

Abstract
To investigate how angiotensin-I converting enzyme (ACE) inhibitors enhance the actions of bradykinin (BK) on B2 receptors independent of blocking BK inactivation, we expressed human somatic ACE and B2 receptors in CHO cells. Bradykinin and its ACE-resistant analogue were the receptor agonists. B2 fused with green fluorescent protein (GFP) and ACE were coprecipitated with antisera to GFP or ACE shown in Western blots. Immunohistochemistry of fixed cells localized ACE by red color and B2-GFP by green. Yellow color on plasma membranes of coexpressing cells also indicated enzyme-receptor complex formation. Using ACE-fused cyan fluorescent protein donor and B2-fused yellow fluorescent protein acceptor, we registered fluorescence resonance energy transfer (FRET) by the enhanced fluorescence of donor upon acceptor photo-bleaching, establishing close (within 10 nm) positions of B2 receptors and ACE. Bradykinin stimulation cointernalized ACE and B2 receptors. We expressed ACE fused to N-terminus of B2 receptors, anchoring only receptors to plasma membranes. Here, in contrast to cells where both ACE and B2 receptors are separately anchored, ACE inhibitors neither enhance activation of chimeric B2, nor resensitize desensitized B2 receptors. Heterodimer formation between ACE and B2 receptors can be a mechanism for ACE inhibitors to augment kinin activity at cellular level.
Keywords: Oligomer, FRET, converting enzyme, peptide receptors, kinins
INTRODUCTION
Historically, the kallikrein-kinin and the renin-angiotensin systems were investigated independently (1–4). A connecting link was revealed when kininase II was found to be identical with angiotensin converting enzyme (ACE), and that by cleaving a C-terminal dipeptide, a single enzyme released angiotensin (Ang) II and inactivated bradykinin (5, 6). These two peptide substrates of ACE, Ang I and bradykinin, frequently have opposing actions on the cardiovascular system. Millions of patients receive ACE inhibitors to treat conditions such as congestive heart failure or diabetic nephropathy, besides hypertension (7–12). The beneficial effects of ACE inhibition on the heart and blood vessels are at least partly mediated by kinins acting via bradykinin B2 receptors, because ACE inhibition reduces kinin degradation (13, 14), but ACE inhibitors have other favorable actions as well. Experiments on isolated organs and cultured cells showed that ACE inhibitors also potentiate bradykinin indirectly at the receptor level (15–17).
In order to explore further how this potentiation occurs, we transfected Chinese hamster ovary (CHO) cells with cDNA of human ACE and B2 receptors. We then used these cells along with cells that express ACE and the B2 receptor as native proteins to examine the mechanism of ACE inhibitors. We found that ACE inhibitors augment the release of signal transduction products by bradykinin not only by inhibiting the degradation of bradykinin, but by indirectly affecting B2 receptors (15–18).
Tachyphylaxis, or desensitization of bradykinin receptors by an agonist, is a well- known phenomenon. Resensitization restores the potential for the receptors to recouple G-proteins to initiate signal transduction. On the basis of experiments, done in part with ACE-resistant bradykinin analogs, we suggested that potentiation of bradykinin and resensitization of its receptors result from crosstalk between ACE and the B2 receptors on the cell membrane. ACE and the hepta-helical G-protein-linked bradykinin receptors can colocalize, and, in so doing, they may form heterodimers (17).
We wanted to establish the proximity of transfected human ACE and bradykinin B2 receptors on the plasma membrane and to determine whether the distance between the enzyme and receptors allows allosteric enhancement of receptor activity (18–20). We transfected CHO cells with cDNA of human ACE and B2 receptors, where the constructs were coupled to green (GFP), monomer red (mRFP), yellow (YFP) or cyan (CFP) fluorescent proteins. We then followed the association of bradykinin B2 receptors and ACE by various techniques, including immunoprecipitation, immunostaining, fluorescence confocal microscopy, fluorescence resonance energy transfer (FRET) measurements and time-lapse photography of live cells. This allowed us to determine how the receptors and ACE associate on the surface of transfected cells and how they respond to bradykinin, an ACE-resistant bradykinin analogue and an ACE inhibitor. For a negative control, we fused ACE to B2 receptors and expressed them as a single protein molecule in CHO cells. Results of our experiments established the association of B2 receptors and ACE on the plasma membrane and indicate a mode of action of ACE inhibitors that may contribute to their successful therapeutic effects (7–11).
METHODS
Materials
CHO cells were obtained from the American Type Culture Collection (Rockville, MD). The cDNA of human ACE and the cDNA encoding human B2 receptors were from Prof. P. Corvol (College de France, Paris) and from Syntex Co (Palo Alto, CA). Mammalian expression vectors pcDNA3 and pcDNA6, geneticin and blasticidin S HCl were purchased from Invitrogen (Carlsbad, CA). Fetal bovine serum was from Atlanta Biologicals (Norcross, GA). [5,6,8,9,11,12,14,15-3H(N)] arachidonic acid ([3H]AA; 250 Ci/mmol) was from American Radiolabeled Chemicals (St. Louis, MO). [prolyl2,3-3,4(n)-3H] bradykinin ([3H]BK, 71Ci/mmol) was from Amersham Biosciences (Piscataway, NJ). Bradykinin, NP-40 detergent, penicillin, the alkaline phosphatase detection kits and other chemicals were purchased from Sigma Chemical Co (St. Louis, MO). Enalaprilat (EPT) was provided by Merck, Sharpe & Dohme Research Division (Whitehouse, NJ). The ACE resistant bradykinin analog (BKan; Arg-Pro-Pro-Gly-Phe-Ser-Pro-Pheψ[CH2NH]Arg) was from EMD Biosciences (La Jolla, CA), Hippuryl-His-Leu (Hip-His-Leu) was from Bachem (Philadelphia, PA). Alexa 568 conjugate and anti-GFP serum were from Molecular Probes (Eugene, Oregon). The vectors for GFP, CFP, and YFP were obtained from Clonetech (Palo, Alto, CA). The cDNA coding for monomer red fluorescent protein (mRFP) was a gift from Dr. Roger Tsien, University of California, San Diego. Rabbit antiserum to human ACE was elicited with ACE purified in this laboratory (18).
Methods
Construction of fluorescent protein expression vectors
B2-pEGFP-N1
The Eco RI-Sac I (1–721) fragment of human bradykinin B2 receptor cDNA was ligated with a PCR amplified Sac I-Bam HI fragment (721–1338) of B2 cDNA into Eco RI-Bam HI sites of pEGFP-N1 (Clontech, Palo Alto, CA). In PCR, through the down stream primer design (5′-CAGGATCCTGTCTGCTCCCTGCCCAGTC-3′), the translation stop codon of B2 was removed, a Bam HI cleavage site was added and the reading frame of B2 cDNA was fused into EGFP.
