluated at the cluster centroids indicate that the first function tends to separate the “severely” (Cluster 3) from the “moderately” (Cluster 1) and “mildly” affected (Cluster 2) groups. Consistent with expected relationships in behavioral performance of asymptomatic mice (Cluster 2), the loading matrix of correlations between the predictors and discriminant functions (Table 3) suggests that the first function measures higher food and water intake, lower step-down latency and higher novel-object contact time. The second function distinguishes “moderately” affected group (Cluster 2), which shows a higher score than the “mild” (Cluster 1) and “severe” (Cluster 3) groups. Consistent with the mean differences (Figure 2), Cluster 2 is distinguished primarily by food intake, step-down latency, activity speed, and sucrose intake.
Table 3.
Discriminant Function Analysis of Cluster Membership Predicted From Behavioral Measures
| Function 1 | Function 2 | |
|---|---|---|
| % Between-Cluster | 68 | 32 |
| Variance | ||
| Accounted For | ||
| Standardized | ||
| Loadings | ||
| Food intake | .62 | .51 |
| Water intake | .54 | .25 |
| Step-down latency | −.48 | .48 |
| Novel object exploration | .42 | .05 |
| Speed | −.06 | .48 |
| Sucrose intake | −.09 | −.28 |
| Floating time | −.11 | −.16 |
| Function at Cluster | ||
| Centroids | ||
| “Mild” (Cluster 2) | 1.53 | −.85 |
| “Moderate” (Cluster 1) | −.18 | 1.35 |
| “Severe” (Cluster 3) | −3.08 | −1.13 |
| Jack-Knifed | 92.4% | |
| Classification | ||
| Accuracy | ||
The disease profile of clusters can be inferred from three types of variables. Cluster 3 is the smallest (about 18% of the sample) and is comprised of animals with the most severe behavioral and brain anomalies. The MRL-lpr mice from this group ate and drank the least, took more time to descend from an elevated platform, and showed impaired exploration of a novel object, excessive immobility in the forced swim test and reduced locomotor speed. More importantly, indices of severe brain atrophy were detected in this group, as evidenced by low brain mass and enlarged ventricles. In comparison to other groups, however, mice from Cluster 3 showed the highest sucrose consumption, lower spleen mass, and serum IL-1beta levels. Taken together, the profile of animals from Cluster 3 (“severely affected”) suggests that factors other than peripheral autoimmunity contribute to the progress of neurodegeneration, which appears as the biological basis for several autoimmunity-associated behavioral deficits. By contrast, the typical markers of autoimmune/inflammatory disease (splenomegaly and high levels of IL-1beta) were profound in mice from Cluster 1 (about 41% of the sample). Except for their blunted responsiveness to sucrose, their performance was relatively better (or comparable in the step-down test) to the performance of animals from Cluster 3. This finding is consistent with our previous studies, in which the detrimental effects of circulating pro-inflammatory cytokines on behavior were observed. As shown in other paradigms (Anisman and Merali 1999), we learned that systemic production of IL-6, IFN-gamma, IL-1, and TNF-alpha impairs the response of experimental animals to palatable stimulation (Sakic et al 2001; Ballok et al 2003; Kwant and Sakic 2004). The third cluster (Cluster 2, about 41% of the sample) displayed higher food and water intake, reduced preference for sucrose, increased floating, lower locomotor speed, but the least profound anxiety-related behaviors (in the step-down and novel object paradigms) and lack of severe brain pathology. Although the spleen was somewhat heavier than in Cluster 3, the mice from this group did not show as high levels of IL-1beta as mice in Cluster 1.
Discussion
In an attempt to understand the causal link between systemic autoimmunity and behavior, our previous studies revealed dissimilar behavioral profiles between MRL-lpr mice and their congenic MRL +/+ controls. However, the possibility that this is due to a subtle difference in their genomes was never completely ruled out. To further minimize the potentially confounding effect of a subtle genetic discrepancy, we presently explored the relationships among autoimmunity, neuropathology, and behavior in a large cohort of inbred MRL-lpr mice. Three neuroimmunologically distinct subpopulations were defined on the basis of their behavioral performance and its relationships to brain morphology and immunologic manifestations. We learned that profound within-group heterogeneity in disease manifestations is largely accounted for by the link between brain pathology and behavoral dysfunction. Although the origin of this relationship needs further study, the present results are consistent with well-documented behavioral heterogeneity in NP-SLE and point to mechanisms others than simple Mendelian inheritance in the etiology of altered behavior. The present findings, however, do not completely exclude the possibility that gene regulation contributes to clinical diversity. Namely, recent epigenetic studies provide evidence that during gametogenesis, development, and aging, heritable modifications in gene expression occur due to potentially reversible changes in DNA methylation and/or chromatin structure (Pikaard 2000; Mechali 2001). These mechanisms have been hypothesized in human inflammatory and psychiatric disorders (Petronis and Petroniene 2000; Petronis et al 2000) and we acknowledge that they may account for diverse subphenotypes in inbred MRL-lpr animals. At the same time, we take the point that they do not negate a principal role of systemic autoimmune processes in affecting brain morphology and function.
