

Abstract
Hermansky–Pudlak Syndrome (HPS) is a genetically heterogeneous disorder in which mutations in one of several genes interrupts biogenesis of melanosomes, platelet dense bodies, and lysosomes. Affected patients have oculocutaneous albinism, a bleeding diathesis, and sometimes develop granulomatous colitis or pulmonary fibrosis. In order to assess the role of HPS genes in melanosome biogenesis, melanocytes cultured from patients with HPS subtypes 1, 2, or 3 were assessed for the localization of various melanocyte proteins. Tyrosinase, Tyrp1, and Dct/Tyrp2 were atypically and distinctly expressed in HPS-1 and HPS-3 melanocytes, whereas only tyrosinase showed an atypical distribution in HPS-2 melanocytes. The HPS1 and AP3B1 (i.e., HPS-2) gene products showed no expression in HPS-1 and HPS-2 melanocytes, respectively, whereas HPS-3 melanocytes exhibited normal expression for both proteins. In normal human melanocytes, the HPS1 protein was expressed as an approximately 80 kDa molecule with both granular and reticular intracellular profiles. In HPS-1, lysosome associated membrane protein 1 (LAMP1), and LAMP3 were localized to abnormal large granules; in HPS-2, all LAMPs exhibited a normal granular expression; and in HPS-3, LAMP1, and LAMP3 exhibited a distinct less granular and more floccular pattern. In contrast, the expressions of Rab 27, transferrin, and cKit were unaffected in all three HPS genotypes. These data demonstrate that the three initially identified subtypes of human HPS exhibit distinct defects in the trafficking of various melanocyte-specific proteins.
Keywords: pigmentation, albinism, tyrosinase, lysosomes, translocation
Abbreviations: HPS, Hermansky—Pudlak Syndrome; LAMP, lysosome associated membrane protein; NHM, normal human melanocytes
The biogenesis of functionally pigmented melanosomes is complex and poorly understood, but a few elements of this cellular process have been elucidated (Orlow, 1998; Raposo and Marks, 2002). Numerous structural proteins, melanogenic enzymes, and regulatory proteins need to be accurately trafficked from their site of synthesis to their melanosomal targets for efficient melanization to occur in this distinct organelle (Marks and Seabra, 2001). Specialized protein complexes function in the selective recruitment of melanosome-destined cargo at sites of synthesis, the translocation of cargo vesicles toward the melanosomes, either directly or via an intermediate organelle, and the efficient recognition, docking, fusion, and incorporation of these specific cargo into the melanosomes. Elucidating these trafficking molecules/chaperones in the melanocyte has been a challenging endeavor.
Hermansky–Pudlak Syndrome (HPS [MIM203300]) is a genetically heterogeneous, autosomal recessive disorder that provides an experiment of nature for understanding how trafficking molecules function in the biogenesis of melanosomes. To date, seven genetic types of the HPS have been identified in humans (Huizing et al, 2002; Li et al, 2003; Zhang et al, 2003) and approximately 15 genetically distinct HPS models exist in mice (Swank et al, 1998; Huizing et al, 2002). HPS presents with oculocutaneous albinism and a bleeding diathesis (Hermansky and Pudlak, 1959; King et al, 2001; Huizing et al, 2002). The albinism results from the inability of cutaneous and ocular melanocytes to create fully pigmented melanosomes due to mistrafficking of melanogenic proteins (Huizing et al, 2002). The bleeding diathesis results from the absence of dense bodies within platelets, compromising clot formation (Witkop et al, 1987; Gahl et al, 1998). Some HPS patients develop lysosomal ceroid storage (Bednar et al, 1964), granulomatous colitis (Schinella et al, 1980), or a fatal pulmonary fibrosis (Harmon et al, 1994; Brantly et al, 2000). Each individual HPS gene is considered to regulate a molecular aspect critical for the biosynthesis of melanosomes, platelet dense bodies, and lysosomes.
HPS-1, HPS-2, and HPS-3 are the three genetic forms of human HPS initially identified. The HPS1 gene was originally mapped and isolated in a population of patients residing in northwestern Puerto Rico (Wildenberg et al, 1995; Oh et al, 1996). HPS1 localizes to chromosome 10q23 and encodes a predicted 79.3 kDa protein with no homology to known proteins, although it contains a short segment predicted to form coiled-coil structures (Oh et al, 1996). The HPS1 protein, whose function remains unknown, is distributed in melanotic cells among uncoated vesicles, early-stage melanosomes, and the cytoplasm as part of a large complex of molecular weight ~ 200 kDa in the cytoplasm and >500 kDa in a membrane-bound form (Oh et al, 2000). Melanocytes from HPS-1 patients are hypopigmented and exhibit unique membranous complexes with tyrosinase-containing compartments (Boissy et al, 1998).
Mutations in the AP3B1 gene cause HPS-2 (Dell’Angelica et al, 1999). AP3B1 is localized to chromosome 5q14.1 and encodes the 140 kDa β3A-subunit of adaptor complex-3 (AP3) involved in vesicle/membrane formation and trafficking (Dell’Angelica et al, 1997, 1998). Melanocytes from HPS-2 patients are hypopigmented and exhibit sequestration of tyrosinase to large endosome/multivesicular body-like structures (Huizing et al, 2001b).
The HPS3 gene was initially identified in a central Puerto Rican HPS isolate (Anikster et al, 2001) and subsequently in other populations (Huizing et al, 2001a). HPS3 is located on chromosome 3q24 and encodes a predicted 113.7 kDa protein with no apparent homology to other proteins (Anikster et al, 2001; Suzuki et al, 2001). The HPS3 protein, however, does contain a putative clathrin binding region (Anikster et al, 2001), suggesting participation in intracellular trafficking. We have recently demonstrated that 50 nm vesicles containing tyrosinase are extensively distributed throughout HPS-3 melanocytes rather than being confined to the trans-Golgi network as in normal human melanocytes (NHM) (Boissy et al, 2004).
Specialized protein complexes function in series along a cellular pathway for trafficking. Multimeric components within these various complexes coordinate cargo recruitment, vesicle budding, cytoskeletal interaction, target recognition, and vesicle fusion (Bonifacino and Glick, 2004). HPS gene products are components of a group of complexes termed BLOC (i.e., biogenesis of lysosome-related organelles complexes) (Falcon-Perez et al, 2002; Chiang et al, 2003; Ciciotte et al, 2003; Li et al, 2003; Martina et al, 2003; Gautam et al, 2004). BLOC facilitate the trafficking of cargo to melanosomes, lysosomes, and platelet dense bodies. But the molecular functions and sites of action of BLOC remain unknown.
In this report, we compare the trafficking patterns of various melanosomal proteins in melanocytes cultured from patients with HPS-1, HPS-2, and HPS-3 to further characterize the various pathways required for correct protein trafficking and assembly of a functional melanosome. The abnormal distribution patterns of proteins transported via endolysosomal pathways distinguish these HPS subtypes and implicate distinct sites of action for the three HPS gene products.
Results
Expression of tyrosinase gene family proteins
Melanocytes from patients with HPS-1, HPS-2, and HPS-3 were evaluated by immunocytochemistry for the expression pattern of tyrosinase gene family proteins [i.e., tyrosinase, tyrosinase-related protein-1 (Tryp1), and DOPAchrome tautomerase/tyrosinase-related protein-2 (Dct/Tyrp2)] (Fig 1). In NHM, all three proteins exhibited intense staining around the perinuclear area with marked granularity. Throughout the lateral areas of the cell body and along the dendrites, the staining remained granular but less dense. In HPS-1 melanocytes, in addition to this normal staining pattern, all three proteins were localized to large granules distributed predominantly throughout the cell body and occasionally within the dendrites (Fig 1).