B2-EYFP-pcDNA6
Human B2 receptor cDNA with a C-terminal EYFP tag was constructed by introducing an EYFP fragment (661–1402) into the Bam HI-Not I sites of B2-pcDNA6 V5-His, which contains a blasticidin selection marker.
ACE-ECFP-N1
We used high fidelity PCR to modify the C-terminus of the two active domains of human ACE cDNA. We eliminated its stop codon and created a BamH I cleavage site to fuse the full-length ACE reading frame with CFP in the vector. Then we ligated an ACE Eco RI-Not I fragment (1–3821 bp) with its C- terminal PCR Not I-Bam HI fragment into the Eco RI-Bam HI sites of pECFP-NI (Clontech, Palo Alto, CA).
B2-mRFP-N1
The mRFP –N1 vector was constructed by replacing CFP with mRFP. A Bam HI-Hind III fragment of mRFP from the mRFP-pRSETB (From Dr. Roger Tsien of the University of California at San Diego) was cloned into the Bam HI/Eco RV sites of pCDNA3 after its Hind III site was filled in with a DNA polymerase I large fragment. The Bam HI-Not I fragment from mRFP-pCDNA3 was used to replace CFP, and the B2 cDNA was then inserted into this vector.
Transfection and selection of CHO/ACE+B2-GFP cells
The cDNA of B2-GFP was transfected into CHO-K1 cells, and the appropriate clones were selected in HAM’s F-12 medium supplemented with 0.5 mg/ml geneticin. Cells were grown to 60–80% confluency prior to transfection. Cells that highly expressed GFP were separated by fluorescence-activated cell sorting with an Elite ESP cell sorter (Coulter Corp.). Binding of [3H]-bradykinin established the functionality of the B2-GFP. Transfection of cDNA of ACE into CHO/B2-GFP cells was done as described above, and clones with highest ACE activity were selected for further study.
Construction of ACE-B2 receptor fusion protein (ACE-B2-pcDNA6)
We used a Hind III – Not I fragment of ACE cDNA (1–3741) that encoded both N- and C- domains and part of the stem region of ACE along with a PCR-amplified full-length human bradykinin B2 receptor cDNA to construct the receptor fusion protein. The B2 receptor cDNA contained a 5’end Not I cleavage site for fusing its reading frame to ACE. Both cDNA fragments were ligated together into the Hind III-Bam HI sites of pcDNA6.
Transfection and selection of CHO/ACE-B2 cells
We used purified ACE-B2 pcDNA6 DNA to transfect CHO cells. Screening for enzyme activity and the B2 receptor response to agonist allowed selection of a stable clone that expressed the human ACE-B2 receptor fusion protein. Western blot analysis of cell lysates with rabbit polyclonal antiserum to ACE detected an ACE-B2 fusion protein of approximately 240 kDa.
ACE Activity
Cells were scraped from culture dishes and centrifuged at 1,000 x g for 10 min at 4°C. The pellet was suspended in 0.5 ml 200 mM HEPES buffer (pH 7.4) containing 300 mM NaCl and 5 μl protease inhibitor cocktail, without sequestering agents and sonicated three times for 15 sec each. The samples were then centrifuged at 100,000 g for 30 min at 4°C. The supernatant (S1) was removed, the re-suspended pellets were solubilized in 0.5 ml HEPES buffer containing 0.1% NP-40 detergent and 10 μl protease inhibitor cocktail, and centrifuged again at 100,000 g for 30 min at 4°C. The final supernatant (S2) was saved for assay of enzyme activity. Samples were incubated at 37°C with 1 mM Hip-His-Leu substrate in 100 mM HEPES, 150 mM NaCl, (pH 7.4). The reaction was stopped with 0.28 M NaOH, and the liberated His-Leu was detected by addition of 100 μl of 20mg/ml o-phthaldiadehyde for 10 min at room temperature. The reaction was stopped with 200 μl 3 M HCl, and fluorescence was measured following excitation at an emission wavelength of 500 nm following excitation at 363 nm (18).
Arachidonic acid ([3H]AA) release
Stimulation of B2 receptors activates phospholipase A2 followed by AA release from cells (15). [3H]AA (250Ci/mmol) was diluted to 1 μCi/ml in HAM’s F-12 cell culture medium containing 0.5% fetal bovine serum and added to CHO cell monolayers (60–90% confluent). After 18–24 h of incubation, the loaded cells were washed with medium containing 0.1% fat-free bovine serum albumin. For potentiation experiments, the cells were treated first with an ACE inhibitor or a B2 receptor antagonist as a control for 10 min at room temperature, then bradykinin or related agonists were added, and the incubation continued for 30 min longer at 37°C. For resensitization experiments, the cells were first desensitized by application of a high concentration of agonist for 30 min at 37°C, then they were treated with either medium alone, a second dose of agonist, or the ACE inhibitor enalaprilat without removing the medium containing the original agonist. Samples were drawn after an additional 5 min incubation at 37°C, and radioactivity was measured. Results from these experiments showed that the receptors were desensitized by bradykinin, but addition of an ACE inhibitor restored the receptor sensitivity to agonist in the medium (18).
Localization of ACE and B2 receptors in unstimulated CHO/ACE+B2-GFP cells
We used immunohistochemistry to establish the localization of ACE and B2 receptors in cultured cells. Near-confluent monolayers of CHO/ACE+B2-GFP cells were grown on coverslips. We then removed the medium from the wells and fixed the cells for 20 min at room temperature with 0.8 ml 4% paraformaldehyde in phosphate buffered saline (PBS) containing 0.12 M sucrose. The fixed cells were washed 3 times, 5 min each with 100 mM glycine in PBS and then with PBS alone for another 3 times with gentle shaking. Non-specific binding was blocked by incubation for 1 h with 5% normal goat serum in PBS containing 0.2% bovine serum albumin and 0.1% Triton X-100. Then the cells were incubated at 4°C overnight with 0. 5 ml of 1:8,000 v/v polyclonal anti-ACE in 2% normal goat serum in PBS and 0.1% Triton X-100. After the incubation with the primary antibody, the cells were washed 3 times with PBS containing 0.1% Triton X-100 and blocked once again for 1 h. Cells were then incubated for 1 h at room temperature with 0.5 ml 1:1,500 v/v goat anti-rabbit secondary antibody (Alexa 568 conjugate) in PBS containing 2% normal goat serum, 1% BSA and 0.1% Triton X-100. Finally, the coverslips were washed 3 times with PBS containing 0.1% Triton X-100, rinsed briefly with distilled water and mounted on glass slides. Fluorescent images were obtained following excitation at wavelengths of 458 nm for GFP and 568 nm for Alexa 568 using a Zeiss LSM 510 confocal microscope. Optical sections (0.5 μm; 16 frame averages) were taken on the vertical (orthogonal, or Z) axis perpendicular to the plane of the cells.