The present behavioral heterogeneity is consistent with the evidence that overt and serological markers of lupus-like disease vary significantly among autoimmune animals (Andrews et al 1978). More importantly, they match qualitative assessments of ventricular size by others, who reported hydrocephalus in approximately 46% of cases, moderate ventricular enlargement in 30%, and no ventricular enlargement in 24% of 13-week old MRL-lpr mice (Denenberg et al 1992). In addition, the MRL-lpr mice with hydrocephalus performed poorer in a variety of other behavioral tasks, despite the evidence that plasma levels of circulating immune factors did not differ among the three groups. Although the number of our statistically-identified subgroups is consistent with a subjective classification used in the above study, the interpretation of clustering solutions depends on methods and distance metrics chosen and, as many multivariate techniques, is also somewhat subjective. However, we made additional steps in validation of the chosen solution by examining the prediction of immune and neuropathological variables.
The cluster with the most disturbed behavior (Cluster 3) had profound, quantifiable ventricular enlargement and reduced brain mass, while the brain morphology of the Cluster 2 mice (showing relatively undisturbed behavioral performance) was least affected. As reported before (Sakic et al 2000b), in some mice CD4+ and CD8+ T-cells extensively infiltrated the choroid plexus, third ventricle, interventricular foramena, meninges, and perivascular spaces around the ventricles. Although there was a high degree of consistency of cell numbers among sections of a single brain, some brains had less than 10 cells per section, while others had more than 100. Based on this heterogeneity we expected that the intensity of infiltration will correlate with the severity of autoimmune disease, brain pathology, or behavioral dysfunction. Although there was a trend for CD8+ (cytotoxic) T-cells to differ among the clusters (Table 2), this was not significant, which suggests that other detrimental factors are involved.
The ventricular enlargement in behaviorally impaired Cluster 3 mice may reflect atrophy of the neuronal tissue and may underlie behavioral deficits in the MRL-lpr substrain. The fact that responsiveness to a palatable sucrose solution was lowest in the Cluster 1 mice leaves the possibility that an early upregulation in the synthesis of neuractive cytokines affects the neuroendocrine system and leads to early changes in motivated behavior (Sakic et al 1997a; Turnbull and Rivier 1999; Dunn et al 1999; Anisman and Merali 1999; Dunn 2000). The lack of significant relationships between immune and neuropatholoical measures (data not shown) leads to a question of terminal factors which induce brain pathology in lupus-like conditions. We consider at least three explanations below.
First, it is possible that the presently selected measures of immune activity at the periphery are not instrumental in the etiology of behavioral deficits, and that this relationship would be detected if other factors (e.g. circulating serum autoantibodies reactive to brain antigens) were measured (Khin and Hoffman 1993; Crimando and Hoffman 1995; Zameer and Hoffman 2001). Despite some controversies and inconsistences in the literature (Harris and Pierangeli 1997), correlational evidence suggests that circulating brain-reactive antibodies (BRA) play a role in etiology of NP-SLE and other brain disorders (Greenwood et al 2002; Jennekens and Kater 2002; Tanaka et al 2003; Schott et al 2003; Lang et al 2003). The second possibility is that more productive correlations would be observed if immune factors were measured in the cerebrospinal fluid (CSF). While circulating autoantibodies and immune complexes may be associated with glomerulonephritis, joint pathology, skin lesions, etc., they may not play a role in CNS pathology. Recent report on pro-apoptotic properties of anti-NR2 receptor antibodies from CSF of a NP-SLE patient (DeGiorgio et al 2001), as well as our observation that CSF (but not serum) from diseased MRL-lpr mice is toxic to cultured neurons is consistent with this notion (Maric et al 2001). If so, antibodies cross-reactive with CNS antigens are likely produced intrathecally and are not present in the peripheral circulation, at least not in quantities sufficient to induce neuronal death. In the light of evidence that B-cells infiltrate brains of MRL-lpr mice (Vogelweid et al 1991; Farrell et al 1997; Zameer and Hoffman 2004) and that the IgG fraction of their CSF is toxic to proliferative brain cells (unpublished work), it is likely that the analysis of BRA in CSF would reveal an important relationship to brain pathology. The third possibility is that circulating cytokines trigger an anti-inflammatory response in the endocrine system by upregulating the synthesis of corticosterone (CORT), which is one of the most potent immunosuppressive mediators in the mammal system (McEwen et al 1997). Sustained binding of CORT to its central receptors renders neurons vulnerable to various metabolic insults (Uno et al 1994; Sapolsky 2000). This neurotoxic mechanism is based on the evidence that onset of autoimmune disease in MRL-lpr mice is accompanied by alterations in the endocrine system, including increased plasma levels of CORT and melatonin (Hu et al 1993; Lechner et al 1996; Lechner et al 2000), imbalanced expression of corticotropin releasing factor (CRF) and arginine vasopressin (AVP) mRNA in the paraventricular nucleus and amygdala (Shanks et al 1997; Shanks et al 1999; Sakic et al 1999), and reduced serum levels of testosterone (Sakic et al 1998a). Both castration and estrogen treatment exacerbate the disease in MRL-lpr mice, while testosterone treatment improves it (Melez et al 1978; Shear et al 1983; Carlsten et al 1989; Carlsten et al 1990). In our recent study (unpublished work) we learned that exogenous CORT exacerbates brain pathology in MRL-lpr mice while exerting beneficial effects on inflammation and autoimmunity. Therefore, it is possible that brain atrophy in autoimmune mice is primarily associated with cytokine-induced activation of the pituitary-adrenal axis, which is also an integral part of an adaptive, anti-inflammatory mechanism (McEwen et al 1997). However, additional damage of periventricular tissue may occur at the later stage of the disease, when the blood-brain barrier becomes more permeable to activated B-cells, which become a source of BRA neurotoxic to periventricular tissues.
In addition to controlling for genetic variability, we controlled for environmental variability by housing and testing the cohorts in nearly identical conditions. However, other influences on behavior (such as health status and maternal behavior) were beyond our control because the offsprings were reared in The Jackson Laboratories. The MRL strain is characterized by hyper-maturity and they reach reproductive age at 7–8 weeks. Therefore, it is possible that some animals were born by younger females in which the disease is less active, while others were born by mothers in which disease symptoms are florid. Experiments in which healthy embryos were transferred to pregnant autoimmune mothers revealed that mice born by autoimmune mothers show impaired learning/memory performance in adulthood (Denenberg et al 1991; Denenberg et al 1996). This implies that the in utero environment and maternal behavior have significant effects on brain development. Therefore, it seems reasonable to hypothesize that behavioral diversity among offspring is associated with dissimilar disease severity in pregnant dams.
Although we detected the subpopulations of MRL-lpr mice, it remains to be elucidated whether they represent distinct disease entities or alternatively, different stages of the same disease process influenced by accelerating or inhibitory factors. Direct examination of immune parameters within the CNS, assessment of endocrine measures, and consideration of maternal and early life influences may all provide insight into the source of the heterogeneity in this model. It is our expectation that further understanding of the pathogenesis of AABS will facilitate understanding not only of NP-SLE, but also of brain disorders in which autoimmune phenomena are observed (Krause et al 2002; Gaughran 2002; Lang et al 2003).
Acknowledgments
This work was supported by funds from the National Institute of Health (1R21 AR49163-01) and Canadian Institutes of Mental Health (MOP 38065) to BS, who is an Intermediate Fellow of the Ontario Mental Health Foundation (OMHF) and recipient of the Father Sean O’Sullivan Research Centre career development award. There are no financial or other conflicts of interest related to this work.
We thank Drs. Judah Denburg and Henry Szechtman for critical feedback in designing the experiment.
References
- Abbott NJ, Mendonca LL, Dolman DE. The blood-brain barrier in systemic lupus erythematosus. Lupus. 2003;12:908–915. doi: 10.1191/0961203303lu501oa. [DOI] [PubMed] [Google Scholar]
- Alexander EL, Murphy ED, Roths JB, Alexander GE. Congenic autoimmune murine models of central nervous system disease in connective tissue disorders. Ann Neurol. 1983;14:242–248. doi: 10.1002/ana.410140211. [DOI] [PubMed] [Google Scholar]
- Andrews BS, Eisenberg RA, Theofilopoulos AN, Izui S, Wilson CB, McConahey PJ, et al. Spontaneous murine lupus-like syndromes. Clinical and immunopathological manifestations in several strains. J Exp Med. 1978;148:1198–1215. doi: 10.1084/jem.148.5.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisman H, Merali Z. Anhedonic and anxiogenic effects of cytokine exposure. Adv Exp Med Biol. 1999;461:199–233. doi: 10.1007/978-0-585-37970-8_12. [DOI] [PubMed] [Google Scholar]
- Ballok DA, Szechtman H, Sakic B. Taste responsiveness and diet preference in autoimmune MRL mice. Behav Brain Res. 2003;140:119–130. doi: 10.1016/s0166-4328(02)00276-0. [DOI] [PubMed] [Google Scholar]
- Boumpas DT, Yamada H, Patronas NJ, Scott D, Klippel JH, Balow JE. Pulse cyclophosphamide for severe neuropsychiatric lupus. Q J Med. 1991;81:975–984. doi: 10.1093/qjmed/81.3.975. [DOI] [PubMed] [Google Scholar]
- Brooks WM, Sabet A, Sibbitt WL, Barker PB, van Zijl PC, Duyn JH, et al. Neurochemistry of brain lesions determined by spectroscopic imaging in systemic lupus erythematosus. J Rheumatol. 1997;24:2323–2329. [PubMed] [Google Scholar]
- Bruyn GAW. Controversies in lupus: Nervous system involvement. Ann Rheum Dis. 1995;54:159–167. doi: 10.1136/ard.54.3.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsten H, Holmdahl R, Tarkowski A, Nilsson LA. Oestradiol- and testosterone-mediated effects on the immune system in normal and autoimmune mice are genetically linked and inherited as dominant traits. Immunology. 1989;68:209–214. [PMC free article] [PubMed] [Google Scholar]
- Carlsten H, Tarkowski A, Holmdahl R, Nilsson LA. Oestrogen is a potent disease accelerator in SLE-prone MRL lpr/lpr mice. Clin Exp Immunol. 1990;80:467–473. doi: 10.1111/j.1365-2249.1990.tb03311.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crimando J, Hoffman SA. Characterization of murine brain-reactive monoclonal IgG autoantibodies. Brain Behav Immun. 1995;9:165–181. doi: 10.1006/brbi.1995.1016. [DOI] [PubMed] [Google Scholar]
- DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med. 2001;7:1189–1193. doi: 10.1038/nm1101-1189. [DOI] [PubMed] [Google Scholar]
- Denburg JA, Behmann SA. Lymphocyte and neuronal antigens in neuropsychiatric lupus: presence of an elutable, immunoprecipitable lymphocyte/neuronal 52 kd reactivity. Ann Rheum Dis. 1994;53:304–308. doi: 10.1136/ard.53.5.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denburg JA, Carbotte RM, Denburg SD. Central nervous system lupus. Rheum Rev. 1993;2:123–132. [Google Scholar]
- Denenberg VH, Mobraaten LE, Sherman GF, Morrison L, Schrott LM, Waters NS, et al. Effects of the autoimmune uterine/maternal environment upon cortical ectopias, behavior and autoimmunity. Brain Res. 1991;563:114–122. doi: 10.1016/0006-8993(91)91522-3. [DOI] [PubMed] [Google Scholar]
- Denenberg VH, Sherman G, Schrott LM, Waters NS, Boehm GW, Galaburda AM, et al. Effects of embryo transfer and cortical ectopias upon the behavior of BXSB-Yaa and BXSB-Yaa plus mice. Dev Brain Res. 1996;93:100–108. doi: 10.1016/0165-3806(96)00010-7. [DOI] [PubMed] [Google Scholar]
- Denenberg VH, Sherman GF, Rosen GD, Morrison L, Behan PO, Galaburda AM. A behavior profile of the MRL/Mp lpr/lpr mouse and its association with hydrocephalus. Brain Behav Immun. 1992;6:40–49. doi: 10.1016/0889-1591(92)90058-v. [DOI] [PubMed] [Google Scholar]
- Dunn AJ. Cytokine activation of the HPA axis. Ann N Y Acad Sci. 2000;917:608–617. doi: 10.1111/j.1749-6632.2000.tb05426.x. [DOI] [PubMed] [Google Scholar]
- Dunn AJ, Wang J, Ando T. Effects of cytokines on cerebral neurotransmission. Comparison with the effects of stress. Adv Exp Med Biol. 1999;461:117–127. doi: 10.1007/978-0-585-37970-8_8. [DOI] [PubMed] [Google Scholar]
- Duprez T, Nzeusseu A, Peeters A, Houssiau FA. Selective involvement of the choroid plexus on cerebral magnetic resonance images: a new radiological sign in patients with systemic lupus erythematosus with neurological symptoms. J Rheumatol. 2001;28:387–391. [PubMed] [Google Scholar]
- Egan O, Royce JR. Litter size and emotionality in two strains of mice. J Comp Physiol Psychol. 1973;82:55–59. doi: 10.1037/h0033809. [DOI] [PubMed] [Google Scholar]
- Farrell M, Sakic B, Szechtman H, Denburg JA. Effect of cyclophosphamide on leucocytic infiltration in the brain of MRL/lpr mice. Lupus. 1997;6:268–274. doi: 10.1177/096120339700600310. [DOI] [PubMed] [Google Scholar]
- Gaughran F. Immunity and schizophrenia: autoimmunity, cytokines, and immune responses. Int Rev Neurobiol. 2002;52:275–302. doi: 10.1016/s0074-7742(02)52013-4. [DOI] [PubMed] [Google Scholar]
- Greenwood DL, Gitlits VM, Alderuccio F, Sentry JW, Toh BH. Autoantibodies in neuropsychiatric lupus. Autoimmunity. 2002;35:79–86. doi: 10.1080/08916930290016547. [DOI] [PubMed] [Google Scholar]
- Griffey RH, Brown MS, Bankhurst AD, Sibbitt RR, Sibbitt WL. Depletion of high-energy phosphates in the central nervous system of patients with systemic lupus erythematosus, as determined by phosphorus-31 nuclear magnetic resonance spectroscopy. Arthritis Rheum. 1990;33:827–833. doi: 10.1002/art.1780330609. [DOI] [PubMed] [Google Scholar]
- Hanly JG, Hong C, Smith S, Fisk JD. A prospective analysis of cognitive function and anticardiolipin antibodies in systemic lupus erythematosus. Arthritis Rheum. 1999;42:728–734. doi: 10.1002/1529-0131(199904)42:4<728::AID-ANR16>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Hanson VG, Horowitz M, Rosenbluth D, Spiera H, Puszkin S. Systemic lupus erythematosus patients with central nervous system involvement show autoantibodies to a 50-kD neuronal membrane protein. J Exp Med. 1992;176:565–573. doi: 10.1084/jem.176.2.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris EN, Pierangeli S. Antiphospholipid antibodies and cerebral lupus. Ann N Y Acad Sci. 1997;823:270–278. doi: 10.1111/j.1749-6632.1997.tb48400.x. [DOI] [PubMed] [Google Scholar]
- Hess DC, Taormina M, Thompson J, Sethi KD, Diamond B, Rao R, et al. Cognitive and neurologic deficits in the MRL/lpr mouse: a clinicopathologic study. J Rheumatol. 1993;20:610–617. [PubMed] [Google Scholar]
- Hildenbrand GLG. Systemic lupus erythematosus: one disease, a thousand faces, a potent management. Heal J. 1992;8:1–2. [Google Scholar]
- Hu Y, Dietrich H, Herold M, Heinrich PC, Wick G. Disturbed immunoendocrine communication via the hypothalamo-pituitary-adrenal axis in autoimmune disease. Int Arch Allergy Immunol. 1993;102:232–241. doi: 10.1159/000236531. [DOI] [PubMed] [Google Scholar]
- Jacobsen S, Petersen J, Ullman S, Junker P, Voss A, Rasmussen JM, et al. A multicentre study of 513 Danish patients with systemic lupus erythematosus. I. Disease manifestations and analyses of clinical subsets. Clin Rheumatol. 1998;17:468–477. doi: 10.1007/BF01451282. [DOI] [PubMed] [Google Scholar]
- Jara LJ, Irigoyen L, Ortiz MJ, Zazueta B, Bravo G, Espinoza LR. Prolactin and interleukin-6 in neuropsychiatric lupus erythematosus. Clin Rheumatol. 1998;17:110–114. doi: 10.1007/BF01452255. [DOI] [PubMed] [Google Scholar]
- Jennekens FG, Kater L. The central nervous system in systemic lupus erythematosus. Part 2. Pathogenetic mechanisms of clinical syndromes: a literature investigation. Rheumatology (Oxford) 2002;41:619–630. doi: 10.1093/rheumatology/41.6.619. [DOI] [PubMed] [Google Scholar]
- Khin NA, Hoffman SA. Brain reactive monoclonal auto-antibodies: production and characterization. J Neuroimmunol. 1993;44:137–148. doi: 10.1016/0165-5728(93)90035-w. [DOI] [PubMed] [Google Scholar]
- Krause I, He XS, Gershwin ME, Shoenfeld Y. Brief report: immune factors in autism: a critical review. J Autism Dev Disord. 2002;32:337–345. doi: 10.1023/a:1016391121003. [DOI] [PubMed] [Google Scholar]
- Kwant A, Sakic B. Behavioral effects of infection with interferon-gamma adenovector. Behav Brain Res. 2004;151:73–82. doi: 10.1016/j.bbr.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Lang B, Dale RC, Vincent A. New autoantibody mediated disorders of the central nervous system. Curr Opin Neurol. 2003;16:351–357. doi: 10.1097/01.wco.0000073937.19076.d5. [DOI] [PubMed] [Google Scholar]
- Lechner O, Dietrich H, Oliveira dos SA, Wiegers GJ, Schwarz S, Harbutz M, et al. Altered circadian rhythms of the stress hormone and melatonin response in lupus-prone MRL/MP-fas(Ipr) mice. J Autoimmun. 2000;14:325–333. doi: 10.1006/jaut.2000.0375. [DOI] [PubMed] [Google Scholar]
- Lechner O, Hu Y, Jafarian-Tehrani M, Dietrich H, Schwarz S, Herold M, et al. Disturbed immunoendocrine communication via the hypothalamo-pituitary-adrenal axis in murine lupus. Brain Behav Immun. 1996;10:337–350. doi: 10.1006/brbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Maric D, Millward JM, Ballok DA, Szechtman H, Barker JL, Denburg JA, et al. Neurotoxic properties of cerebrospinal fluid from behaviorally impaired autoimmune mice. Brain Res. 2001;920:183–193. doi: 10.1016/s0006-8993(01)03060-8. [DOI] [PubMed] [Google Scholar]
- Martinez-Cordero E, Rivera Garcia BE, Aguilar Leon DE. Anticardiolipin antibodies in serum and cerebrospinal fluid from patients with systemic lupus erythematosus. J Investig Allergol Clin Immunol. 1997;7:596–601. [PubMed] [Google Scholar]
- McEwen BS, Biron CA, Brunson KW, Bulloch K, Chambers WH, Dhabhar FS, et al. The role of adrenocorticoids as modulators of immune function in health and disease: neural, endocrine and immune interactions. Brain Res Rev. 1997;23:79–133. doi: 10.1016/s0165-0173(96)00012-4. [DOI] [PubMed] [Google Scholar]
- Mechali M. DNA replication origins: from sequence specificity to epigenetics. Nat Rev Genet. 2001;2:640–645. doi: 10.1038/35084598. [DOI] [PubMed] [Google Scholar]
- Melez KA, Reeves JP, Steinberg AD. Modification of murine lupus by sex hormones. Ann Immunol (Paris) 1978;129:707–714. [PubMed] [Google Scholar]
- Monleon S, D’Aquila P, Parra A, Simon VM, Brain PF, Willner P. Attenuation of sucrose consumption in mice by chronic mild stress and its restoration by imipramine. Psychopharmacology (Berl ) 1995;117:453–457. doi: 10.1007/BF02246218. [DOI] [PubMed] [Google Scholar]
- Muscat R, Willner P. Suppression of sucrose drinking by chronic mild unpredictable stress: a methodological analysis. Neurosci Biobehav Rev. 1992;16:507–517. doi: 10.1016/s0149-7634(05)80192-7. [DOI] [PubMed] [Google Scholar]
- Perry S, Miller F. Psychiatric aspects of systemic lupus erythematosus. In: Lahita RG, editor. Systemic lupus erythematosus. 2 ed. New York: Churchill Livingstone; 1992. pp. 845–863. [Google Scholar]
- Petronis A, Gottesman II, Crow TJ, DeLisi LE, Klar AJ, Macciardi F, et al. Psychiatric epigenetics: a new focus for the new century. Mol Psychiatry. 2000;5:342–346. doi: 10.1038/sj.mp.4000750. [DOI] [PubMed] [Google Scholar]
- Petronis A, Petroniene R. Epigenetics of inflammatory bowel disease. Gut. 2000;47:302–306. doi: 10.1136/gut.47.2.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikaard CS. The epigenetics of nucleolar dominance. Trends Genet. 2000;16:495–500. doi: 10.1016/s0168-9525(00)02113-2. [DOI] [PubMed] [Google Scholar]
- Porsolt RD, Le Pichon M, Jalfre M. Depression: a new animal model sensitive to antidepressant treatments. Nature. 1977;266:730–732. doi: 10.1038/266730a0. [DOI] [PubMed] [Google Scholar]
- Ramos PC, Mendez MJ, Ames PRJ, Khamashta MA, Hughes GRV. Pulse cyclophosphamide in the treatment of neuropsychiatric systemic lupus erythematosus. Clin Exp Rheumatol. 1996;14:295–299. [PubMed] [Google Scholar]
- Romesburg HC. Cluster analysis for researchers. Belmont, CA: Lifetime Learning Publications; 2003. [Google Scholar]
- Sakic B, Denburg JA, Denburg SD, Szechtman H. Blunted sensitivity to sucrose in autoimmune MRL-lpr mice: a curve-shift study. Brain Res Bull. 1996;41:305–311. doi: 10.1016/s0361-9230(96)00190-6. [DOI] [PubMed] [Google Scholar]
- Sakic B, Gurunlian L, Denburg SD. Reduced aggressiveness and low testosterone levels in autoimmune MRL-lpr males. Physiol Behav. 1998a;63:305–309. doi: 10.1016/s0031-9384(97)00422-8. [DOI] [PubMed] [Google Scholar]
- Sakic B, Kolb B, Whishaw IQ, Gorny G, Szechtman H, Denburg JA. Immunosuppression prevents neuronal atrophy in lupus-prone mice: evidence for brain damage induced by autoimmune disease? J Neuroimmunol. 2000a;111:93–101. doi: 10.1016/s0165-5728(00)00364-7. [DOI] [PubMed] [Google Scholar]
- Sakic B, Laflamme N, Crnic LS, Szechtman H, Denburg JA, Rivest S. Reduced corticotropin-releasing factor and enhanced vasopressin gene expression in brains of mice with autoimmunity-induced behavioral dysfunction. J Neuroimmunol. 1999;96:80–91. doi: 10.1016/s0165-5728(99)00021-1. [DOI] [PubMed] [Google Scholar]
- Sakic B, Maric I, Koeberle PD, Millward JM, Szechtman H, Maric D, et al. Increased TUNEL-staining in brains of autoimmune Fas-deficient mice. J Neuroimmunol. 2000b;104:147–154. doi: 10.1016/s0165-5728(99)00277-5. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Braciak TA, Richards CD, Gauldie J, Denburg JA. Reduced preference for sucrose in autoimmune mice: a possible role of interleukin-6. Brain Res Bull. 1997a;44:155–165. doi: 10.1016/s0361-9230(97)00107-x. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Denburg JA. Neurobehavioral alteration in autoimmune mice. Neurosci Biobehav Rev. 1997b;21:327–340. doi: 10.1016/s0149-7634(96)00018-8. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Denburg JA, Gorny G, Kolb B, Whishaw IQ. Progressive atrophy of pyramidal neuron dendrites in autoimmune MRL-lpr mice. J Neuroimmunol. 1998b;87:162–170. doi: 10.1016/s0165-5728(98)00085-x. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Denburg SD, Denburg JA. Immunosuppressive treatment prevents behavioral deficit in autoimmune MRL-lpr mice. Physiol Behav. 1995;58:797–802. doi: 10.1016/0031-9384(95)00135-6. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Gauldie J, Denburg JA. Behavioral effects of infection with IL-6 adenovector. Brain Behav Immun. 2001;15:25–42. doi: 10.1006/brbi.1999.0576. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Keffer M, Talangbayan H, Stead R, Denburg JA. A behavioral profile of autoimmune lupus-prone MRL mice. Brain Behav Immun. 1992;6:265–285. doi: 10.1016/0889-1591(92)90048-s. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Talangbayan H, Denburg SD, Carbotte RM, Denburg JA. Behaviour and immune status of MRL mice in the postweaning period. Brain Behav Immun. 1994a;8:1–13. doi: 10.1006/brbi.1994.1001. [DOI] [PubMed] [Google Scholar]
- Sakic B, Szechtman H, Talangbayan H, Denburg SD, Carbotte RM, Denburg JA. Disturbed emotionality in autoimmune MRL-lpr mice. Physiol Behav. 1994b;56:609–617. doi: 10.1016/0031-9384(94)90309-3. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch Gen Psychiatry. 2000;57:925–935. doi: 10.1001/archpsyc.57.10.925. [DOI] [PubMed] [Google Scholar]
- Schott K, Schaefer JE, Richartz E, Batra A, Eusterschulte B, Klein R, et al. Autoantibodies to serotonin in serum of patients with psychiatric disorders. Psychiatry Res. 2003;121:51–57. doi: 10.1016/s0165-1781(03)00137-9. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Rochas M, Weller B, Sheinkman A, Tal I, Golan D, et al. High association of anticardiolipin antibodies with psychosis. J Clin Psychiatry. 1998;59:20–23. doi: 10.4088/jcp.v59n0105. [DOI] [PubMed] [Google Scholar]
- Senecal JL. Lupus: The Disease with 1,000 Faces, 2nd ed. Calgary: Lupus Canada; 1991. [Google Scholar]
- Shanks N, Moore PM, Perks P, Lightman SL. Endocrine correlates of murine systemic lupus erythematosus in the MRL lpr/lpr model. Ann N Y Acad Sci. 1997;823:252–255. doi: 10.1111/j.1749-6632.1997.tb48397.x. [DOI] [PubMed] [Google Scholar]
- Shanks N, Moore PM, Perks P, Lightman SL. Alterations in hypothalamic-pituitary-adrenal function correlated with the onset of murine SLE in MRL +/+ and lpr/lpr mice. Brain Behav Immun. 1999;13:348–360. doi: 10.1006/brbi.1998.0535. [DOI] [PubMed] [Google Scholar]
- Shear HL, Wofsy D, Talal N. Effects of castration and sex hormones on immune clearance and autoimmune disease in MRL/Mp-lpr/lpr and MRL/Mp-+/+ mice. Clin Immunol Immunopathol. 1983;26:361–369. doi: 10.1016/0090-1229(83)90120-4. [DOI] [PubMed] [Google Scholar]
- Sherman GF, Galaburda AM, Behan PO, Rosen GD. Neuroanatomical anomalies in autoimmune mice. Acta Neuropathol (Berl ) 1987;74:239–242. doi: 10.1007/BF00688187. [DOI] [PubMed] [Google Scholar]
- Sibbitt WL, Haseler LJ, Griffey RH, Hart BL, Sibbitt RR, Matwiyoff NA. Analysis of cerebral structural changes in systemic lupus erythematosus by proton MR spectroscopy. AJNR Am J Neuroradiol. 1994;15:923–928. [PMC free article] [PubMed] [Google Scholar]
- Sibbitt WL, Sibbitt RR. Magnetic resonance spectroscopy and positron emission tomography scanning in neuropsychiatric systemic lupus erythematosus. Rheum Dis Clin North Am. 1993;19:851–868. [PubMed] [Google Scholar]
- Sibbitt WL, Sibbitt RR, Griffey RH, Eckel C, Bankhurst AD. Magnetic resonance and computed tomographic imaging in the evaluation of acute neuropsychiatric disease in systemic lupus erythematosus. Ann Rheum Dis. 1989;48:1014–1022. doi: 10.1136/ard.48.12.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Matsunaga H, Kimura M, Tatsumi K, Hidaka Y, Takano T, et al. Autoantibodies against four kinds of neurotransmitter receptors in psychiatric disorders. J Neuroimmunol. 2003;141:155–164. doi: 10.1016/s0165-5728(03)00252-2. [DOI] [PubMed] [Google Scholar]
- Tapanes FJ, Vasquez M, Ramirez R, Matheus C, Rodriguez MA, Bianco N. Cluster analysis of antinuclear autoantibodies in the prognosis of SLE nephropathy: are anti-extractable nuclear antibodies protective? Lupus. 2000;9:437–444. doi: 10.1191/096120300678828604. [DOI] [PubMed] [Google Scholar]
- Theofilopoulos AN. Murine models of lupus. In: Lahita RG, editor. Systemic lupus erythematosus. 2 ed. New York: Churchill Livingstone; 1992. pp. 121–194. [Google Scholar]
- Tincani A, Brey R, Balestrieri G, Vitali C, Doria A, Galeazzi M, et al. International survey on the management of patients with SLE. 2. The results of a questionnaire regarding neuropsychiatric manifestations. Clin Exp Rheumatol. 1996;14:S23–S29. [PubMed] [Google Scholar]
- Toubi E, Khamashta MA, Panarra A, Hughes GRV. Association of antiphospholipid antibodies with central nervous system disease in systemic lupus erythematosus. Am J Med. 1995;99:397–401. doi: 10.1016/s0002-9343(99)80188-0. [DOI] [PubMed] [Google Scholar]
- Tsai CY, Wu TH, Tsai ST, Chen KH, Thajeb P, Lin WM, et al. Cerebrospinal fluid interleukin-6, prostaglandin E2 and autoantibodies in patients with neuropsychiatric systemic lupus erythematosus and central nervous system infections. Scand J Rheumatol. 1994;23:57–63. doi: 10.3109/03009749409103028. [DOI] [PubMed] [Google Scholar]
- Tsao BP. The genetics of human systemic lupus erythematosus. Trends Immunol. 2003;24:595–602. doi: 10.1016/j.it.2003.09.006. [DOI] [PubMed] [Google Scholar]
- Turnbull AV, Rivier CL. Regulation of the hypothalamic-pituitary-adrenal axis by cytokines: actions and mechanisms of action. Physiol Rev. 1999;79:1–71. doi: 10.1152/physrev.1999.79.1.1. [DOI] [PubMed] [Google Scholar]
- Uno H, Eisele S, Sakai A, Shelton S, Baker E, DeJesus O, et al. Neurotoxicity of glucocorticoids in the primate brain. Horm Behav. 1994;28:336–348. doi: 10.1006/hbeh.1994.1030. [DOI] [PubMed] [Google Scholar]
- van Dam AP, Wekking EM, Callewaert JAC, Schipperijn AJM, Oomen HAPC, Dejong J, et al. Psychiatric symptoms before systemic lupus erythematosus is diagnosed. Rheumatol Int. 1994;14:57–62. doi: 10.1007/BF00300248. [DOI] [PubMed] [Google Scholar]
- Vogelgesang SA, Heyes MP, West SG, Salazar AM, Sfikakis PP, Lipnick RN, et al. Quinolinic acid in patients with systemic lupus erythematosus and neuropsychiatric manifestations. J Rheumatol. 1996;23:850–855. [PubMed] [Google Scholar]
- Vogelweid CM, Johnson GC, Besch-Williford CL, Basler J, Walker SE. Inflammatory central nervous system disease in lupus-prone MRL/lpr mice: comparative histologic and immunohistochemical findings. J Neuroimmunol. 1991;35:89–99. doi: 10.1016/0165-5728(91)90164-3. [DOI] [PubMed] [Google Scholar]
- Wekking EM. Psychiatric symptoms in systemic lupus erythematosus - an update. Psychosom Med. 1993;55:219–228. doi: 10.1097/00006842-199303000-00011. [DOI] [PubMed] [Google Scholar]
- Zameer A, Hoffman SA. Immunoglobulin binding to brain in autoimmune mice. J Neuroimmunol. 2001;120:10–18. doi: 10.1016/s0165-5728(01)00412-x. [DOI] [PubMed] [Google Scholar]
- Zameer A, Hoffman SA. B and T cells in the brains of autoimmune mice. J Neuroimmunol. 2004;146:133–139. doi: 10.1016/j.jneuroim.2003.10.052. [DOI] [PubMed] [Google Scholar]