Figure 1. Melanocytes cultured from patients with Hermansky–Pudlak Syndrome (HPS)-1, HPS-2, and HPS-3 exhibit specific alterations in the distribution of tyrosinase gene family members.
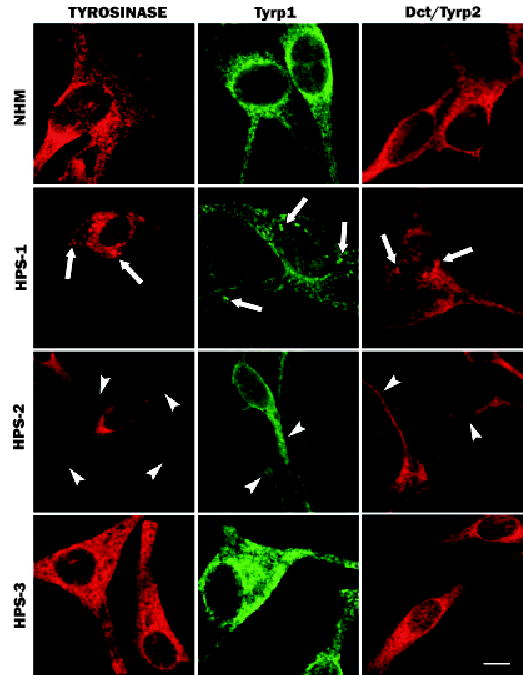
Cultures of melanocytes derived from an unaffected individual (normal human melanocytes (NHM)) and from patients with HPS-1, HPS-2, or HPS-3 were immunostained for tyrosinase, tyrosinase-related protein-1 (Tyrp1), or DOPAchrome tautomerase/tyrosinase-related protein-2 (Dct/Tyrp2). The NHM exhibited a granular staining pattern with prominent perinuclear localization for all three proteins. HPS-1 melanocytes exhibited a similar granular staining pattern with additional large aggregates (arrows) for all three proteins throughout the melanocytes. HPS-2 melanocytes exhibited a relative restriction of tyrosinase to the cell body with limited or no expression in the dendrites (arrowheads); Tyrp1 and Dct/Tyrp2 exhibited normal expression patterns. HPS-3 melanocytes displayed a floccular staining pattern within the cell body and dendrites for tyrosinase, Tyrp1, and Dct/Tyrp2 expression. Scale bar =10 μm.
In HPS-2 melanocytes, tyrosinase was predominantly restricted in its expression to the cell body with minimal expression along the dendrites (Fig 1). In contrast, the localization patterns for both Tyrp1 and Dct/Tyrp2 appeared normal, with marked expression in the dendrites (Fig 1). In HPS-3 melanocytes, both tyrosinase and Tyrp1 exhibited an expression pattern subtlety different from NHM. Although expression of these proteins occurred in both the cell body and dendrites, the pattern was less granular and more floccular compared with that in NHM (Fig 1). This differential stain pattern in HPS-3 melanocytes was most noticeable throughout the dendrites where the immunofluorescence appeared more homogeneously distributed in HPS-3 melanocytes and heterogeneous (i.e., granular) in NHM.
The co-localization of tyrosinase and Tyrp1 in large granules, characteristic of HPS-1 melanocytes, was quite heterogeneous (Figs 2a–c). Some large granules contained one protein selectively, but more frequently a large granule contained both proteins. Occasionally, tyrosinase and Tyrp1 exhibited discrete, separate areas of localization within the granules, with partial overlap. As previously described, the granules represent large membranous complexes containing melanosomes (Boissy et al, 1998). Dihydroxyphenylalanine (DOPA) histochemistry of HPS-1 melanocytes revealed melanin reaction product in a subset of cisternae of the membranous profiles and within some of the associated melanosomes (Fig 2d), consistent with the heterogeneity in localization of tyrosinase and Tyrp1 observed on light microscopy.
Figure 2. Hermansky–Pudlak Syndrome (HPS)-1 melanocytes exhibit a variation in co-localization of tyrosinase and tyrosinase-related protein-1 (Tyrp1) within characteristic large granules/membranous complexes.
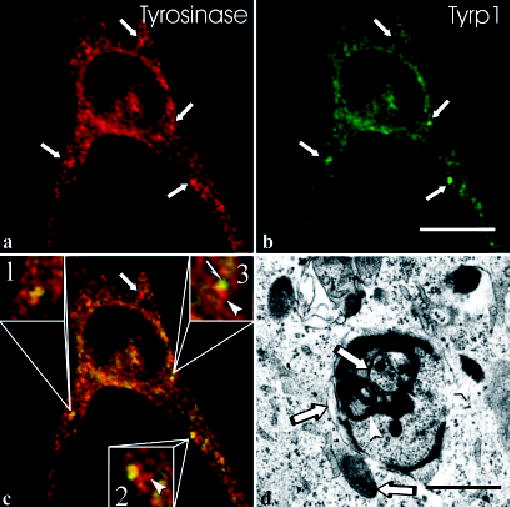
In HPS-1 melanocytes, the characteristic large granules (arrows) are positive for tyrosinase [red in (a)] and Tyrp1 [green in (b)]. The merged image (c) demonstrates that some large granules contained tyrosinase only (arrow), some show equal co-localization of tyrosinase and Tyrp1 (inset #1), some display prominent co-localization of tyrosinase and Tyrp1 with areas containing tyrosinase only (arrowhead, inset #2), and some show polar expression of tyrosinase (arrowhead, inset #3) and Tyrp1 (half-arrow) with an intervening area of co-localization. Electron microscopic evaluation of HPS-1 melanocytes treated for DOPA histochemistry (d) demonstrates that, within a membranous complex, some cisternae and melanosomes express (arrowheads) or lack (arrows) reaction product, indicating the presence and absence of tyrosinase, respectively. Scale bars =10 (b) and 1.5 (d) μm.
Expression of HPS gene family proteins
Melanocytes from patients with HPS-1, HPS-2, and HPS-3 were evaluated by immunocytochemistry for expression of the HPS1 protein and the β3A and μ3A subunits of AP3 (Fig 3). In NHM, HPS1 showed a distinct perinuclear pattern with dramatically reduced expression along the dendrites (Fig 3). HPS-2 and HPS-3 melanocytes exhibited a similar pattern of expression. In contrast, HPS1 was absent from melanocytes of HPS-1 patients bearing either the 16 base pair duplication (Fig 3) or the S396 del C (not shown) mutations in HPS1.
Figure 3. Melanocytes cultured from patients with Hermansky–Pudlak Syndrome (HPS)-1, HPS-2, and HPS-3 exhibit specific alterations in the distribution of HPS gene products.
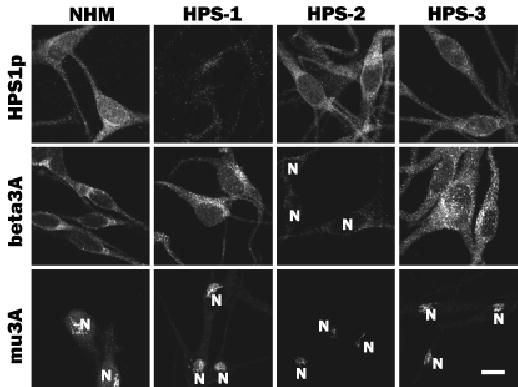
Cultures of melanocytes derived from an unaffected individual (normal human melanocytes (NHM)) and patients with HPS-1, HPS-2, or HPS-3 were immunostained for HPS1 (HPS-p) and the β3A (beta3A) and μ3A (mu3A) subunits of AP3. Expression of HPS1 protein (HPS1p) was similar in NHM, HPS-2 melanocytes, and HPS-3 melanocytes but markedly diminished in HPS-1 melanocytes. Expressions of both β3A and μ3A were normal in NHM, HPS-1 melanocytes, and HPS-3 melanocytes but markedly diminished in HPS-2 melanocytes. N, nucleus. Scale bar =10 μm.