Coimmunoprecipitation of B2-GFP and ACE
We added 3 μl rabbit polyclonal anti-GFP antibodies or 2 μl antiserum to human ACE to the final cell lysates (S2). Each tube contained 400 μl of 100 mM HEPES (pH 7.4) containing 0.5% NP-40, 150 mM NaCl and 5 μl protease inhibitors; we then added one of the following: wild type-CHO (30 μg protein), CHO/ACE (165 μg), CHO/B2-GFP (30 μg), or CHO/ACE+B2-GFP (30 μg). The samples were kept overnight at 4°C, then rotated for 2 h at 4°C with protein A beads (15 μl). The beads were washed 8 times with 100 mM HEPES buffer (pH 7.4) containing 0.5% NP-40 and 150 mM NaCl. The proteins were eluted with Laemmli buffer and separated on 10% SDS-PAGE. The proteins were then electro-transferred to nitrocellulose membranes and probed with anti-ACE or anti-GFP for 1h at 1:4,000 or 1:2,000 (v/v) dilution. Goat anti-rabbit antibodies coupled to alkaline phosphatase were the secondary antibodies (1:20,000) used to detect the reaction, the bands were developed with an alkaline phosphatase detection kit.
Fluorescence resonance energy transfer (FRET)
CHO cells (5×105 cells/30 mm dish) were cotransfected with cDNA of the donor CFP fusion protein (ACE-CFP) and cDNA of the acceptor YFP fusion protein (B2-YFP) using 10 μl of superfectin. Twenty-four hours after transfection, the cells were cultured with G418 (0.5 mg/mL) and blasticidin S (5 μg/ml) in HAM’s F-12 medium (10% FBS, 1% penicillin/streptomycin). The cultures were maintained for one to two weeks until no more cells died. The cells were then grown on glass cover slips and fixed with 2% para-formaldehyde in PBS with 0.12 M sucrose. The fixed cells were viewed with a Zeiss LSM-510 META confocal imaging system equipped with a 50 mW argon laser and 25 × objective. Cells that expressed either ACE-CFP or B2-YFP protein were imaged with a Zeiss META detector. An image stack was generated with the 458 nm laser line spanning an emission wavelength range from 462.9 to 602 nm with bandwidths of 10.7 nm (pinhole of 1.66 Airy units and a vertical [Z] resolution of <2.0 μm). These spectra served as CFP and YFP reference emission signatures. We used them in a linear unmixing algorithm (Zeiss AIM software) to separate the fluorescence contribution of CFP and YFP (on a pixel by pixel basis) in images of cells that coexpressed ACE-CFP and B2-YFP protein (21). Pre-bleach CFP and YFP images were collected using the argon laser with a 458 nm/514 nm dual dichroic. A selected region of interest (ROI) on the plasma membrane was irradiated with the 514 nm laser line (100% intensity) for 55 s (200 iterations) to photo-bleach YFP. Post-bleach images were captured immediately. FRET in the ROI was evidenced by an increase in CFP fluorescence intensity (donor dequenching) following YFP (acceptor) photo-bleaching (21, 22).
Movement of ACE and B2 receptor in stimulated cells
Time-lapse photographs of ACE-GFP and B2-mRFP movements in living cells were taken using confocal microscopy. CHO cells stably expressing ACE-GFP and B2-mRFP were treated with medium for 3 min and then 1 μM BKan (an ACE-resistant kinin agonist) was added. Pictures were taken every 30 sec from just prior to BKan addition (0 min) to 10 min after and then every 2 min from 10 to 46 min. In related experiments CHO cells expressing ACE-GFP and B2-mRFP were treated with medium or 1μM BKan for 1, 10, or 30 min at 37°C. Cells were fixed with 4% paraformaldehyde + 0.12M sucrose in PBS for 20 min at room temperature, washed 3 times with PBS, rinsed with distilled H2O and viewed under a confocal microscope.
Protein Assay was done as described previously (23).
Statistics
Data are expressed as mean ±SEM (n≥3). Statistical evaluation was performed by 1-way ANOVA for matched values.
RESULTS
[3H]AA release from CHO/ACE+B2-GFP cells
We wanted to determine the effects of ACE inhibitors on ACE and B2-GFP receptors that are presumably colocalized within the transfected CHO cells. Accordingly, we examined whether enalaprilat could resensitize modified B2 receptors that were first desensitized by an agonist, bradykinin (18, 19), or by an ACE-resistant analogue (BKan). This would distinguish the ACE inhibitor effects on the B2-GFP receptors from the inhibition of kinin inactivation (17).
Fig 1. shows that 1 μM enalaprilat resensitizes B2 receptors to 1 μM bradykinin (A) and 1 μM BKan (B). CHO/ACE+B2-GFP cells were first exposed to 1 μM agonists for 30 min at 37°C to desensitize B2 receptors, then peptide ligand, enalaprilat, or medium alone was added without removing the first agonist. The second application of 1 μM bradykinin or BKan released no more [3H]AA than baseline (medium alone) because the B2 receptors were desensitized by the primary application of agonists. However, adding 1 μM enalaprilat enhanced basal [3H]AA release by approximately 125% (Figure A) or 106% (Figure B) without additional peptide agonist administration, indicating resensitization of the receptors to bradykinin or Bkan remaining in the medium. Enalaprilat alone does not activate the B2 receptors, and in absence of ACE expression, the cells are not resensitized (15, 17, 18).
Figure 1. Resensitization of B2-GFP receptors to bradykinin by enalaprilat (EPT).
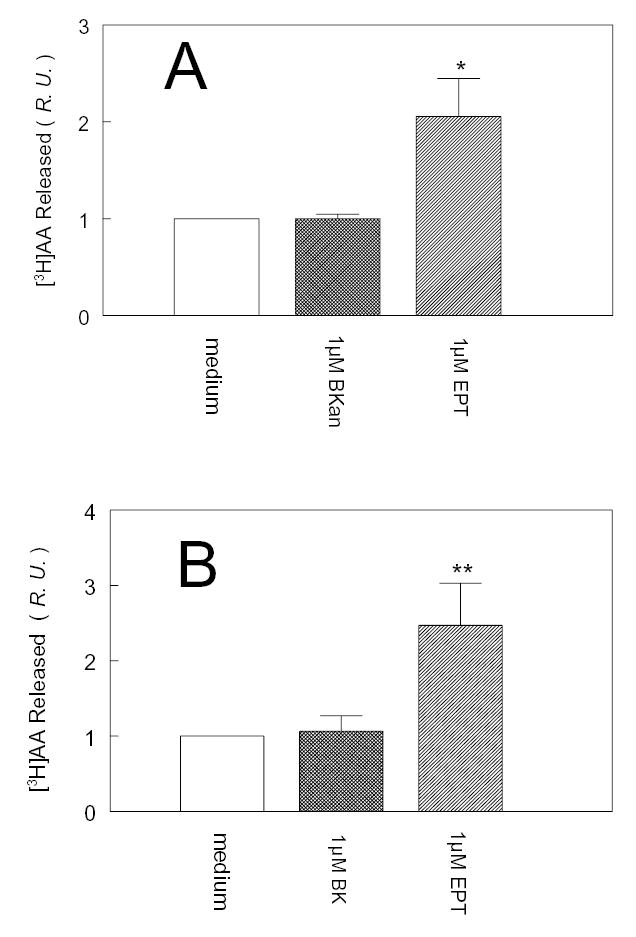
After CHO/ACE+B2-GFP cells were stimulated and desensitized with 1μM bradykinin (BK) (A) or 1μM bradykinin analogue (BKan) (B) for 30 min at 37°C, then they were treated with either medium alone, a second dose of 1μM BK (A) or 1M BKan (B), or 1μM EPT for 5 min at room temperature. Ordinate: relative amount of [3H] arachidonic acid ([3H]AA) released; Baseline = 1. Data are mean ± SEM. In (A), n=4, *, p<0.01; compared with 1μM BK treatment; in (B); n=4, **, p<0.005, compared with 1μM BKan treatment. R.U. = relative unit. The second dose of agonist added to desensitized cells in absence of EPT was inactive. EPT sensitized the B2 receptors to agonist in the medium.
Colocalization of B2-GFP and ACE: immunoprecipitation
We used immunoprecipitation to explore further the relationship between ACE and bradykinin B2 receptors. Fig. 2 shows that B2-GFP and ACE are coprecipitated from lysates of unstimulated CHO/ACE+B2-GFP cells with the appropriate antibodies. The ACE activity measured in S2 lysates of CHO/ACE cells, was approximately 2.4 μmol/h/mg protein, while in the CHO/ACE+B2-GFP cells, it was about 13 μmol/h/mg. Western blots of the two cell lysates with antibodies against purified human ACE agreed with directly measured enzyme activity and indicated greater ACE expression in CHO/ACE+B2-GFP than in CHO/ACE cells. However, Western blots of B2 (CHO/B2-GFP) and combined B2 and ACE transfected cell lines (CHO/ACE+B2-GFP) when developed with antibodies against GFP demonstrated similar B2 receptor expression.
Figure 2. Coimmunoprecipitation of B2-GFP and ACE in CHO/ACE+B2-GFP cells.
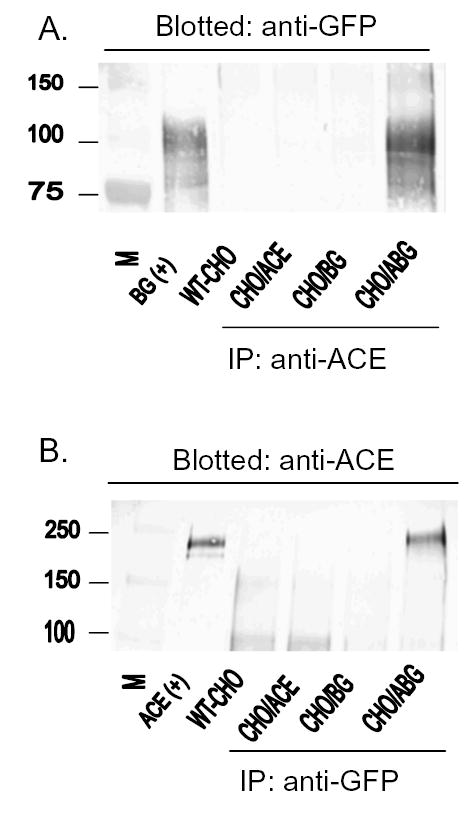
A: Immunoblotting with anti-GFP antiserum (1:2,000 v/v). Attempts for immunoprecipitates (IP) of S2 lysates from various types of cells: CHO (WT-CHO), CHO/ACE, CHO/B2-GFP (CHO/BG) and CHO/ACE+B2-GFP (CHO/ABG) cells. Antiserum to ACE precipitated the protein only from the S2 cell lysates of cells that express ACE, and anti-GFP reacted with the coprecipitate of ACE and B2-GFP in a Western blot of CHO-ABG cells. B: Proteins immunoblotted on gels with anti-ACE antiserum (1:4,000 v/v) after immunoprecipitation with anti-GFP. The antisera were applied in reverse order, but conditions were the same as in A. Note that the molecular mass of human ACE is about 180 kDa and of B2-GFP is about 100 kDa. ACE and B2 were coprecipitated with antisera either to human ACE or GFP when they were from transfected cells expressing both human ACE and B2 bradykinin receptors.
Fig. 2A shows fractions from wild-type CHO (WT-CHO), CHO/ACE, CHO/B2-GFP (CHO/BG) and CHO/ACE+B2-GFP (CHO/ABG) where the ACE- B2 receptor complex was immunoprecipitated with anti-ACE, and immunoblotted with anti-GFP antiserum (1:2,000). We detected an immunoprecipitate only in the S2 fraction of CHO/ABG cells that express both ACE and B2 receptors. Fig 2B shows the results when the same proteins were immunoprecipitated by anti-GFP and the resulting precipitate was immunoblotted with antiserum to ACE.
When the cell lysates were immunoprecipitated by anti-GFP and then blotted by anti-ACE, only CHO/ACE+B2-GFP yielded a band (right lane) corresponding to ACE of about 180 kDa (upper panel) or B2-GFP band (100 kDa) of the positive control (2B). The results were similar when immunoprecipitation and blotting was done in the reverse order. Coimmunoprecipitation of ACE and B2-GFP suggests that these two proteins form a complex in CHO cells. We found no bands in the three control cell lines, WT CHO, CHO/ACE and CHO/BG, after attempted immunoprecipitation under the same conditions.
Colocalization of ACE and B2 receptors: immunohistochemistry
Fig 3 shows the colocalization of ACE and B2-GFP in CHO/ACE+B2-GFP cells by immunohistochemistry. The green color is due to GFP, and the red to an Alexa 568- conjugated secondary antibody that recognized the primary ACE antibody. Panel A shows colocalization of B2-GFP (green, left) and ACE (red, middle), and the overlapping of both (yellow, right). Panel B shows the combined images of the cells plus the vertical (Z axis) sections of the cells in the upper figure. In the lower figure, in the enlarged vertical section, the predominant yellow color indicates that ACE and B2 receptors colocalize on the cell membrane.
Figure 3. Colocalization of ACE and B2-GFP in fixed CHO/ACE+B2-GFP cells.
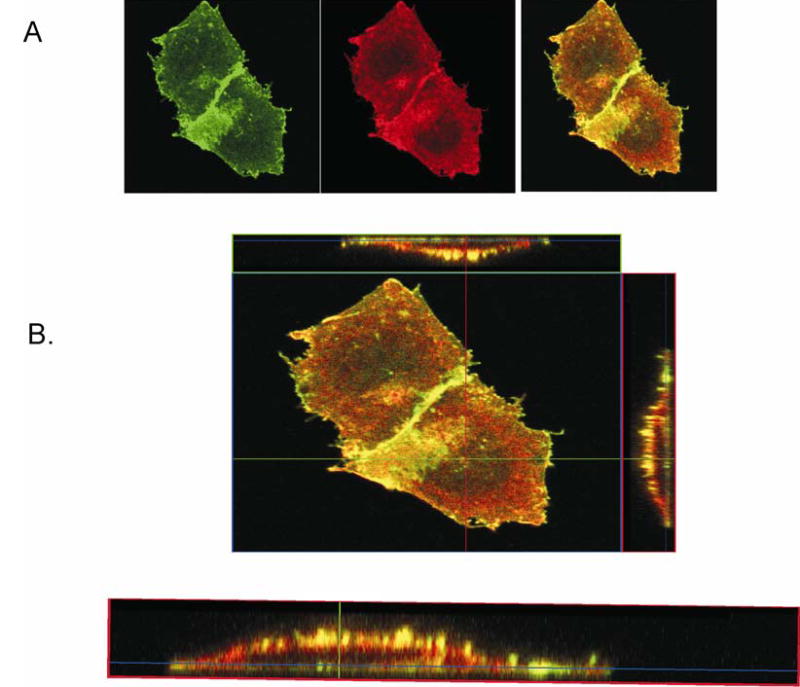
The green color is due to GFP, and the red to Alexa 568 conjugated secondary IgG that recognizes the primary antibody against ACE. A: B2-GFP (green, left) and ACE (red, middle), are colocalized (yellow, right). B, upper: Enlarged view of panel A. B, lower: cross-sectional images of cells viewed in the vertical (Z) plane showing extensive colocalization (yellow) on the cell membrane.
FRET between ACE and B2 receptors
Two μm optical slices were visualized by confocal microscopy through the middle of a CHO cell that coexpressed ACE-CFP and B2-YFP. The two fluorescent proteins, CFP and YFP were excited with an argon laser using a 458 nm/514 nm dual dichroic filter. A Zeiss LSM-510 META detector performed linear unmixing of CFP and YFP emission spectra. Fig 4 shows a representative example of 9 separate experiments. Pre-bleach images were collected (A, B, C). YFP fluorescence was photobleached by scanning with a 514 nm laser for 55 s, then post-bleach images were collected (D, E, F). The white rectangle points to the region of interest (ROI) where photo-bleaching was done. During FRET the donor molecule (CFP) is less than optimally bright, because the acceptor molecule (YFP) adsorbs some of the donor's energy. When the acceptor molecule is inactivated (bleached), the donor appears brighter, as seen in bleached regions where the blue coloring is brighter. This type of FRET measurement is less prone to artifacts caused by spillover of light from one channel to another in the microscope. The increased donor brightness following photo-bleaching of the receptor indicates energy transfer between donor and recipient (21, 22); it is taken as evidence for a close active association between the two proteins (17, 19, 24).
Figure 4. FRET between ACE and B2 receptors.
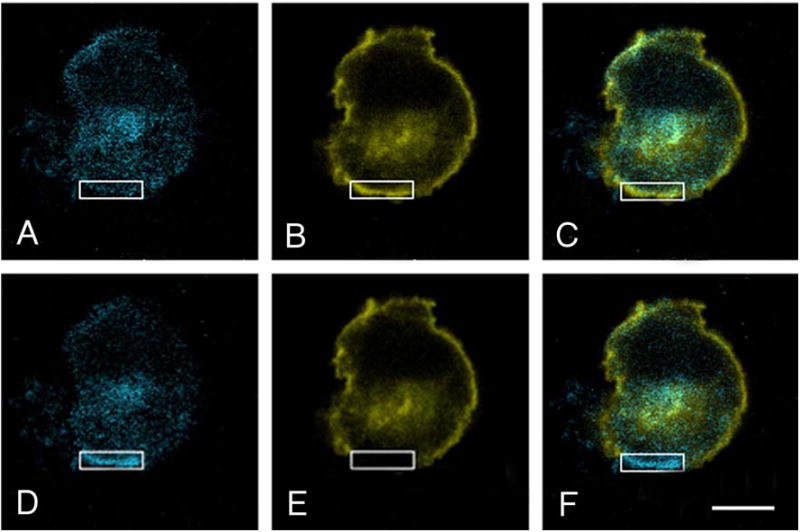
Confocal microscopy of CHO/ACE-CFP+B2-YFP cells shows a 2 μm optical slice in an x,y plane through the middle of a CHO cell coexpressing ACE-CFP and B2-YFP. Linear unmixing of CFP and YFP emission spectra was done with the Zeiss LSM-510 META detector. Pre-bleach images were collected (A, B, C; note region of interest [ROI] outlined in white; Bar = 10 μm). YFP fluorescence was photo-bleached in the ROI by scanning with a 514 nm laser, and post-bleach images were collected (D, E, F). Note increase in donor intensity (A and D) following bleaching of receptor (B and G). This representative experiment of 9 indicates energy transfer (FRET) between the donor ACE enzyme and the acceptor, B2 receptor, and it follows that ACE and B2 receptors colocalize within 10 nm.
Movement of ACE and B2 receptors by agonist stimulation
Prior to adding a B2 receptor agonist, ACE-GFP and B2-mRFP were diffusely distributed along the cell membrane (Fig 5A). Addition of 1μM BKan initiated the movement to colocalize ACE and B2 receptors into plasma membrane domains (patches) within 1 min (5B, arrowheads) with more areas of colocalization by 2 min (5C, arrowheads). After 10 min, the colocalization on the membrane begins to diminish (5D) and by 30 min has subsided on the membrane. At that time, in fixed cells, which allows higher resolution photography, colocalized ACE and B2 receptors (yellow) appear to be concentrated in endosomes, presumably because of internalization (Fig 6).
Fig 5. Bradykinin stimulates movements of ACE and B2 receptors.
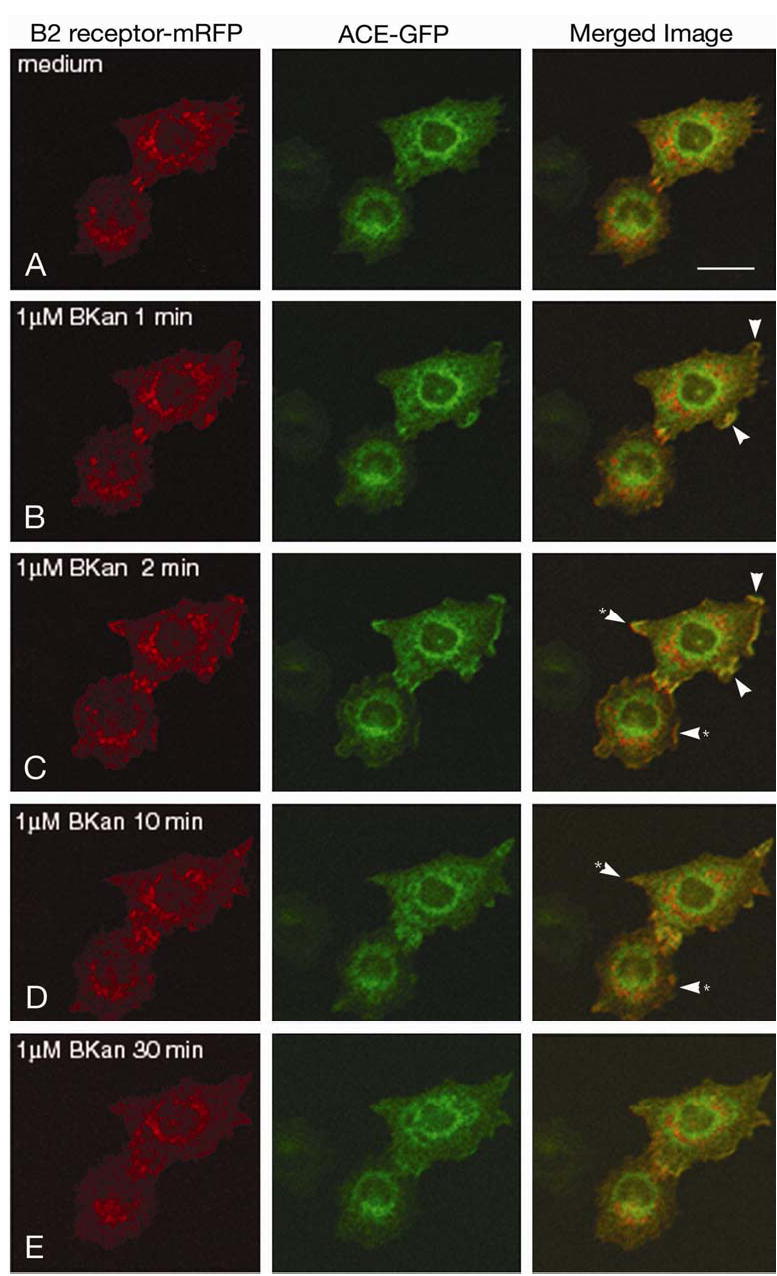
CHO/ACE-GFP+B2-mRFP cells. A. ACE-GFP and B2-mRFP are diffusely distributed on the cell membrane prior to adding an agonist. B. Within one min after 1 μM BKan was added, ACE and B2 receptors have begun to colocalize on the cell surface membrane (arrowheads). C. At two min after stimulation, numerous patches of colocalized ACE and B2 receptor are evident on the surface (arrowheads). D. Within ten min, ACE and B2 receptors are largely divergent from the colocalized patches (*arrowheads, compared to areas in C). E. Thirty min after stimulation, colocalized ACE and B2 receptors appear once again diffusely distributed at the surface. At this time, co-internalization of ACE and B2 is also evident (see Fig 6). Bar = 20 μm.
Fig 6. Internalized ACE-GFP and B2-mRFP.
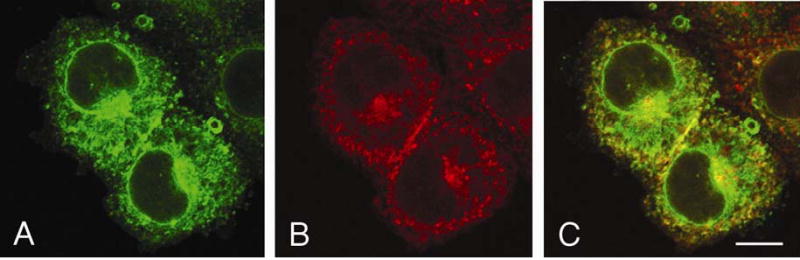
CHO cells were fixed 30 min after stimulation with 1 μM BKan. ACE and B2 appear colocalized (yellow) and cointernalized from the plasma membrane. Bar = 10 μm.
ACE movement did not occur in CHO/ACE-GFP cells that lack B2 receptors. The agonist (1 μM Bkan) failed to cause internalization of ACE even after 1 h. Our observation of agonist-induced formation of ACE-B2 complexes on the membrane followed by endocytosis supports the idea that ACE and B2 receptors form hetero-oligomers (17). As a positive control, we found that addition of 1 μM BKan to CHO/B2-mRFP cells caused B2 receptors to move to surface active domains within 1 min; these complexes were then internalized within 10–30 min (not shown).
ACE-B2 fusion protein in CHO cells
As noted before, ACE must be expressed with B2 receptors on the cell surface, and the enzyme should be in close proximity to the B2 receptors in order for ACE inhibitors to resensitize them (17). As a control, we constructed and expressed an ACE-B2 fusion protein in CHO cells to see if such a construct could account for the observed potentiation and resensitization by ACE inhibitors, or if ACE must be anchored separately to the cell membrane where it would complex with B2 receptors to react to ACE inhibitors. ACE remained catalytically active in CHO/ACE-B2 fusion protein-expressing cells. The bound enzyme (S2 fraction of cells) hydrolyzed Hip-His-Leu 0.3 μmol/h/mg protein and was 98% inhibited by enalaprilat (100 nM). Based on the number of ACE molecules per cell (the same as the number of B2 receptors) the specific activity of the fused ACE enzyme was the same as that determined for wild-type enzyme on the cell surface (19). In saturation binding experiments CHO/ACE-B2 live cells bound specifically [3H]-bradykinin indicating approximately 5 x 104 B2 receptors per cell (data not shown). Thus, both components of the fusion protein remained active on the cell surface. Fig 7 shows the results of potentiation and resensitization experiments. Bradykinin stimulated release of AA from these cells (Fig. 7A) that was blocked by HOE 140. Enalaprilat (1 μM) added before BKan was, failed to release additional AA, showing that the ACE inhibitor did not potentiate activation of B2 receptors (Fig. 7B). Enalaprilat did not resensitize the B2 receptor in cells expressing the fused protein after it was activated (and desensitized) by BKan (Fig 7C). These results contrast with what we obtained when the enzyme and receptor were separately expressed on plasma membrane (Fig. 1).
Fig 7. Lack of potentiation and resensitization in CHO cells expressing ACE-B2 receptor fusion protein.
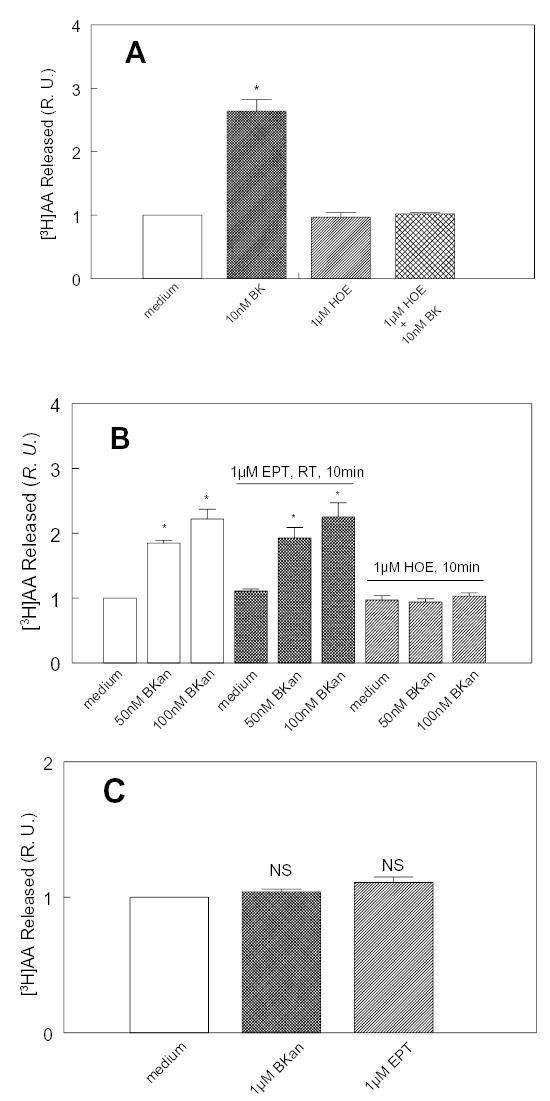
A: Receptor (R) function. Cells were stimulated with 10 nM bradykinin in absence or in presence of HOE 140 and the released [3H] AA was measured after 30 min. Bradykinin stimulated a 3 fold increase in AA release that was blocked by HOE 140, indicating that the receptors in the fusion protein were functional. (*p < 0.001 v. medium control, n = 6). B: Lack of potentiation. Cells were exposed to 50 or 100 nM BKan with or without pretreating them with enalaprilat (EPT). The same relative amount of AA was released is in the presence or absence of EPT. HOE 140 (1 μM) blocked the effect of BKan on B2 receptors (*, p < 0.001 v. medium control, n = 6). C: Lack of resensitization. Cells were pre-incubated with 1 μM BKan for 30 min at 37°C to desensitize the receptors. Then medium or 1 μM BKan or 1 μM EPT was added for 5 min at 37°C. The second dose of BKan did not stimulate the release of AA, indicating that the receptors were desensitized. EPT did not stimulate AA release, thus it did not resensitize the ACE- B2 fusion receptors to BKan in the medium (NS, not significant v. medium control, n = 9, R.U. = relative unit). In the ACE-B2 fusion protein, where only the receptors are attached to the plasma membrane, but not ACE; ACE inhibitor neither potentiated bradykinin nor resensitized the B2 receptors fused to it, although both the B2 receptor and ACE (see Results) remained active.
DISCUSSION
Our aim was to determine if there is a close spatial relationship between ACE and bradykinin B2 receptors. We believe that this relationship can contribute to the well-documented clinical benefits of ACE inhibitors, specifically by enhancing the effect of bradykinin on its B2 receptors (15, 16). That potentiation of bradykinin by these agents extends beyond inhibition of ACE (kininase II) was shown not only with transfected cell lines, but also by numerous experiments with isolated tissues and cultures of primary cells that naturally express ACE and B2 receptors. In bioassay, bradykinin enhanced the isotonic concentration of smooth muscle in isolated guinea pig ileum and adding ACE inhibitor, augmented it even at the point of maximum contraction caused by agonist alone (15). The positive inotropic effect of bradykinin on isolated left atrial preparations of guinea pig heart was increased by ACE inhibitor and the tissue was resensitized to agonist present in the tissue (15). Cultured, bovine (18) or human arterial endothelial cells, which naturally express ACE and B2 receptors, were resensitized to bradykinin, by ACE inhibitor and by Ang 1–9 and Ang 1–7. Thus, this type of interaction between ACE and the B2 receptor may occur naturally within components of the cardiovascular system. It was suggested that ACE inhibitors are exogenous, while the peptide metabolites of Ang I, Ang 1–9 and 1–7, are the endogenous allosteric enhancers of B2 receptor agonist activities (15, 19).
The first potent ACE inhibitors were peptides derived from snake venoms (25); these were called bradykinin potentiating peptides or factors. However, reports published in 1970 and a more recent one claimed that potentiation can occur independent of blocking bradykinin inactivation by ACE or kininase II (26, 27). An initial report on captopril showed that that this ACE inhibitor enhanced the effects of bradykinin stepwise on an isolated organ at much higher concentrations than needed for complete inhibition of ACE or kininase II (28).
Bradykinin and Lys-bradykinin (kallidin) are converted from B2 receptor agonists to B1 receptor ligands after carboxypeptidase M of plasma membrane or carboxypeptidase N of blood plasma, two human enzymes from this laboratory, cleaves off the C-terminal Arg (29). ACE inhibitors, as discussed earlier, amplify B2 receptor activity indirectly via ACE and thereby initiate a different signal transduction pathway involving protein kinase C (PKC), while activation of B2 receptors by bradykinin alone does not (30). In contrast, ACE inhibitors directly activate B1 receptors in human and bovine endothelial cells to release NO and thereby inhibit PKC (31–33). That was demonstrated with different ACE inhibitors, but not with lisinopril.
Many receptors form homo- and heterodimers, which enhance their efficacy and broadens the range of their potential agonists (34–36). We suggested heterodimer formation between ACE and B2 receptors expressed on plasma membranes (17), and that it may occur also in human fibroblasts with neprilysin (37). We tested for ACE–bradykinin receptor heterodimer formation in human cells with various methods. For example, we used ACE-resistant bradykinin analogue peptides as agonists to distinguish between potentiation and blocking of kinin inactivation by ACE, kininase II (17, 19). We determined that the inhibitors acted on B2 receptors, only indirectly via ACE. As reported above, we found that human ACE and B2 receptors are close enough together on the plasma membrane for ACE inhibitors to become allosteric (20) enhancers. They could induce a favorable conformational change in receptors that results in cross-talk between enzymes and peptide receptors.
Because fusion of GFP to B2 receptors increases the molecular weight of the receptors, we tested transfected CHO cells and confirmed that an ACE inhibitor did still potentiate bradykinin and its ACE-resistant analogue, Bkan. They resensitized GFP-coupled B2 receptors that were desensitized to bradykinin, just as observed with native B2 receptors.
We found that 1 μM enalaprilat potentiates didansyl-lysyl-bradykinin (didansyl-kallidin; 0.3–1 μM) on B2-GFP receptors. This agonist is 97% resistant to human ACE (19), yet the release of [3H] AA from CHO cells increased about five-fold. These results (not shown) are similar to those obtained in experiments with other kinins, provided the cells expressed both ACE and B2 receptors (19).
We report here that ACE and B2 receptors were similarly located on the cell membrane and that they could be coprecipitated with the appropriate antisera. Coimmunoprecipitation indicated complex formation between ACE and B2 receptor. The immunohistochemistry also showed the close proximity of the two proteins in CHO/ACE+B2-GFP cells. Under the microscope the B2-GFP receptors were green, while ACE appeared red from the anti-rabbit Alexa 568 IgG conjugate bound to the primary rabbit antibody to human ACE. The overlap appeared as yellow on the plasma membrane.
Coimmunoprecipitation and colocalization studies seemed to confirm that ACE and B2 receptors form heterodimer or -oligomers on the plasma membrane (17). To support this conclusion we used FRET microscopy with acceptor photobleaching between ACE-CFP (donor) and B2-YFP (acceptor). Photobleaching of YFP can convert it to CFP which could cause errors in determining FRET (38). We measured that conversion in our system and found that is was negligible. Thus, the increase in CFP intensity upon photobleaching of YFP indicated a close association between the ACE and B2 receptors (i.e. ≤ 10 nm). Such a close association is indicative of hetero-oligomer formation.
We established that ACE and B2 receptors move together on the membrane and into the cell after bradykinin stimulation. Time-lapse photography of living cells and studies of fixed cells showed that ACE and B2 receptors become localized in surface membrane domains when Bkan was added to the medium. Both proteins were then internalized together on a time scale similar to that for internalization of the stimulated B2 receptor alone.
The C-terminal domain of ACE usually anchors it to the plasma membrane in CHO cells (39–43), but in the fusion protein it was linked directly to the N-domain of B2 receptors to form a chimeric ACE. Although both enzyme and receptors remained active at the 1:1 ratio of ACE to B2, ACE inhibitor neither potentiated BKan nor resensitized the receptors. We take these findings to indicate that both proteins have to be attached to the membrane for ACE inhibitors to affect a more favorable conformational change in B2 receptors.
Ramiprilat induces phosphorylation of a Ser residue in the cytosolic portion of ACE, which after appropriate time for protein synthesis, enhances the expression of ACE and cyclooxygenase 2 in cultured cells (44). We expressed a truncated form of human ACE in cells, where 17 amino acids of the cytosolic C-terminal end of C-domain were removed, so that the mutant ACE lacked the crucial Ser1270, the site of phosphorylation (44). This deletion did not affect the resensitization of the B2 bradykinin receptors by added ACE inhibitor (17), after the cholesterol content of the plasma membrane was depleted. Consequently, the resensitization process that requires phosphorylation of the Ser1270 residue may not be the absolute requirement. Given the short time course of our experiments, it is also unlikely that de novo protein synthesis is involved.
A complex formation between the enzyme and the receptor should be a bimolecular reaction, where the kinetics would be altered if one reactant is expressed in excess. If ACE is in excess, ACE inhibitors could enhance activation of B2 receptors by kinins to a greater extent because the reaction is converted to a pseudo-first order one. However, if the cells express many more B2 receptors than ACE, indirect augmentation of bradykinin will be lacking.
Efficient use of FRET technique depends on donor v. acceptor ratio (24), and it is accelerated by higher acceptor presence independent of the absolute concentration of the two reactants. This favorable ratio between donor-acceptor fluorescent proteins is reversed if no bleaching is employed.
ACE inhibitors have multiple entry points to affect the renin-angiotensin (40, 41, 45) and the kallikrein-kinin systems (6, 13–15, 30–33, 46–50). Besides inhibiting the hydrolysis of ACE substrates, complex formation between ACE and the bradykinin receptors may contribute to the potentiation of B2 receptor agonists by ACE inhibitors, (15, 51) and some endogenous peptides (19).
Acknowledgments
This work was supported by National Institutes of Health (NIH) Grants HL36473 and HL68580. The authors are grateful for the advice of Dr. A.R. Johnson-Zeiger, for the editorial assistance of Ms. C. Sanders, and for the cooperation of Dr. M.-L. Chen.
References
- 1.Webster, M. E. (1970) Kallikreins in glandular tissues. In Bradykinin, Kallidin and Kallikrein. Handbook of Experimental Pharmacology. (Erdös, E. G., ed) Vol. XXV pp. 131–155, Springer Verlag, Heidelberg [Google Scholar]
- 2.Werle E. Discovery of the most important kallikreins and kallikrein inhibitors. In: Erdös EG, editor. Handbook of Experimental Pharmacology. XXV. Springer-Verlag; Heidelberg: 1970. pp. 1–6. [Google Scholar]
- 3.Beraldo WT, Andrade SP. Discovery of bradykinin and the kallikrein-kinin system. In: Farmer SG, editor. The Kinin System. Academic Press Ltd.; San Diego, CA: 1997. pp. 1–8. [Google Scholar]
- 4.Page IH. Hypertension Research. A Memoir 1920–1960. Pergamon Press; New York: 1988. pp. 1–167. [Google Scholar]
- 5.Yang HYT, Erdös EG. Second kininase in human blood plasma. Nature. 1967;215:1402–1403. doi: 10.1038/2151402a0. [DOI] [PubMed] [Google Scholar]
- 6.Yang HYT, Erdös EG, Levin Y. A dipeptidyl carboxypeptidase that converts angiotensin I and inactivates bradykinin. Biochim Biophys Acta. 1970;214:374–376. doi: 10.1016/0005-2795(70)90017-6. [DOI] [PubMed] [Google Scholar]
- 7.Gavras HP, Faxon DP, Berkoben J, Brunner HR, Ryan TJ. Angiotensin converting enzyme inhibition in patients with congestive heart failure. Circulation. 1978;58:770–776. doi: 10.1161/01.cir.58.5.770. [DOI] [PubMed] [Google Scholar]
- 8.Couture R, Girolami JP. Putative roles of kinin receptors in the therapeutic effects of angiotensin 1-converting enzyme inhibitors in diabetes mellitus. Europ J Pharmacol. 2004;500:467–485. doi: 10.1016/j.ejphar.2004.07.045. [DOI] [PubMed] [Google Scholar]
- 9.Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–153. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]
- 10.Pfeffer M. Angiotensin-converting enzyme (ACE) inhibitors in the prevention of cardiovascular disease: Clinical evidence. In: Giles TD, editor. Angiotensin-Converting Enzyme (ACE): Clinical and Experimental Insights. Health Care Communications, Inc.; Fort Lee, NJ: 2001. pp. 219–225. [Google Scholar]
- 11.Solomon SD, Skali H, Bourgoun M, Fang J, Ghali JK, Martelet M, Wojciechowski D, Ansmite B, Skards J, Laks T, Henry D, Packer M, Pfeffer MA. Effect of angiotensin-converting enzyme or vasopeptidase inhibition on ventricular size and function in patients with heart failure: the Omapatrilat Versus Enalapril Randomized Trial of Utility in Reducing Events (OVERTURE) echocardiographic study. Am Heart J. 2005;150:257–262. doi: 10.1016/j.ahj.2004.09.056. [DOI] [PubMed] [Google Scholar]
- 12.Bosch J, Lonn E, Pogue J, Arnold JM, