

Summary
Hermansky–Pudlak syndrome (HPS) is an autosomal recessive disorder resulting from mutations in a family of genes required for efficient transport of lysosomal-related proteins from the trans-Golgi network to a target organelle. To date, there are several genetically distinct forms of HPS. Many forms of HPS exhibit aberrant trafficking of melanosome-targeted proteins resulting in incomplete melanosome biogenesis responsible for oculocutaneous albinism observed in patients. In HPS-1, melanosome-targeted proteins are localized to characteristic membranous complexes, which have morphologic similarities to macroautophagosomes. In this report, we evaluated the hypothesis that HPS-1-specific membranous complexes comprise a component of the lysosomal compartment of melanocytes. Using indirect immunofluorescence, an increase in co-localization of misrouted tyrosinase with cathepsin-L, a lysosomal cysteine protease, occurred in HPS-1 melanocytes. In addition, ribophorin II, an integral endoplasmic reticulum protein that is also a component of macroautophagosomes, and LC3, a specific marker of macrophagosomes, demonstrated localization to membranous complexes in HPS-1 melanocytes. At the electron microscopic level, the membranous complexes exhibited acid phosphatase activity and localization of exogenously supplied horseradish peroxidase (HRP)-conjugated gold particles, indicating incorporation of lysosomal and endosomal components to membranous complexes, respectively. These results confirm that membranous complexes of HPS-1 melanocytes are macroautophagosomal representatives of the lysosomal compartment.
Keywords: macroautophagosomes, Hermansky—Pudlak syndrome, melanocytes, trafficking
Introduction
Little is currently known of the genes responsible for efficient transport of proteins to lysosomal-related organelles, which include the melanosome and platelet dense body (Huizing et al., 2002). However, a collection of genes responsible for Hermansky–Pudlak syndrome (HPS) has been found to be of importance in the formation and trafficking of vesicles from the trans-Golgi network to lysosomal-related organelles. HPS patients present with congenital albinism, a bleeding diathesis, and a pulmonary disorder resulting from immature pigment granules, absent platelet dense bodies, and dysfunctional lamellar bodies, respectively (Boissy et al., 1998). Currently, seven human genetic forms of HPS (i.e. HPS-1 to -7) have been identified and result from mutations in one of six HPS genes (HPS1, HPS3, HPS4, HPS5, HPS6, HPS7) or in an adaptor complex gene (ADTβ3A) (Huizing et al., 2002; Li et al., 2003; Zhang et al., 2003). Analysis of the pathophysiology of melanocytes from patients with various forms of HPS has provided data culminating in a better understanding of the trafficking of tyrosinase-related proteins to the melanosome (Richmond et al., 2005).
Melanocytes from patients with HPS-1 are distinct in that they exhibit relatively large membranous complexes frequently enclosed by a double membrane (Boissy et al., 1998; Natsuga et al., 2005). In HPS-1 melanocytes, tyrosinase, tyrosinase related protein-1 (Tyrp1), and dopa-chrome tautomerase (DCT/Tyrp2) are localized to these membranous complexes, rather than being efficiently targeted to the melanosome (Boissy et al., 1998; Richmond et al., 2005). The origin and function of the membranous complexes are currently unknown. Upon electron microscopic examination, however, these HPS-1-specific membranous complexes morphologically resemble macroautophagic vacuoles (i.e. macroautophagosomes).
Macroautophagy is a dynamic process involving the rearrangement of subcellular membranes to sequester cytoplasm and organelles for delivery to the lysosomal compartment, where delineated material is degraded and recycled (Klionsky and Emr, 2000; Yoshimori, 2004). Autophagocytic turnover of specific cellular components or autophagy-mediated cell death occurs throughout various cellular events, which include molecular recycling, tissue development, and a variety of diseases (Gutierrez et al., 2004; Kisen et al., 1993; Lum et al., 2005; Stromhaug and Klionsky, 2001; Tanaka et al., 2000). The macroautophagosome is a multilamellar organelle believed to be derived from the sequestration of cytoplasmic material by elements of the endoplasmic reticulum (Dunn, 1990a) to which hydrolytic enzymes are targeted via pre-existing lysosomes (Dunn, 1990b). In this study, indirect immunofluorescence and electron microscopy were utilized to assess whether the membranous complexes characteristic of HPS-1 melanocytes represent macroautophagosomal entities of the lysosomal compartment.
Results
Characteristics of HPS-1-specific membranous complexes
Melanocytes from patients with HPS-1 exhibit membranous complexes (Figure 1) primarily localized to the perinuclear area and occasionally within dendrites. Many membranous complexes have profiles similar to macro-autophagosomes characterized as heterogeneous vesicles containing degradative cytoplasmic material and organelles delineated by one to several membranes. Occasionally, profiles of rough endoplasmic reticulum were juxtapositioned peripherally around the membranous complexes. DOPA incubation demonstrated that tyrosinase-containing melanosomes and cisternae were components of the membranous complexes (Figure 1).
Figure 1.

Ultrastructure of melanocytes cultured from patients with HPS-1 treated (A) without or (B–D) with DOPA histochemistry. (A) Low magnification image of perinuclear area containing a Golgi apparatus (G), demonstrating several membranous complexes, i.e. macroautophagosomes (arrows), heterogeneous in size and content and stage 2 melanosomes (arrowheads). (B) Developing membranous complex demonstrating a circular cisterna containing DOPA reaction product (arrow) and profiles of endoplasmic reticulum peripherally juxtapositioned (arrowheads) to the developing macroautophagosome. (C and D) Mature macroautophagosomes (arrows) containing membrane cisterna with DOPA reaction product (arrowhead) and unpigmented (black asterisk) and pigmented (white asterisk) melanosomes. Bars: A, 0.6 μm; B and D, 0.1 μm; C, 0.3 μm.
Co-localization of tyrosinase related proteins with cathepsin-L, ribophorin II, and LC3
Melanocytes from normal and HPS-1 patients were evaluated by indirect immunocytochemistry for expression pattern and co-localization of tyrosinase, Tyrp1, cathepsin-L, ribophorin II, and LC3. LC3 is currently the only highly specific marker of the autophagosome (Yoshimori, 2004). The expression pattern of tyrosinase in normal melanocytes was granular with only a slight increase in intensity of the perinuclear region as compared with the periphery (Figure 2). In contrast, the expression pattern of tyrosinase in HPS-1 melanocytes was largely confined to the perinuclear region of HPS-1 melanocytes, showing a dramatic decrease in the peripheral intensity (Figure 2). The expression pattern of cathepsin-L was rather diffuse and very similar in both normal and HPS-1 melanocyte lines, however, the perinuclear intensity of cathepsin-L appeared increased in HPS-1 melanocytes (Figure 2). Upon visual examination of the perinuclear tyrosinase or Tyrp1 staining of each cell line, an increase in co-localization with cathepsin-L can be observed in HPS-1 melanocytes as compared with normal melanocytes, particularly within large granular aggregates unique to the HPS-1 melanocyte (inset to Figure 2). Quantatively, only 8% of tyrosinase co-localized with cathepsin-L (SD = 2, n = 44) in normal melanocytes whereas in HPS-1 melanocytes 32% of tyrosinase co-localized with cathepsin-L (SD = 9, n = 44), representing a 2.6-fold increase as compared with control (Figure 3).
Figure 2.
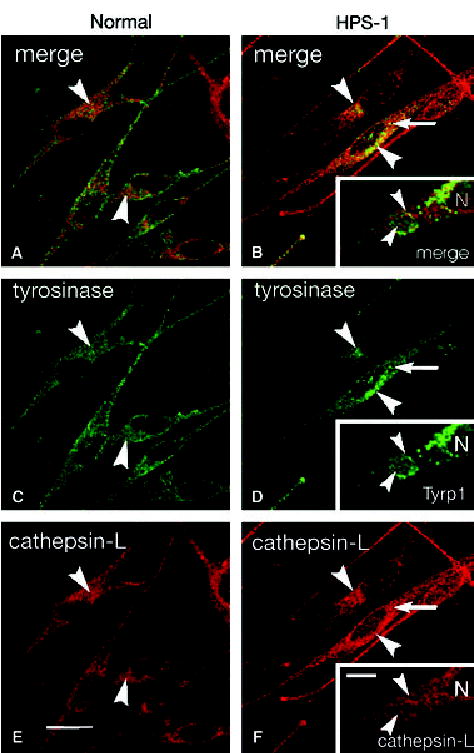
Immunofluorescence of (A, C and E) normal and (B, D and F) HPS-1 melanocytes immunolabeled for tyrosinase (C and D) or Tyrp1 (inset to D) (green) and cathepsin-L (red). (Normal and HPS-1 images were captured from the confocal microscope using identical settings for each primary antibody used.) Minimal co-localization of tyrosinase and cathepsin-L occurred in the perinuclear area (arrowheads) of normal melanocytes where significant co-localization was apparent in HPS-1 melanocytes in the perinuclear area (arrowheads) and in large clusters (arrow). Higher magnification of large perinuclear cluster in HPS-1 melanocytes (insets) demonstrated heterogeneity in localization of Tyrp1 and cathepsin-L throughout the organelle with focal areas of colocalization (arrowheads) within the membranous complex. N, nucleus; bars, 10.0 μm.
Figure 3.

Percentage of tyrosinase that co-localized with ER and lysosomal markers in HPS-1 melanocytes. Both ribophorin II and cathepsin-L exhibited an increase in colocalization with tyrosinase in HPS-1 melanocytes compared with control melanocytes. The increase in co-localization of tyrosinase with ribophorin II indicates endoplasmic reticulum sequestering of tyrosinase-positive vesicles for degradation. The increase in co-localization of ribophorins II and tyrosinase in HPS-1 melanocytes was likely not dramatic because of the transient presence of ribophorins II only along the limiting membrane of the membranous complex, while the tyrosinase is scattered throughout the entire lumen. The increase in co-localization of tyrosinase with cathepsin-L supports the hypothesis that membranous complexes are indeed representatives of the lysosomal compartment.
The expression pattern of ribophorin II was almost entirely limited to the perinuclear region in both HPS-1 and normal cell lines (Figure 4). However, intense perinuclear regions of ribophorin II were periodically noted in HPS-1 melanocytes along with a more readily apparent reticular pattern (Figure 4). These intense ribophorin II regions often demonstrated co-localization with tyrosinase and Tyrp1 along the outer limiting membranes of HPS-1 membranous complexes, albeit minimal (Figure 4e, f). Although infrequent, some membranous complexes were observed to demonstrate a greater amount of co-localization of ribophorin II with tyrosinase and Tyrp1 (asterisked arrow in Figure 4). Quantitatively, the co-localization of tyrosinase with ribophorin II was 0.1% in normal melanocytes (SD = 0.1, n = 69) and 2.3% in HPS-1 melanocytes (SD = 1.7, n = 53) (Figure 3).
Figure 4.

Immunofluorescence of (A and B) normal and (C–F) HPS-1 melanocytes immunolabeled for tyrosinase (green in A and C) or Tyrp1 (green in E and F) and ribophorin II (red) (B and D represent images A and C in which the green channel was removed during photo reproduction. All images were captured from the confocal microscope using identical settings for each primary antibody.) Minimal expression of ribophorin II occurred in the perinuclear area (arrowheads) of normal melanocytes where significantly more expression with a more prominent reticular pattern was apparent in HPS-1 melanocytes (arrowheads). Higher magnification of co-labeled HPS-1 melanocytes (E and F) demonstrating minimal co-localization of Tyrp1 with ribophorin II at peripheral edges of macroautophagosomes (arrows). On rare occasions, extensive co-localization of Tyrp1 and ribophorin II was observed in a macroautophagosome (arrow with asterisk). N, nucleus; Bar, 20.0 μm.
The specific staining pattern of LC3 was diffuse throughout the cell body of both normal and HPS-1 melanocyte lines (Figure 5). However, only in HPS-1 melanocytes was LC3 intensely localized to small and large aggregates within the perinuclear area. Concentrated perinuclear Tyrp1-positive structures, representing membranous complexes, almost always were found to co-localize with the perinuclear LC3 aggregates (Figure 5e). Quantitatively, the co-localization of LC3 with Tyrp1, or visa versa, was significantly increased in HPS-1 melanocytes compared with the normal human melanocytes (Figure 5e). Specifically, 37.2% (SD = 10.4, n = 64) versus 55.4% (SD = 16.3, n = 41) of LC3 co-localized with Tyrp1 in normal versus HPS-1 melanocytes (Figure 5e). Conversely 4.9% (SD = 2.8, n = 64) versus 12.8% (SD = 4.9; n = 41) of Tyrp1 co-localized with LC3 in normal versus HPS-1 melanocytes (Figure 5e).
Figure 5.
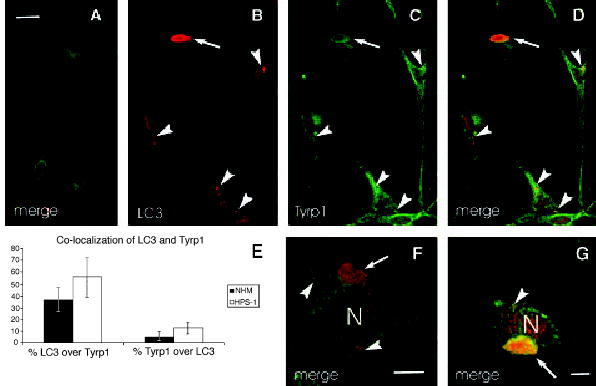
Immunofluorescence of (A) normal and (B–D, F and G) HPS-1 melanocytes immunolabeled for Tyrp1 (green) and LC3 (red). (All images were captured from the confocal microscope using identical settings for each primary antibody.) LC3 was localized to small (arrowheads) and large (arrows) membranous complexes in HPS-1 melanocytes where co-localization with Tyrp1 was apparent. (E) Graph depicting percentage of co-localization of LC3 and Tyrp1 in normal human (NHM) and HPS-1 melanocytes demonstrating increased co-localization in the HPS-1 melanocytes. N, nucleus; bars: A–D, 15 μm; E and F, 5.0 μm.
Acid phosphatase electron microscopy
Normal and HPS-1 melanocytes were processed for acid phosphatase electron microscopy. Non-specific staining was found in the nucleus and along the plasma membrane of normal and HPS-1 melanocytes stained for acid phosphatase (Figure 6). This non-specific staining was also present in melanocytes that were stained without substrate (β-glycerophosphate). Acid phosphatase-specific staining was observed within lysosomes of normal and HPS-1 melanocytes and membranous complexes of HPS-1 melanocytes only when substrate was included in the protocol (Figure 6).
Figure 6.
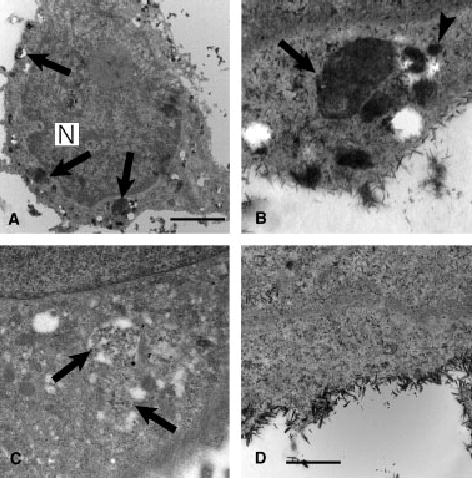
Ultrastructural histochemistry for acid phosphatase activity in HPS-1 melanocytes. Electron dense reaction product was predominantly localized to macroautophagosomes (arrows) as observed with low (A) and high (B) magnification. Smaller vesicles of approximately 50 nm in diameter in the vicinity of the macroautophagosome also exhibited reaction product (arrowhead) and putatively represent lysosomes. For negative controls, HPS-1 melanocytes treated (C) without the acid phosphatase stain exhibiting a membranous complex lacking histochemical staining (arrows) or (D) without the substrate that also demonstrates crystalloid, non-specific, plasma membrane staining. N, nucleus; bars: A, 2.5 μm; B–D, 0.7 μm.
Horseradish peroxidase-conjugated gold electron microscopy
HPS-1 melanocytes were exposed to a prolonged pulse of 14 h with HRP:gold (i.e. 14 h) and then chased for 2 h. This long incubation period was designed to insure delivery of exogenously supplied HRP:gold to the terminal compartment in the phagocytic pathway of HPS-1 melanocytes, i.e. the putative macromelanosome/membranous complexes. Preliminary experiments (not shown) demonstrated that relatively short periods of incubation (i.e. <6 h) resulted in no apparent internalization of HRP:gold within melanocytes, suggesting that the phagocytic capacity of melanocytes is relatively prolonged. Therefore, a pulse period of 14 h followed by a chase period of 2 h was used. Using these time parameters, perinuclear membranous complexes observed in HPS-1 melanocytes were found to contain HRP:gold particles (Figure 7). In contrast, PEG-stabilized gold, which is less efficiently internalized compared with HRP-stabilized gold (Morris and Saelinger, 1990) was not observed in the membranous complexes (Figure 7e). Other vesicles containing HRP:gold particles were presumed to be endosomes and/or lysosomes and were found surrounding the membranous complexes. In addition, HRP:gold was associated with early endosomes derived from the plasma membrane (Figure 7).
Figure 7.

Ultrastructural analysis of late events in the trafficking of exogenous gold-labeled horseradish peroxidase (HRP-Au18). HPS-1 melanocytes were incubated in HRP-Au18 for 14 h, chased for 2 h, and then processed for electron microscopy as described in Materials and methods. (A) Low magnification view of HPS-1 melanocyte demonstrating HRP-Au18 being internalized at the plasma membrane (white arrowheads), within small vesicles of the endosome/lysosome lineage (black arrowheads), and within macroautophagosomes (arrows). (B–D) Higher magnification demonstrated selective localization of HRP-Au18 to vesicles of the endosome/lysosome lineage (arrowheads) and macroautophagosomes (arrows). (E) HPS-1 melanocytes incubated in gold labeled polyethylene glycol (PEG-Au18), using the same pulse/chase procedures used for HRP-Au18, demonstrated a lack of labeling in the macroautophagosome (arrow). Bars: A = 0.6 μm; B–E = 0.4 μm.
A shorter pulse period and variable chase periods were used to characterize the temporal profile of the HRP:gold uptake and transport. This technique confirmed that the initial localization of HRP:gold particles occurs in early endosomes and not macroautophagosomes (Figure 8). Subsequently, prolonged chase periods yielded HRP:gold within late endosomes and macroautophagosomes (Figure 8).
Figure 8.

Ultrastructural analysis of early events in the trafficking of exogenously gold-labeled horseradish peroxidase (HRP-Au18). HPS-1 melanocytes were incubated in HRP-Au18 for 6 h, chased for either 2 or 4 h, and then processed for electron microscopy as described in Materials and methods. After a 2 h chase (A and C), HRP-Au18 could be found associated with the plasma membrane (black arrowheads), small early endosome-like compartments in the periphery of the cell (white arrowheads), and the vicinity of macroautophagosomes (black arrowhead with asterisk). In contrast, HRP-Au18 could not be found within the macroautophagosomes (arrows). After a 4 h chase (B and D), HRP-Au18 could be found in larger endosome-like compartments (black arrowheads) prominent in the Golgi area (G) and within the macroautophagosomes (arrow). Bars = 0.5 μm.
Discussion
Although a complete understanding of intracellular trafficking mechanisms for lysosomal-related organelle proteins such as tyrosinase has yet to be achieved, components of this process have been defined over recent years. In normal melanocytes, tyrosinase-related proteins are trafficked from the Golgi apparatus to the melanosome through a route that includes intermediate compartments (Boissy et al., in press; Huizing et al., 2002; Raposo and Marks, 2002). Transporting vesicles ultimately dock and fuse with the limiting membrane of the Stage II melanosome, depositing the critical enzymes into the lumen of the melanosome for melanin synthesis (Raposo and Marks, 2002; Setaluri, 2000). The HPS-1 protein has been found to be in close association with tubulovesicular structures, small non-coated vesicles, and nascent and early-stage melanosomes within the cytoplasm of the melanocyte and, therefore, proposed to be required for the docking and fusion of tyrosinase-related protein cargo vesicles to the melanosome (Oh et al., 2000; Richmond et al., 2005). When the HPS-1 protein is defective, such as in HPS-1 melanocytes, these transporting vesicles containing tyrosinase and other related proteins cannot efficiently fuse with the melanosome, leading to a decrease in melanin synthesis manifested as oculocutaneous albinism. As a consequence, the tyrosinase-related protein cargo is localized in membranous complexes within the HPS-1 melanocyte (Boissy et al., 1998; Richmond et al., 2005).
Although the role of the membranous complex has yet to be fully elucidated, our findings suggest that it is a component of the lysosomal compartment. We hypothesized that as the orphaned vesicles containing tyrosinase-related proteins accumulate within the cytoplasm, they are ultimately enclosed by elements of the endoplasmic reticulum in the formation of segregated, membranous compartments. The resulting multivesicular structures ultimately fuse with lysosomes and degrade the tyrosinase-related protein cargo contained within the limiting membranes. If this is true, the membranous complexes specific to HPS-1 melanocytes are macroautophagosomes, which represent one mechanism by which intracellular contents are degraded (Dunn, 1990b; Yoshimori, 2004). This process is distinct from other mechanisms of intracellular degradation, which include proteasome hydrolysis via ubiquitin targeting (Hershko, 1988) and microautophagosomal degradation, a process by which invagination of the limiting membrane of individual lysosomes occurs (Mortimore et al., 1989).
The data suggesting membranous complexes are macroautophagosomes were gathered from a variety of independent observations and results. Morphologically, the membranous complexes specific to HPS-1 melanocytes exhibit features of macroautophagosomes. The membranous complexes frequently consist of a double, outer limiting membrane indicative of endoplasmic reticulum encasing cytoplasmic components destined to be incorporated into macroautophagosomes. In addition, profiles of smooth and rough endoplasmic reticulum often appear surrounding and juxtapositioned to the HPS-1 membranous complexes, which is consistent with the proposed origin of these complexes from the endoplasmic reticulum to form macroautophagosomes. The contents of HPS-1 membranous complexes contain melanosomes, vesicles and tubules with and without DOPA reactivity (i.e. tyrosinase), occasional mitochondria, and some cytosol. These inclusions frequently exhibit a degraded appearance suggesting lysosomal-mediated breakdown.
Indirect immunofluorescence was used to assess the difference between the co-localization of tyrosinase and Tyrp1 with cathepsin-L in normal and HPS-1 melanocytes. Cathepsin-L, a versatile lysosomal cysteine protease that functions to degrade a variety of proteins including the PrP(Sc) of scrapie-infected cells (Luhr et al., 2004), has been previously shown to be localized predominately to lysosomes and degradative autophagosomes (Dunn, 1990b). Data suggest that there is a significant increase in the co-localization of tyrosinase with cathepsin-L in HPS-1 melanocytes. This data indicate that the membranous complexes, to which much of the tyrosinase in HPS-1 melanocytes is sequestered, must also contain cathepsin-L. This is likely because of the fusion of the endoplasmic reticulum-derived multivesicular structure with lysosomes, forming a macroautophagosome described as a membranous complex.
Because macroautophagosomes are believed to originate from the endoplasmic reticulum, the co-localization of tyrosinase and Tyrp1 with ribophorin II, an integral endoplasmic reticulum protein, was also assessed. Ribophorin II is one of at least four trans-membrane proteins comprising the oligosaccharyltransferase of the endoplasmic reticulum (Sanjay et al., 1998) and has been previously demonstrated to reside in the outer limiting membrane of nascent macroautophagosomes (Dunn, 1990a). Although only a marginal amount of co-localization between tyrosinase and ribophorin II was detected quantitatively in HPS-1 melanocytes, the expression pattern of ribophorin II in HPS-1 melanocytes periodically consisted of small intense packets in the perinuclear region. Although these small concentrated regions of ribophorin II were not observed to co-localize extensively with tyrosinase related proteins, they were in close proximity to perinuclear tyrosinase and Tyrp1 granules and may indicate the initial engulfing of these vesicles by the endoplasmic reticulum. It has been demonstrated that endoplasmic reticulum membranous components of forming macroautophagosomes are rapidly integrated into the complex where membrane proteins, such as ribophorins II, are quickly degraded (Dunn, 1990a). This rapid transitional stage in the formation of macroautophagosomes could explain the relatively minimal increase in co-localization of ribophorin II and tyrosinase in HPS-1 melanocytes demonstrated in this report. Even though the formation of macroautophagosomes is dramatically increased in HPS-1 melanocytes when compared with normal melanocytes, it is important to note that only a small portion of the endoplasmic reticulum would still be expected to be involved in macroautophagosome formation at any given time. Therefore, even under conditions of accelerated macroautophagosomy, the vast majority of tyrosinase would be contained within the lumen of the membranous complex and not co-localize with the small subset of ribophorins II that is transiently localized in the ER-derived limiting membrane.
LC3 is currently the only highly specific marker of the macrophagosome (Yoshimori, 2004). LC3 is a mammalian homologue of the yeast Atg8, which is a specific constituent of the autophagosomal membrane (Kabeya et al., 2000). LC3 exists in two forms, an 18 kDa cytosolic form (LC3-I) and a 16 kDa processed form (LC3-II) located on the autophagosomal membrane (Kabeya et al., 2000). Upon stimulation, the c-terminal arginine residue of LC3-I is proteolytically removed exposing the penultimate glycine residue of LC3-II that in turn becomes conjugated to phosphatidylethanolamine associated with the autophagic membrane inducing elongation/enlargement of the macroautophagosome (Reggiori and Klionsky, 2002). Co-localization of LC3 with tyrosinase related proteins in the membranous complexes characteristic of HPS-1 melanocytes confirms that these structures are macroautophagosomes responsible for the degradation of the wayward cargo.
Secondary lysosomes and macroautophagosomes contain acid phosphatase, a lysosomal enzyme that in part functions to degrade contents such as collagen within these structures (Dunn, 1990b; Yajima, 1986). Acid phosphatase activity was assessed in HPS-1 melanocytes at the electron microscopic level using the technique developed by Gomori (1952). Upon inspection of the membranous complexes at the EM level following treatment with Gomori’s chloride media, acid phosphatase activity was identified within the lumen of the membranous complex. This result is consistent with the fusion of lysosomes with HPS-1-specific membranous complexes, forming macroautophagosomes.
Another hallmark of lysosomal/phagosomal complexes is the subsequent localization of exogenously supplied horseradish peroxidase (HRP) (Nichols, 1982). Previous work has indicated that HRP:gold is endocytosed by eukaryotic cells and accumulates within the lysosomes following an adequate chase period (Nichols, 1982). HPS-1 melanocytes demonstrated HRP:gold particles in various endosomal compartments and ultimately in the lumen between the outer double membrane and their inner vesicles of the specific membranous complexes. These results further corroborate our hypothesis that the HPS-1 specific membranous complexes are indeed components of the lysosomal compartment.
When taken together, these observations are consistent with the proposed hypothesis that programmed vesicular degradation via macroautophagosomes occurs in HPS-1. We propose that upon encountering a block in their fusion with stage II melanosomes, tyrosinase-containing cargo vesicles are dispersed throughout the cytosol where they are corralled by endoplasmic reticulum, forming a macroautophagosome with a double limiting membrane. Lysosomes then fuse with these membranous complexes in order to degrade the misrouted tyrosinase and other related proteins contained therein.
Materials and methods
Cell culture
Melanocyte cultures were developed using forearm skin biopsies from normal human volunteers in a protocol approved by the University of Cincinnati Institutional Review Board and from patients with HPS-1 enrolled in a protocol approved by the National Institute of Child Health and Human Development and the National Human Genome Research Institute Institutional Review Boards to study the clinical and molecular aspects of HPS. After written informed consent was obtained, a 2-mm punch biopsy was taken and placed immediately in melanocyte growth medium with 2× antibiotic/antimycotic solution. Skin samples were placed in trypsin (2.5 mg/l) and incubated for 2 h at 37°C. After replacing the trypsin with MCDB-153 medium (Irvine Scientific, Santa Anna, CA, USA), the tissue was gently vortexed for 30 s to separate the dermis from a suspension of epidermal cells. The epidermal cells were seeded into a 2-cm2 well in MCDB-153 medium as previously described (Medrano and Nordlund, 1990). The MCDB-153 growth medium was supplemented with 0.6 ng/ml basic fibroblast growth factor, 8 nM 12-O-tetradecanoylphorbol-13-acetate (TPA), 5 μg/ml insulin, 5 μg/ml transferrin, 1.0 μg/ml alpha-tocopherol, 30 μg/ml crude pituitary extract (Clonetic Laboratories, San Diego, CA, USA), 0.5 μg/ml hydrocortisone, and 4% fetal calf serum. Cultures were fed with fresh medium twice weekly. Fibroblasts were eliminated by incubating cultures for 3–4 d in the presence of 10 μg/ml geneticin (G418 sulfate) (Halaban and Alfano, 1984). Cultures from the second to the fifth passage were used for the experiments described herein.
Indirect immunofluorescence microscopy
Established cultures of melanocytes, maintained in regular MCDB-153 growth media, were plated on gelatin-coated Lab-Tek (Nunc, Napterville, IL, USA) chamber slides at 2.5 × 104 cells per 0.9 cm2 well and processed for indirect immunofluorescence the next day. The cells were fixed in 2% paraformaldehyde in tris-buffered saline (TBS) for 10 min at room temperature, made permeable with 100% HPLC-grade methanol for 3 min at room temperature, washed with 1% bovine serum albumin (BSA) in TBS for 3 × 5 min, and then blocked with 5% normal goat serum in 1% BSA in TBS for 60 min at room temperature. The cells were then incubated for 60 min at room temperature with primary antibodies diluted in 1% BSA in TBS. The primary antibodies used in this study included rabbit polyclonal antiserum to ribophorin II (Santa Cruz Biotechnology, Santa Cruz, CA, USA) (1:100), rabbit polyclonal antiserum to Cathepsin-L (Biomol Research Laboratories, Plymouth Meeting, PA, USA) (1:100), rabbit polyclonal antiserum to LC3 (gift from T. Yoshimori) (1:500), mouse monoclonal antiserum to Tyrp1 (Mel-5; Signet Laboratories, Dedham, MA, USA) (1:200), and mouse monoclonal antiserum to tyrosinase (Research Diagnostics, Flanders, NJ, USA) (1:100). Cells were washed with 1% BSA in TBS for 3 × 5 min and then incubated for 45 min at 37°C with a secondary antibody solution diluted in 1% BSA in TBS. The secondary antibodies included Alexa Fluor® 546 goat anti-rabbit IgG and Alexa Fluor® 488 goat anti-mouse IgG (Molecular Probes, Eugene, OR, USA) (1:250). After washing, coverslips were applied to the sides using Gel/Mount (Biomeda, Foster City, CA, USA), an aqueous mounting medium with anti-fading agents, and observed and digitally photographed with a Zeiss (Thornwood, NY, USA) LSM 510 confocal microscope using identical image capture settings for the normal/HS-1 pairs stained with the various respective antibodies.
Analysis of co-localization
Paired digital images attained from a Zeiss LSM 510 confocal microscope were analyzed for co-localization using MetaMorph® (Universal Imaging, Downingtown, PA, USA) image analysis. Thresholds for each antibody were established using digital images of melanocytes stained with only secondary antibody that were attained using identical settings as their primary antibody stained counterparts. Individual pixel discrimination was 0.14 μm.
Electron microscopy
Established cultures of melanocytes, maintained in regular MCDB-153 growth media, were plated on gelatin-coated Lab-Tek (Nunc, Napterville, IL, USA) chamber slides at 2.5 × 104 cells per 0.9 cm2 well and processed for either dihydroxyphenylalanine (DOPA) or acid phosphatase histochemistry the next day. For routine and DOPA electron microscopy, the cells were fixed in half-strength Karnovsky’s fixative (Karnovsky, 1965) and processed for DOPA histochemistry as previously described (Boissy et al., 1998). For acid phosphatase histocytochemistry, the cells were fixed with quarter-strength Karnovsky’s fixative for 10 min at room temperature, washed with 0.2 M sodium cacodylate buffer for 5 min at room temperature, and then washed with 5 × 10−3 M sodium acetate for 5 min at room temperature. The cells were then incubated with Gomori’s chloride medium (Gomori, 1952) as modified by De Jong et al. (1979) for 2 × 60 min at 40° C. Cells for routine and histocytochemical evaluation were then washed with 0.2 M sodium cacodylate buffer for 3 × 5 min, treated with 1.0% osmium tetroxide containing 1.5% potassium ferrocyanide (Karnovsky, 1971) for 30 min at room temperature, and then washed a final time with 0.2 M sodium cacodylate buffer for 3 × 5 min. The cells next underwent a serial dehydration in ethanol that included staining with 0.5% uranyl acetate in 75% ethanol for 30 min at room temperature. After reaching dehydration in 100% ethanol, the cells were embedded in Eponate 12. Areas of the Epon cast were cut out and mounted on Epon pegs and sectioned on a RMC MT 6000-XL ultramicrotome. Ultrathin sections were then stained with aqueous solutions of 2% uranyl acetate and 0.3% osmium tetroxide for 15 min each and then viewed and digitally photographed in a ZEOL 1230 transmission electron microscope (All tissue-processing supplies were purchased from Ted Pella, Tustin, CA, USA.).
Horseradish peroxidase conjugated gold electron microscopy
IgG:gold colloids
Eighteen nanometer gold sols (Au18) were prepared by the reduction of chloroauric acid with sodium citrate (Morris and Saelinger, 1990). Briefly, 20 ml of 0.5% chloroauric acid was added to 1000 ml of doubly distilled water. The solution was heated to a boil and 25 ml of freshly prepared 1% sodium citrate was added. Upon addition of the sodium citrate, the solution changed color from yellow, to clear, to blue, to purple and finally to red. The red color indicated that the reaction was completed. After the solution cooled to 23°C, the pH was adjusted to 7.5 with a 1 N potassium carbonate solution. The amount of HRP (Sigma Chemical Co., St Louis, MO, USA) required to stabilize 1000 ml of the Au18 was determined as described previously (Morris and Saelinger, 1990). In most instances, it required about 10 mg of HRP to stabilize the 1000 ml of the gold sols, converting them into gold colloids. Thirty minutes after the addition of the HRP, 10 ml of a 2% polyethylene glycol (PEG) solution was added to assure that the colloids were stabilized. Finally, the gold colloids were filter sterilized and stored at 4°C until needed. Prior to use, the HRP:Au18 solution was centrifuged at 30 000 g for 1 h and the pellet resuspended in MCDB-153 growth media at one-tenth the original volume (i.e. 10× solution).
Cell processing
Established cultures of melanocytes, maintained in regular MCDB-153 growth media, were plated on gelatin-coated Lab-Tek (Nunc, Napterville, IL, USA) chamber slides at 2.5 × 104 cells per 0.9 cm2 well and processed for HRP:Au18 electron microscopy the next day. In the initial experiment, cells were first pulsed with a 10× solution of HRP conjugated to 18 nm colloidal gold for 14 h at 37°C and then chased for 2 h with regular MCDB-153 growth media at 37°C. This extensive pulse period allowed sufficient time for phagocytized label to be transported to the terminal compartment, i.e. macrophagosomes. In a subsequent experiment, cells were pulsed with a 10× solution of HRP:Au18 for 6 h and then chased for 2 or 4 h. This pulse/chase regimen allowed for the evaluation of cellular profiles associated with the transport of the exogenously supplied label. In order to establish a negative control, cells were also pulsed with PEG conjugated to 18 nm colloidal gold and then chased as described above. The cells were then fixed with one-half strength Karnovsky’s fixative (Karnovsky, 1965), washed with 0.2 M sodium cacodylate buffer for 3 × 5 min, dehydrated in serial dilutions of ethanol, and further processed as described above.
Acknowledgments
The authors wish to thank William A. Gahl, MD, of the National Institutes of Health, for skin biopsies from patients with HPS-1 and Tamotsu Yoshimori, PhD, of the National Institute of Genetics in Mishima, Japan for anti-LC3 antiserum. This work was supported in part by Grants R01 AR45429 and T35 DK060444 from the National Institutes of Health.
References
- Boissy RE, Zhao Y, Gahl WA. Altered protein localization in melanocytes from Hermansky–Pudlak syndrome: support for the role of the HPS gene production in intracellular trafficking. Lab Invest. 1998;78:1037–1048. [PubMed] [Google Scholar]
- Boissy RE, Huzing M, Gahl WA. Chapter 7: Biogenesis of Melanosomes. The Pigmentary System: Physiology and Pathophysiology. In: Nordlund JJ, Boissy RE, Hearing VJ, King RA, Oetting WS, Ortonne J-P, editors. 2nd edn. Oxford, UK: Blackwell Publishing; in press. [Google Scholar]
- De Jong AS, Hak TJ, Van Duijn P, Daems WT. A new dynamic model system for the study of capture reactions for diffusable compounds in cytochemistry. II Effect of the composition of the incubation medium on the trapping of phosphate ions in acid phosphatase cytochemistry. Histochem J. 1979;11:145–161. doi: 10.1007/BF01002992. [DOI] [PubMed] [Google Scholar]
- Dunn WA. Studies on the mechanisms of autophagy: formation of the autophagic vacuole. J Cell Biol. 1990a;110:1923–1933. doi: 10.1083/jcb.110.6.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn WA. Studies on the mechanisms of autophagy: Maturation of the autophagic vacuole. J Cell Biol. 1990b;110:1935–1945. doi: 10.1083/jcb.110.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomori G. Chicago, IL: University of Chicago Press; 1952. Microscopic Histochemistry; pp. 189–194. [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- Halaban R, Alfano FD. Selective elimination of fibroblasts from cultures of normal human melanocytes. In Vitro. 1984;20:447–450. doi: 10.1007/BF02619590. [DOI] [PubMed] [Google Scholar]
- Hershko A. Ubiquitin-mediated protein degradation. J Biol Chem. 1988;263:15237–15240. [PubMed] [Google Scholar]
- Huizing M, Boissy RE, Gahl WA. Hermansky–Pudlak syndrome: vesicle formation from yeast to man. Pigment Cell Res. 2002;15:405–419. doi: 10.1034/j.1600-0749.2002.02074.x. [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Uneo T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnovsky MJ. A formaldehyde-glutaraldehyde fixative of high osmolarity for use in electron microscopy. J Cell Biol. 1965;27:137A. [Google Scholar]
- Karnovsky MJ. Use of Ferrocyanide-Reduced Osmium Tetroxide in Electron Microscopy. (Chicago: Proceedings of the 14th Annual Meeting of American Society for Cell Biology); 1971. p. 146. [Google Scholar]
- Kisen GO, Tessitore L, Costelli P, Gordon PB, Schwarze PE, Baccino FM, Segler PO. Reduced autophagic activity in primary rat hepatocellular carcinoma and ascites hepatoma cells. Carcinogenesis. 1993;14:2501–2505. doi: 10.1093/carcin/14.12.2501. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhang Q, Oiso N, et al. Hermansky–Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1) Nat Genet. 2003;35:84–89. doi: 10.1038/ng1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhr KM, Nordstrom EK, Low P, Kristensson K. Cathepsin B and L are involved in degradation of prions in GT1-1 neuronal cells. Neuroreport. 2004;15:1663–1667. doi: 10.1097/01.wnr.0000134931.81690.34. [DOI] [PubMed] [Google Scholar]
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005;120:237–248. doi: 10.1016/j.cell.2004.11.046. [DOI] [PubMed] [Google Scholar]
- Medrano EE, Nordlund JJ. Successful culture of adult human melanocytes obtained from normal and vitiligo donors. J Invest Dermatol. 1990;95:441–445. [PubMed] [Google Scholar]
- Morris RE, Saelinger CB. Visualization of intracellular trafficking of proteins by the biotinyl-ligand: avidin-gold method. In: Wilchek M, Bayer EA, editors. In Methods in Enzymology (Avidin–Biotin Technology) Vol. 184. Orlando, FL: Academic Press; 1990. pp. 379–388. [PubMed] [Google Scholar]
- Mortimore GE, Poso AR, Lardeux BR. Mechanism and regulation of protein degradation in liver. Diabetes Metab Rev. 1989;5:49–70. doi: 10.1002/dmr.5610050105. [DOI] [PubMed] [Google Scholar]
- Natsuga K, Akiyama M, Shimizu T, Suzuki T, Ito S, Tomita Y, Tanaka J, Shimizu H. Ultrastructural features of trafficking defects are pronounced in melanocytic nevus in Hermansky-Pudlak Syndrome Type 1. J Invest Dermatol. 2005;125:154–158. doi: 10.1111/j.0022-202X.2005.23743.x. [DOI] [PubMed] [Google Scholar]
- Nichols BA. Uptake and digestion of horseradish peroxidase in rabbit alveolar macrophages. Formation of pathway connecting lysosomes to cell surface. Lab Invest. 1982;47:235–246. [PubMed] [Google Scholar]
- Oh J, Liu ZX, Feng GH, Raposo G, Spritz RA. The Hermansky–Pudlak syndrome (HPS) protein is part of a high molecular weight complex involved in biogenesis of early melanosomes. Hum Mol Genet. 2000;9:375–385. doi: 10.1093/hmg/9.3.375. [DOI] [PubMed] [Google Scholar]
- Raposo G, Marks MS. The dark side of lysosome-related organelles: specializations of the endocytic pathway for melanosome biogeneisi. Traffic. 2002;3:237–248. doi: 10.1034/j.1600-0854.2002.030401.x. [DOI] [PubMed] [Google Scholar]
- Reggiori F, Klionsky DJ. Autophagy in the eukaryotic cell. Eukaryot Cell. 2002;1:11–12. doi: 10.1128/EC.01.1.11-21.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond B, Huizing M, Knapp J, Koshoffer A, Zhao Y, Gahl WA, Boissy RE. Melanocytes derived from patients with Hermansky–Pudlak syndrome types 1, 2, and 3 have distinct defects in cargo trafficking. J Invest Dermatol. 2005;124:420–427. doi: 10.1111/j.0022-202X.2004.23585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjay A, Fu J, Kreibich G. DAD1 is required for the function and the structural integrity of the oligosaccharyltransferase complex. J Biol Chem. 1998;273:26094–26099. doi: 10.1074/jbc.273.40.26094. [DOI] [PubMed] [Google Scholar]
- Setaluri V. Sorting and targeting of melanosomal membrane proteins: Signals, pathways, and mechanisms. Pigment Cell Res. 2000;13:128–134. doi: 10.1034/j.1600-0749.2000.130302.x. [DOI] [PubMed] [Google Scholar]
- Stromhaug PE, Klionsky DJ. Approaching the molecular mechanism of autophagy. Traffic. 2001;2:524–531. doi: 10.1034/j.1600-0854.2001.20802.x. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Guhde G, Suter A, Eskelinen EL, Hartmann D, Lüllmann-Rauch R, Janssen PML, Blanz J, von Figura K, Saftig P. Accumulation of autophagic vacuoles and cardiomyopathy in LAMP-2-deficient mice. Nature. 2000;406:902–906. doi: 10.1038/35022595. [DOI] [PubMed] [Google Scholar]
- Yajima T. Acid phosphatase activity and intracellular collagen degradation by fibroblasts in vitro. Cell Tissue Res. 1986;245:253–260. doi: 10.1007/BF00213929. [DOI] [PubMed] [Google Scholar]
- Yoshimori T. Autophagy: a regulated bulk degradation process inside cells. Biochem Biophys Res Common. 2004;313:453–458. doi: 10.1016/j.bbrc.2003.07.023. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Zhao B, Li W, et al. Ru2 and Ru encode mouse orthologs of the genes mutated in human Hermansky-Pudlak syndrome types 5 and 6. Nat Genet. 2003;33:145–153. doi: 10.1038/ng1087. [DOI] [PubMed] [Google Scholar]