The expression of the β3A and μ3A subunits of AP3 in NHM occurred with a distinct localized area in the cell body (Fig 3). The μ3A subunit exhibited a compact, tubular-network profile, unilateral to the nucleus. In contrast, the β3A subunit exhibited a more diffuse staining pattern, with a more intense focus localized unilateral to the nucleus. Similarly, HPS-1 and HPS-3 melanocytes displayed normal expression for both β3A and μ3A (Fig 3). In contrast, HPS-2 melanocytes exhibited an absent or dramatically reduced expression of β3A and the μ3A, respectively.
Expression of the HPS1 protein in NHM was further evaluated (Fig 4). Western blotting showed that HPS1 antibodies recognized an approximately 80 kDa protein in melanocytes derived from both a Caucasian and an African-American donor; this protein was absent from melanocytes of an HPS-1 patient (Fig 4a). In addition, the HPS1 antibodies exhibited, by routine fluorescent microscopy, prominent cell body staining with reduced dendritic staining in NHM (Fig 4b) that was absent from HPS-1 melanocytes (Fig 4c). Further analysis of HPS1 protein in NHM by confocal microscopy demonstrated prominent expression in the perinuclear area of the cell body (Fig 4d) with a relative reduction of staining in the Golgi area and the dendrites (Fig 4d). The perinuclear staining appeared predominantly granular (Fig 4d, arrows) with a reticular staining pattern apparent among the granular profiles (Fig 4d, arrowheads).
Figure 4. The Hermansky–Pudlak Syndrome (HPS)1 protein is expressed in normal human melanocytes (NHM) and absent in HPS-1 melanocytes.
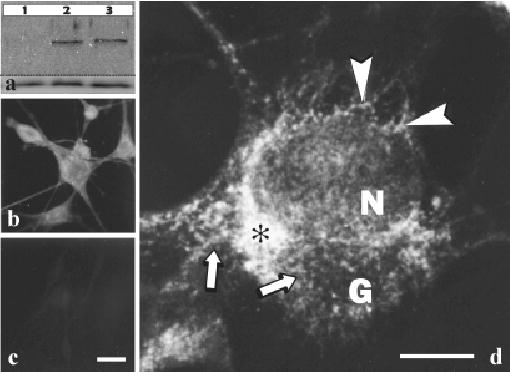
(a) Lysates of cultured melanocytes derived from an HPS-1 patient with the 16 bp duplication (lane 1), a normal Caucasian (lane 2), and a normal African/American (lane 3) individual were processed for western blot analysis using HPS1 antiserum (upper blot) and actin antibody (lower blot). A band of ~ 80 kDa was present in both controls and absent in the HPS-1 samples. Cultured melanocytes derived from a Caucasian individual (b and d) and an HPS-1 patient with the 16 bp duplication (c) were processed for indirect immunofluorescent cytochemistry using HPS1 antiserum. Immunostaining as observed by routine fluorescent microscopy was present predominantly in the cell body of control melanocytes (b) and absent in the HPS-1 melanocytes (c). As observed by confocal microscopy (d), the staining pattern for the HPS-1 protein in NHM was prominent in the perinuclear area of the cell body (asterisk) with a relative reduction in staining in the Golgi area (G) and the dendrites. The perinuclear staining appeared predominantly granular (arrows) with a reticular staining pattern (arrowheads) apparent among the granular profiles. N, nucleus. Scale bars =20 (c) and 7.5 (d) μm.
Expression of various melanocyte-expressed proteins
Melanocytes of patients with HPS-1, HPS-2, and HPS-3 were evaluated by immunocytochemistry for the expression pattern of LAMP 1–3, Rab27, transferrin, and cKit (Fig 5). In NHM, LAMP 1–3 exhibited a granular staining pattern within the cell body and throughout the dendrites. HPS-1 melanocytes exhibited normal localization for LAMP2, but LAMP1 and LAMP3 showed additional expression in large granules similar to that observed for the tyrosinase gene family members. HPS-2 melanocytes displayed normal localization of LAMP 1–3. HPS-3 melanocytes exhibited a floccular distribution pattern for LAMP1 and LAMP3, similar to that observed for tyrosinase, Tyrp1, and Dct/Tyrp2, and normal expression for LAMP2. Expression of Rab27, transferrin, and cKit exhibited normal profiles in melanocytes of the three HPS subtypes.
Figure 5. Melanocytes cultured from patients with Hermansky–Pudlak Syndrome (HPS)-1, HPS-2 and HPS-3 were evaluated for expression of various proteins.
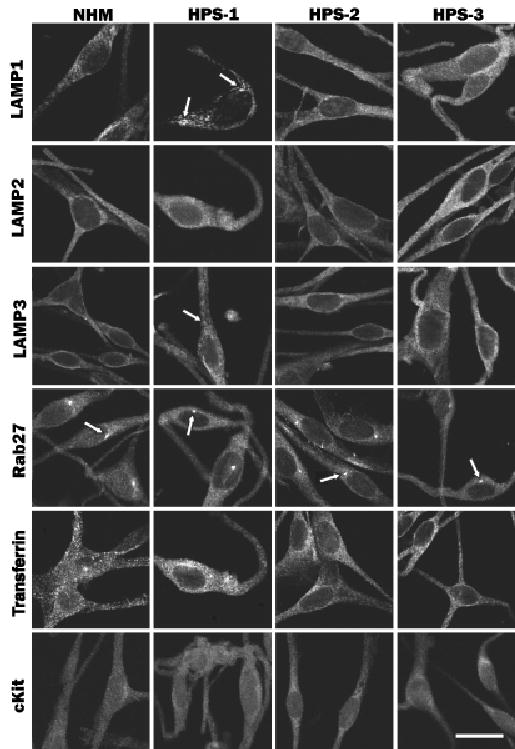
Normal human melanocytes (NHM) and HPS-1, HPS-2, or HPS-3 melanocytes were immunostained for LAMP 1–3, Rab 27, transferrin, and cKit. Expression of LAMP1 and LAMP3 was granular and that of LAMP2 was diffuse throughout the NHM. HPS-1 melanocytes exhibited, in addition to the normal localization for LAMP 1–3, localization to large granules (arrows) for LAMP1 and LAMP3. HPS-2 melanocytes exhibited normal localization for LAMP 1–3. HPS-3 melanocytes exhibited a more floccular distribution pattern for LAMP1 and LAMP3 and a normal distribution of LAMP2. Expression of Rab27, transferrin, and cKit in NHM exhibited a uniform pattern throughout the melanocytes with a distinct centriole localization (arrows), a punctate pattern throughout the melanocytes, and a diffuse pattern throughout the melanocytes, respectively. HPS-1, HPS-2, and HPS-3 melanocytes exhibit a normal staining pattern for Rab27, transferrin, and cKit. Scale bar =20 μm.
Discussion
The cellular pathways through which cargo proteins are directed, from their site of synthesis to melanosomes, have yet to be clearly delineated. The most prominent such cargos are the tyrosinase gene family proteins tyrosinase, Tyrp1 and Dct/Tyrp2. Tyrosinase is a glycoprotein synthesized in the rough endoplasmic reticulum that travels through the Golgi for carbohydrate modification (Ujvari et al, 2001; Francis et al, 2003; Watabe et al, 2004). Although it is already enzymatically mature by the time it reaches the trans-Golgi, tyrosinase has been shown with DOPA histochemistry and electron microscopy to bud off the trans-Golgi into vesicles that can be clathrin coated and travel a short distance to premelanosomes (Novikoff et al, 1968; Maul, 1969; Maul and Brumbaugh, 1971). Tyrosinase may also traffic through structures resembling multivesicular bodies en route to the premelanosome (Turner et al, 1976). Tyrp1 and Dct/Tyrp2 are also glycoproteins that traverse the Golgi apparatus (Tsukamoto et al, 1992; Jimenez-Cervantes et al, 1994); Tyrp1 may also travel through an organelle resembling a large multivesicular body (Orlow et al, 1993). It is unknown whether tyrosinase, Tyrp1, and Dct/Tyrp2 travel together in the same or independent pathways to early-stage melanosomes, defined as containing the melanofilament molecule Silver/Pmel17 (Berson et al, 2001, 2003).
Analysis of melanocytes cultured from patients with HPS-1, HPS-2, and HPS-3 revealed a normal distribution for proteins that do not reside in melanosomes (i.e., transferrin and cKit). In addition, a normal distribution was observed for Rab27, a family of molecules that regulate exocytosis of various cell-specific organelles (Izumi et al, 2003), in which Rab27a associates with the melanosome to facilitate its transport to and capture at the terminal ends of dendrites (Strom et al, 2002). In contrast, mutations in HPS1 and HPS3 did cause a distinctly aberrant localization of the tyrosinase family proteins. In the absence of HPS1, the tyrosinase-related proteins, as well as LAMP1 and LAMP3, were sequestered in large membranous complexes resembling macroautophagosomes (Boissy et al, 1998).
Macroautophagy is a dynamic process involving rearrangement of subcellular membranes to sequester cytoplasm and organelles for delivery to the lysosome or vacuole where the sequestered cargo is degraded and recycled (Dunn, 1990; Seglen and Bohley, 1992; Klionsky and Emr, 2000). The incorporation of tyrosinase, Tyrp1, and Dct/ Tyrp2 into macroautophagosomes in HPS-1 melanocytes suggests that the HPS1 protein targets these cargos to melanosomes, and the cargos move by default to macroautophagosomes when HPS1 is dysfunctional. Precedent for such default pathways is provided by the mutant huntingtin protein, which becomes aberrantly localized to large autophagosomes in Huntington’s disease (Kegel et al, 2000). In addition, the endogenous antigenic proteins presented by MHC class II traffic through autophagosomes (Nimmerjahn et al, 2003).
Consistent with the role of HPS-1 in melanosomal targeting of cargo is the ultrastructural localization of HPS1 to the cytoplasmic face of melanosomes (Oh et al, 2000) (Boissy, personal observation). HPS1 appears to be a predominantly cytosolic protein that can be associated with membranes, possibly as a multimeric complex (Dell’Angelica et al, 2000; Oh et al, 2000). HPS1 also appears to interact with the HPS4 protein (Anderson et al, 2003) in a transient or indirect manner (Suzuki et al, 2002) as part of the biogenesis of lysosome-related organelles complex-3 (BLOC-3) (Chiang et al, 2003; Martina et al, 2003).
Mutations in HPS3 also result in aberrant localization of all three tyrosinase family members but with a different distribution than what occurs in HPS-1 melanocytes. This suggests distinct roles for the HPS3 and HPS1 proteins. HPS-3 melanocytes exhibit a diffuse, less granular distribution for the three tyrosinase gene family proteins, as well as for LAMP1 and LAMP3. Recent evidence indicates that tyrosinase is localized in 50 nm vesicles that are abnormally scattered throughout the cell body and dendrites of HPS3 melanocytes (Boissy et al, 2004), as if by default of a normal, HPS3-mediated pathway. This suggests that the HPS3 protein regulates the targeting of vesicles to a recipient site and, in the absence of the HPS3 protein, these cargo vesicles are widely distributed throughout the cell body and dendrites of the mutant cells. In addition, LAMP1 and LAMP2 exhibit a greater perinuclear concentration in HPS-3 fibroblasts compared with normal cells and frequently fail to extend into the cell periphery (Helip-Wooley et al, 2004). The recognition step in trafficking regulated by HPS3 appears to be independent of HPS1, since the morphologies of the two HPS subtypes differ.
The HPS3 protein contains a putative clathrin binding domain, a possible tyrosine phosphorylation site at codon 295, and several putative tyrosine-based sorting signals, but no potential di-leucine sorting signals (Anikster et al, 2001) that could function in trafficking cargo. In normal murine melanocytes, the HPS3 protein exhibits a cytoplasmic distribution and is not associated with the type 1 IP3 receptor, Rab3A, or the AP3 complex (Suzuki et al, 2001). In NHM, the HPS3 protein interacts with clathrin (Helip-Wooley et al, 2004), and may function in the BLOC-2 complex (Di Pietro et al, 2004; Gautam et al, 2004) that also contains the HPS5 and HPS6 proteins (Zhang et al, 2003).
In contrast to HPS1 and HPS3, the AP3B1 gene (associated with HPS-2) affects only tyrosinase without any apparent influence on the cellular distribution of Tyrp1 or Dct/ Tyrp2. AP3 is one of four adaptor complexes, i.e., heterotetrameric complexes involved in vesicle/membrane formation and trafficking (Dell’Angelica et al, 1999; Robinson and Bonifacino, 2001). Adaptor complexes play a role in the formation of coated vesicles, as well as in the selection of cargo for these vesicles. AP3 contains δ, μ3, and σ3subunits, in addition to β3A. The known mutations in the AP3B1 gene cause loss of the β3A, destabilization of the δ, μ3, and σ3 subunits (Fig 3) (Huizing et al, 2001b), and thus loss of functional AP3. The β3A subunit binds clathrin, whose rigid triskelion structure causes outpouching from an existing membrane (Dell’Angelica et al, 1998). In addition, AP3 contains putative di-leucine and tyrosine-based signal motifs for sorting in the β3A and the μ3A subunits, respectively (Robinson and Bonifacino, 2001). Tyrosinase interacts with AP3 via its di-leucine motif (Honing et al, 1998) and is retained in multivesicular body/late endosome-like structures in HPS-2 melanocytes (Huizing et al, 2001b). Apparently, neither Tyrp1 nor Dct/Tyrp2 express the requisite di-leucine motif required to bind AP3, accounting for their normal distribution in HPS-2 melanocytes. In contrast, AP3B1 gene mutations result in impaired trafficking of tyrosinase to melanosomes. HPS-2 fibroblasts also traffic LAMP 1–3 through the plasma membrane in an enhanced fashion, suggesting that the plasma membrane provides a default pathway that operates when normal AP3 function is blocked in this cell type (Dell’Angelica et al, 1999). This default pathway does not appear to occur in HPS-2 melanocytes, suggesting that an AP3-independent route via the plasma membrane is not an alternate for tyrosinase in melanocytes.
Taken together, these data suggest that the gene products of HPS1, AP3, and HPS3 regulate distinctly different steps in the trafficking pathway for tyrosinase gene family proteins from the Golgi to melanosomes. This proposed assessment for the cellular roles of these proteins is based primarily on morphologic data; however, biochemical analysis is required to substantiate this model. The HPS1 protein is essential for the accurate deposition of these proteins to the melanosomes. In the absence of HPS1, tyrosinase, Tyrp1, and Dct/Tyrp2 are trafficked to macrophagosomes, presumably for default degradation. The HPS1 protein is primarily a cytosolic protein that can be associated with the limiting membrane of the melanosome (Oh et al, 2000) (Boissy, personal observations). This positions HPS1, along with its partner HPS4, at the recognition/docking/fusion interface between incoming cargo vesicles and melanosomes (Fig 6). The HPS3 protein also regulates the accurate trafficking of tyrosinase gene family members to melanosomes. As described above, however, the HPS3-regulated step is distinct from that regulated by the HPS1/HPS4 complex. We propose that HPS3, along with its complex partners HPS5 and HPS6, functions upstream of HPS1/HPS4 (Fig 6). In contrast, the protein defective in HPS-2 (i.e., the β3A subunit of AP3) regulates the trafficking of tyrosinase only. We propose that this selective step occurs within an endosome/multivesicular-like sorting body used for transit between the Golgi and melanosomes (Fig 6). It is uncertain whether Tyrp1 and Dct/Tyrp2 travel through a similar intermediate compartment and whether they use an alternate adaptor-like molecule to do so. In addition, it is uncertain whether Tyrp1 and Dct/Tyrp2 are sorted through similar or distinct pathways with respect to each other.
Figure 6. Model for involvement of Hermansky–Pudlak Syndrome (HPS1), HPS2, and HPS3 in the trafficking of tyrosinase gene family members from the Golgi to the melanosome.
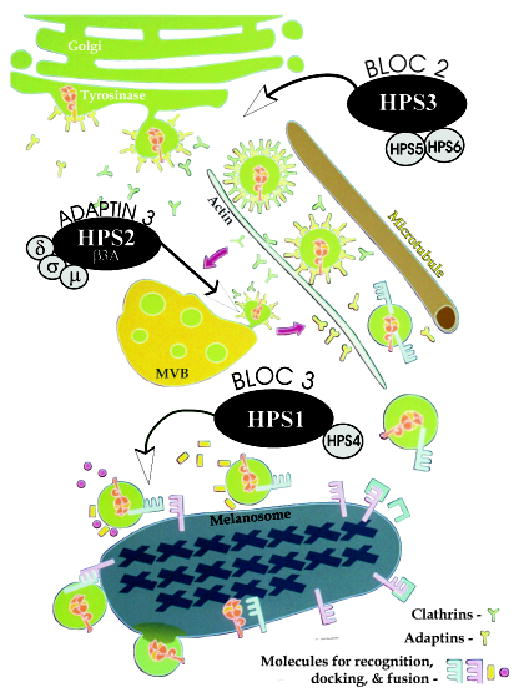
Tyrosinase, tyrosinase-related protein-1 (Tyrp1), and DOPAchrome tautomerase/tyrosinase-related protein-2 (Dct/Tyrp2) are recruited from the Golgi apparatus into clathrin-coated vesicles to be trafficked along cytoskeletal element to Stage II melanosomes in the perinuclear area. Biogenesis of lysosome-related organelles complexes (BLOC)-2, composed in part of HPS3, HPS5, and HPS6, facilitates this early cargo trafficking event. In the absence of HPS3, cargo vesicles are aberrantly trafficked beyond the perinuclear area. Tyrosinase, and possibly Tyrp1 and Dct/Tyrp2, transit through multivesicular body (MVB)/late endosome-like structures on route to melanosomes. Adaptin3 facilitates the recruitment of tyrosinase from the MVB. In the absence of the β3A subunit of AP3, tyrosinase is aberrantly retained in the MVB. Ultimately, vesicles containing tyrosinase, Tyrp-1, and Dct/Tyrp2 recognize, dock, and fuse with Stage II melanosomes. BLOC-3, composed in part of HPS1 and HPS4, facilitates this later cargo trafficking event. In the absence of HPS1, cargo vesicles are aberrantly trafficked to macroautophagosomes.
The comparative analysis of the patterns of melanocyte protein distribution in HPS-1, HPS-2, and HPS-3 described herein provides a glimpse into the elaborate mechanism responsible for targeting cargo from the Golgi apparatus to melanosomes. Some HPS molecules regulate trafficking of tyrosinase gene family members coordinately en route to the melanosomes, whereas other HPS molecules assist the trafficking of selective melanocyte proteins. Melanin synthesis generates several toxic intermediates. Segregation of tyrosinase gene family members during post-translational translocation may be important to prevent inappropriate melanization from occurring in non-melanosomal structures prior to accumulation in the melanosomal microdomain.
Materials and Methods
Cell culture
Cultures of melanocytes were developed from neonatal foreskins or skin biopsies. Normal human neonatal foreskins (from African/American and Caucasian infants) were obtained from the nursery of University Hospital in Cincinnati after routine circumcision using a protocol approved by the University of Cincinnati Institutional Review Board. Three HPS-1 patients, two HPS-2 patients, and three HPS-3 patients were enrolled in a protocol approved by the National Institute of Child Health and Human Development and the National Human Genome Research Institute Institutional Review Boards to study the clinical and molecular aspects of HPS. All procedures for obtaining human tissue were conducted according to Declaration of Helsinki principles. Patient numbers corresponded to those of a master list of enrolled subjects. Two of the HPS-1 patients were homozygous for the 16 base pair duplication and one for the S396 del C mutation in the HPS1 gene. The two HPS-2 patients were brothers and compound heterozygous for a 63 base pair deletion and an L580R mutation in the β3A gene (Dell’Angelica et al, 1999). Two of the three HPS-3 patients were homozygous for a 3.9 kb deletion encompassing exon 1 of the HPS3 gene, and the third HPS-3 patient was compound heterozygous for the 3.9 kb deletion and a I243insA. After written informed consent was obtained, a 4 mm punch biopsy was taken from each patient, and half was placed immediately in melanocyte growth medium with 2 × antibiotic/antimycotic solution and shipped by express mail to Cincinnati, Ohio. All skin samples were placed in trypsin (2.5 mg per L) and incubated for 2 h at 37°C. The trypsin was replaced with MCDB-153 medium, and the tissue was gently vortexed for 30 s to separate the dermis as a single piece and to produce an epidermal cell suspension. The epidermal cells were seeded in a T-25 cm2 flask (for foreskins) or a 2 cm2 well (for skin biopsies) in MCDB-153 medium (Irvine Scientific, Santa Anna, California) as previously described (Medrano and Nordlund, 1990). The MCDB-153 growth medium was supplemented with 0.6 ng per mL basic fibroblast growth factor, 8 nM 12-O-tetrad-ecanoylphorbol-13-acetate (TPA), 5 μg per mL insulin, 5 μg per mL transferrin, 1.0 μg per mL α-tocopherol, 30 μg per mL crude pituitary extract (Clonetic Laboratories, San Diego, California), 0.5 μg per mL hydrocortisone, 20 μg per mL catalase from bovine liver, and 10% heat-inactivated fetal calf serum. Cultures were fed with fresh medium twice weekly. Catalase was omitted from the medium after Day 6. Fibroblasts were eliminated by incubating cultures for 3–4 d in the presence of 100 μg per mL geneticin (G418 sulfate) (Halaban and Alfano, 1984). Cultures from the second to the fifth passage were used for the experiments described herein.
Immunofluorescent microscopy
Established cultures of melanocytes, maintained in regular MCDB-153 growth media, were plated on gelatin-coated Lab-Tek (Nunc, Naperville, Illinois) chamber slides at 2.5 × 104 cell per 0.9 cm2 well and processed for indirect immunofluorescence the next day. The cells were fixed in 2% formaldehyde in phosphate-buffered saline (PBS) for 10 min at room temperature, blocked in 1% bovine serum albumin (BSA) in PBS for 5 min at room temperature, and incubated in primary antibody solution diluted in 0.2% Saponin in PBS containing 0.1% BSA for 60 min at room temperature. The primary antibodies used in this study included rabbit polyclonal antiserum to tyrosinase provided by R. King and W.S. Oetting (hPEP1; University of Minnesota, Minneapolis) (1:300), mouse monoclonal antiserum to Tyrp1 (Mel-5; Signet Laboratories, Dedham, Massachusetts), (1:50), rabbit polyclonal antiserum to Dct/Tyrp2 provided by V. Hearing (PEP8; National Institutes of Health), (1:200), mouse monoclonals antisera to LAMP1 (H4A3), LAMP2 (H4B4), and LAMP3 (H5C6) (Developmental Studies Hybridoma Bank (Iowa City, Iowa) (1:300), rabbit polyclonal antiserum to the HPS1 protein (Dell’Angelica et al, 2000) (1:300), rabbit polyclonal antisera to β3A and μ3A provided by M. S. Robinson (Cambridge Institute for Medical Research, Cambridge, Great Britain) (both 1:300), rabbit polyclonal antiserum to Rab27 (610595; BD Transduction Laboratories, Lexington, Kentucky) (1:300), rabbit polyclonal antiserum to transferrin (0061; Dako, Santa Barbara, CA) (1:300), and mouse monoclonal antiserum to c-Kit (CD117; Novocastra Laboratories, Newcastle upon Tyne, UK) (1:50). Cells were then washed and incubated in secondary antibody solution diluted in 0.2% Saponin in PBS containing 0.1% BSA for 60 min at room temperature. The secondary antibodies consisted of Cy-2-conjugated donkey anti-mouse and Cy-3-conjugated donkey anti-rabbit (Jackson Immunoresearch, West Grove, Pennsylvania). After washing, coverslips were applied to the slides using Fluoromount G (Southern Biotechnologies, Birmingham, Alabama) and observed and digitally photographed with either a Leitz (Rockleigh, New Jersey) Dialu Fluorescent microscopy or a Zeiss (Thornwood, New York) LSM 510 confocal microscope.
DOPA histochemistry and electron microscopy
Established cultures of HPS-1 melanocytes, maintained in regular MCDB-153 growth medium, were seeded into Lab-Tek chamber slides (Nunc) coated with 1% pig gelatin, and grown to 90% confluence. Cultured melanocytes were fixed in wells with half-strength Karnovsky’s fixative (Karnovsky, 1965) in 0.2 M sodium cacodylate buffer at pH 7.2 for 30 min at room temperature. For DOPA histochemistry, fixed cells were incubated in a 0.1% solution of L-DOPA twice for 2.5 h. The cells were washed three times in buffer and treated with 1.0% osmium tetroxide containing 1.5% potassium ferrocyanide (Karnovsky, 1971) for 30 min. The cells were washed, stained en bloc with 0.5% uranyl acetate for 30 min, dehydrated, and embedded in Eponate 12. Areas of the Epon cast were cut out and mounted on Epon pegs and sectioned on an RMC MT 6000-XL ultramicrotome. Ultrathin sections were then stained with aqueous solutions of uranyl acetate (2%) and lead citrate (0.3%) for 15 min each and then viewed and digitally photographed in a ZEOL 1230 transmission electron microscope. (All tissue-processing supplies were purchased from Ted Pella, Tustin, California.)
Western blot analysis
Total cellular proteins, from an NHM line and a line of melanocytes derived from a patient with HPS-1 (homozygous for a 16-base pair duplication in HPS1), were extracted using RIPA buffer. Equal amounts of protein were fractionated on a 10% sodium dodecyl sulfate polyacrylamide gel. The proteins were transferred to a nitrocellulose membrane and incubated with 10% non-fat dry milk in PBS and Tween-20 (PBST, pH 7.4 with 0.2% Tween-20) for 1 h at room temperature. The membrane was probed with the rabbit polyclonal antiserum to the HPS1 protein (Dell’Angelica et al, 2000) (1:1000) or a monoclonal antibody to actin (Santa Cruz Biotechnology, Santa Cruz, California) (1:500). Primary antiserum/antibody was diluted in 2% nonfat milk/PBST and incubated at 4°C for 3 h. The bands of interest were visualized by indirect immuno-enzymatic staining using an alkaline phosphatase-labeled secondary antiserum followed by BCIP/NBT substrate (Kirkegaard and Perry, Gaithersburg, Maryland).
Footnotes
This work was supported in part by Grant 5 R01 AR45429 from the National Institutes of Health (to R. E. B.). The authors thank Dr Prashiela Manga for critical review of the manuscript and Ms Joan Griggs for assistance in preparation of the manuscript.
References
- Anderson PD, Huizing M, Claassen DA, White J, Gahl WA. Hermansky–Pudlak syndrome type 4 (HPS-4): Clinical and molecular characteristics. Hum Genet. 2003;113:10–17. doi: 10.1007/s00439-003-0933-5. [DOI] [PubMed] [Google Scholar]
- Anikster Y, Huizing M, White J, Bale S, Gahl WA, Toro J. Mutation of a new gene causes a unique form of Hermansky–Pudlak syndrome in genetic isolate of central Puerto Rico. Nat Genet. 2001;28:376–380. doi: 10.1038/ng576. [DOI] [PubMed] [Google Scholar]
- Bednar B, Hermansky F, Lojda Z. Vascular pseudohemophilia associated with ceroid pigmentophagia in albinos. Am J Pathol. 1964;45:283–294. [PMC free article] [PubMed] [Google Scholar]
- Berson JF, Harper DC, Tenza D, Raposo G, Marks MS. Pmel17 initiates premelanosome morphogenesis within multivesicular bodies. Mol Biol Cell. 2001;12:3451–3464. doi: 10.1091/mbc.12.11.3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson JF, Theos AC, Harper DC, Tenza D, Raposo G, Marks MS. Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J Cell Biol. 2003;161:521–533. doi: 10.1083/jcb.200302072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissy RE, Richmond B, Huizing M, Helip-Wooley A, Zhao Y, Koshoffer A, Gahl WA. Melanocyte specific proteins are aberrantly trafficked in melanocytes of Hermansky–Pudlak syndrome type 3. Am J Pathol in press. 2004 doi: 10.1016/S0002-9440(10)62247-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissy RE, Zhao Y, Gahl WA. Altered protein localization in melanocytes from Hermansky–Pudlak syndrome: Support for the role of the HPS gene product in intracellular trafficking. Lab Invest. 1998;78:1037–1048. [PubMed] [Google Scholar]
- Bonifacino JS, Glick BS. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- Brantly M, Avila NA, Shotelersuk V, Lucero C, Huizing M, Gahl WA. Pulmonary function and high-resolution CT findings in patients with an inherited form of pulmonary fibrosis, Hermansky–Pudlak syndrome, due to mutations in HPS-1. Chest. 2000;117:129–136. doi: 10.1378/chest.117.1.129. [DOI] [PubMed] [Google Scholar]
- Chiang PW, Oiso N, Gautam R, Suzuki T, Swank RT, Spritz RA. The Hermansky–Pudlak Syndrome 1 (HPS1) and HPS4 proteins are components of two complexes, BLOC-3 and BLOC-4, involved in the biogenesis of lysosome-related organelles. J Biol Chem. 2003;278:20332–20337. doi: 10.1074/jbc.M300090200. [DOI] [PubMed] [Google Scholar]
- Ciciotte SL, Gwynn B, Moriyama K, Huizing M, Gahl WA, Bonifacino JS, Peters LL. Cappuccino, a mouse model of Hermansky–Pudlak syndrome, encodes a novel protein that is part of the pallidin-muted complex (BLOC-1) Blood. 2003;101:4402–4407. doi: 10.1182/blood-2003-01-0020. [DOI] [PubMed] [Google Scholar]
- Dell’Angelica EC, Aguilar RC, Wolins N, Hazelwood S, Gahl WA, Bonifacino JS. Molecular characterization of the protein encoded by the Hermansky–Pudlak syndrome Type 1 gene. J Biol Chem. 2000;275:1300–1306. doi: 10.1074/jbc.275.2.1300. [DOI] [PubMed] [Google Scholar]
- Dell’Angelica EC, Klumperman J, Stoorvogel W, Bonifacino JS. Association of the AP-3 adaptor complex with clathrin. Science. 1998;280:431–434. doi: 10.1126/science.280.5362.431. [DOI] [PubMed] [Google Scholar]
- Dell’Angelica EC, Ooi CE, Bonifacino JS. β3A-adaptin, a subunit of the adaptor-like complex AP-3. J Biol Chem. 1997;272:15078–15084. doi: 10.1074/jbc.272.24.15078. [DOI] [PubMed] [Google Scholar]
- Dell’Angelica EC, Shotelersuk V, Aguilar RC, Gahl WA, Bonifacino JS. Altered trafficking of lysosomal proteins in Hermansky–Pudlak syndrome due to mutations in the β3A subunit of the AP-3 adaptor. Mol Cell. 1999;3:11–21. doi: 10.1016/s1097-2765(00)80170-7. [DOI] [PubMed] [Google Scholar]
- Di Pietro SM, Falcon-Perez JM, Dell’Angelica EC. Characterization of BLOC-2, a Complex Containing the Hermansky–Pudlak Syndrome Proteins HPS3, HPS5 and HPS6. Traffic. 2004;5:276–283. doi: 10.1111/j.1600-0854.2004.0171.x. [DOI] [PubMed] [Google Scholar]
- Dunn WA., Jr Studies on the mechanisms of autophagy: Maturation of the autophagic vacuole. J Cell Biol. 1990;110:1935–1945. doi: 10.1083/jcb.110.6.1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falcon-Perez JM, Starcevic M, Gautam R, Dell’Angelica EC. BLOC-1, a novel complex containing the pallidin and muted proteins involved in the biogenesis of melanosomes and platelet-dense granules. J Biol Chem. 2002;277:28191–28199. doi: 10.1074/jbc.M204011200. [DOI] [PubMed] [Google Scholar]
- Francis E, Wang N, Parag H, Halaban R, Hebert DN. Tyrosinase maturation and oligomerization in the endoplasmic reticulum require a melanocyte-specific factor. J Biol Chem. 2003;278:25607–25617. doi: 10.1074/jbc.M303411200. [DOI] [PubMed] [Google Scholar]
- Gahl WA, Brantly M, Kaiser-Kupfer MI, et al. Genetic defects and clinical characteristics of patients with a form of oculocutaneous albinism (Hermansky–Pudlak syndrome) N Engl J Med. 1998;338:1258–1264. doi: 10.1056/NEJM199804303381803. [DOI] [PubMed] [Google Scholar]
- Gautam R, Chintala S, Li W, et al. The Hermansky–Pudlak syndrome 3 (cocoa) protein is a component of the biogenesis of lysosome-related organelles complex-2 (BLOC-2) J Biol Chem. 2004;279:12935–12942. doi: 10.1074/jbc.M311311200. [DOI] [PubMed] [Google Scholar]
- Halaban R, Alfano FD. Selective elimination of fibroblasts from cultures of normal human melanocytes. In Vitro Cell Dev Biol. 1984;20:447–450. doi: 10.1007/BF02619590. [DOI] [PubMed] [Google Scholar]
- Harmon KR, Witkop CJ, White JG, et al. Pathogenesis of pulmonary fibrosis: Platelet-derived growth factor precedes structural alterations in the Hermansky–Pudlak syndrome. J Lab Clin Med. 1994;123:617–627. [PubMed] [Google Scholar]
- Helip-Wooley A, Westbroek W, Dorward H, Mommaas M, Boissy R, Gahl WA, Huizing M. Hermansky Pudlak syndrome type 3 protein interacts with clathrin and functions in trafficking of lysosome-related organelles. Mol Biol Cell in press. 2004 [Google Scholar]
- Hermansky F, Pudlak P. Albinism associated with hemorrhagic diathesis and unusual pigmented reticular cells in the bone marrow: Report of two cases with histochemical studies. Blood. 1959;14:162–169. [PubMed] [Google Scholar]
- Honing S, Sandoval IV, von Figura K. A di-leucine-based motif in the cytoplasmic tail of LIMP-II and tyrosinase mediates selective binding of AP-3. EMBO J. 1998;17:1304–1314. doi: 10.1093/emboj/17.5.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizing M, Anikster Y, Fitzpatrick DL, et al. Hermansky–Pudlak syndrome type 3 in Ashkenazi Jews and other non-Puerto Rican patients with hypopigmentation and platelet storage pool deficiency. Am J Hum Genet. 2001a;69:1022–1032. doi: 10.1086/324168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huizing M, Boissy RE, Gahl WA. Hermansky–Pudlak syndrome: Vesicle formation from yeast to man. Pigment Cell Res. 2002;15:405–419. doi: 10.1034/j.1600-0749.2002.02074.x. [DOI] [PubMed] [Google Scholar]
- Huizing M, Sarangarajan R, Strovel E, Zhao Y, Gahl WA, Boissy RE. AP-3 mediates tyrosinase but not TRP-1 trafficking in human melanocytes. Mol Biol Cell. 2001b;12:2075–2085. doi: 10.1091/mbc.12.7.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi T, Gomi H, Kasai K, Mizutani S, Torii S. The roles of Rab27 and its effectors in the regulated secretory pathways. Cell Struct Funct. 2003;28:465–474. doi: 10.1247/csf.28.465. [DOI] [PubMed] [Google Scholar]
- Jimenez-Cervantes C, Solano F, Kobayashi T, Urabe K, Hearing VJ, Lozano JA, Garcia-Borron JC. A new enzymatic function in the melanogenic pathway: The DHICA oxidase activity of tyrosinase related protein-1 (TRP1) J Biol Chem. 1994;269:17993–18001. [PubMed] [Google Scholar]
- Karnovsky MJ. A formaldehyde–glutaraldehyde fixative of high osmolality for use in electron microscopy. J Cell Biol. 1965;27137 (abstr) [Google Scholar]
- Karnovsky MJ. Use of ferrocyanide-reduced osmium tetroxide in electron microscopy. J Cell Biol. 1971;51146 (abstr) [Google Scholar]
- Kegel KB, Kim M, Sapp E, McIntyre C, Castano JG, Aronin N, DiFiglia M. Huntingtin expression stimulates endosomal–lysosomal activity, endosome tubulation, and autophagy. J Neurosci. 2000;20:7268–7278. doi: 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RA, Hearing VJ, Creel DJ, Oetting WS. Albinism. In: Scriver CR, Beaudet AL, Valle DL, Sly WS, editors. The Metabolic and Molecular Bases of Inherited Disease. 8th edn. Vol 4. New York: McGraw-Hill; 2001. pp. 5587–5627. [Google Scholar]
- Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhang Q, Oiso N, et al. Hermansky–Pudlak syndrome type 7 (HPS-7) results from mutant dysbindin, a member of the biogenesis of lysosome-related organelles complex 1 (BLOC-1) Nat Genet. 2003;35:84–89. doi: 10.1038/ng1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marks MS, Seabra MC. The melanosome: Membrane dynamics in black and white. Nat Rev Mol Cell Biol. 2001;2:738–748. doi: 10.1038/35096009. [DOI] [PubMed] [Google Scholar]
- Martina JA, Moriyama K, Bonifacino JS. BLOC-3, a protein complex containing the Hermansky–Pudlak syndrome gene products HPS1 and HPS4. J Biol Chem. 2003;278:29376–29384. doi: 10.1074/jbc.M301294200. [DOI] [PubMed] [Google Scholar]
- Maul GG. Golgi–melanosome relationship in human melanoma in vitro. J Ultrastruct Res. 1969;26:163–176. doi: 10.1016/s0022-5320(69)90042-2. [DOI] [PubMed] [Google Scholar]
- Maul GG, Brumbaugh JA. On the possible function of coated vesicles in melanogenesis of the regenerating fowl feather. J Cell Biol. 1971;48:41–48. doi: 10.1083/jcb.48.1.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medrano EE, Nordlund JJ. Successful culture of adult human melanocytes from normal and vitiligo donors. J Invest Dermatol. 1990;95:441–445. [PubMed] [Google Scholar]
- Nimmerjahn F, Milosevic S, Behrends U, Jaffee EM, Pardoll DM, Bornkamm GW, Mautner J. Major histocompatibility complex class II-restricted presentation of a cytosolic antigen by autophagy. Eur J Immunol. 2003;33:1250–1259. doi: 10.1002/eji.200323730. [DOI] [PubMed] [Google Scholar]
- Novikoff A, Albala A, Biempica L. Ultrastructural and cytochemical observations on B16 and Harding-Passey melanomas. The origin of premelanosomes and compound melanosomes. J Histochem Cytochem. 1968;16:299–319. doi: 10.1177/16.5.299. [DOI] [PubMed] [Google Scholar]
- Oh J, Bailin T, Fukai K, et al. Positional cloning of a gene for Hermansky–Pudlak syndrome, a disorder of cytoplasmic organelles. Nat Genet. 1996;14:300–306. doi: 10.1038/ng1196-300. [DOI] [PubMed] [Google Scholar]
- Oh J, Liu Z-X, Feng GH, Raposo G, Spritz RA. The Hermansky–Pudlak syndrome (HPS) protein is part of a high molecular weight complex involved in biogenesis of early melanosomes. Hum Mol Genet. 2000;9:375–385. doi: 10.1093/hmg/9.3.375. [DOI] [PubMed] [Google Scholar]
- Orlow SJ. The biogenesis of melanosomes. In: Nordlund JJ, Boissy RE, Hearing VJ, King RA, Ortonne J-P, editors. The Pigmentary System, Physiology and Pathophysiology. New York: Oxford University Press; 1998. pp. 97–106. [Google Scholar]
- Orlow SJ, Boissy RE, Moran DJ, Pifko-Hirst S. Subcellular distribution of tyrosinase and tyrosinase-related protein-1: Implications for melanosomal biogenesis. J Invest Dermatol. 1993;100:55–64. doi: 10.1111/1523-1747.ep12354138. [DOI] [PubMed] [Google Scholar]
- Raposo R, Marks MS. The dark side of lysosome-related organelles: Specialization of the endocytic pathway for melanosome biogenesis. Traffic. 2002;3:237–248. doi: 10.1034/j.1600-0854.2002.030401.x. [DOI] [PubMed] [Google Scholar]
- Robinson MS, Bonifacino JS. Adaptor-related proteins. Curr Opin Cell Biol. 2001;13:444–453. doi: 10.1016/s0955-0674(00)00235-0. [DOI] [PubMed] [Google Scholar]
- Schinella RA, Greco MA, Cobert BL, Denmark LW, Cox RP. Hermansky–Pudlak syndrome with granulomatous colitis. Ann Intern Med. 1980;92:20–23. doi: 10.7326/0003-4819-92-1-20. [DOI] [PubMed] [Google Scholar]
- Seglen PO, Bohley P. Autophagy and other vacuolar protein degradation mechanisms. Experientia. 1992;48:158–172. doi: 10.1007/BF01923509. [DOI] [PubMed] [Google Scholar]
- Strom M, Hume AN, Tarafder AK, Barkagianni E, Seabra MC. A family of Rab27-binding proteins. Melanophilin links Rab27a and myosin Va function in melanosome transport. J Biol Chem. 2002;277:25423–25430. doi: 10.1074/jbc.M202574200. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Li W, Zhang Q, et al. The gene mutated in cocoa mice, carrying a defect of organelle biogenesis, is a homologue of the human Hermansky–Pudlak syndrome-3 gene. Genomics. 2001;78:30–37. doi: 10.1006/geno.2001.6644. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Li W, Zhang Q, et al. Hermansky–Pudlak syndrome is caused by mutations in HPS4, the human homolog of the mouse light-ear gene. Nat Genet. 2002;30:321–324. doi: 10.1038/ng835. [DOI] [PubMed] [Google Scholar]
- Swank RT, Novak EK, McGarry MP, Rusiniak ME, Feng L. Mouse models of Hermansky–Pudlak syndrome: A review. Pigment Cell Res. 1998;11:60–80. doi: 10.1111/j.1600-0749.1998.tb00713.x. [DOI] [PubMed] [Google Scholar]
- Tsukamoto K, Jackson IJ, Urabe K, Montague P, Hearing VJ. A second tyrosinase related protein, TRP-2, is a melanogenic enzyme termed dopachrome tautomerase. EMBO J. 1992;11:519–526. doi: 10.1002/j.1460-2075.1992.tb05082.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner WA, Jr, Taylor JD, Tchen TT. The role of multivesicular bodies in melanosome formation. Pigment Cell. 1976;3:33–45. doi: 10.1016/s0022-5320(75)80004-9. [DOI] [PubMed] [Google Scholar]
- Ujvari A, Aron R, Eisenhaure T, et al. Translation rate of human tyrosinase determines its N-linked glycosylation level. J Biol Chem. 2001;276:5924–5931. doi: 10.1074/jbc.M009203200. [DOI] [PubMed] [Google Scholar]
- Watabe H, Valencia JC, Yasumoto K, et al. Regulation of tyrosinase processing and trafficking by organellar pH and by proteasome activity. J Biol Chem. 2004;279:7971–7981. doi: 10.1074/jbc.M309714200. [DOI] [PubMed] [Google Scholar]
- Wildenberg SC, Oetting WS, Almod¢var C, Krumwiede M, White JG, King RA. A gene causing Hermansky–Pudlak syndrome in a Puerto Rican population maps to chromosome 10q2. Am J Hum Genet. 1995;57:755–765. [PMC free article] [PubMed] [Google Scholar]
- Witkop CJJ, Krumwiede M, Sedano H, White JG. Reliability of absent platelet dense bodies as a diagnostic criterion for Hermansky–Pudlak syndrome. Am J Hematol. 1987;26:305–311. doi: 10.1002/ajh.2830260403. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Zhao B, Li W, et al. Ru2 and Ru encode mouse orthologs of the genes mutated in human Hermansky–Pudlak syndrome types 5 and 6. Nat Genet. 2003;33:145–153. doi: 10.1038/ng1087. [DOI] [PubMed] [Google Scholar]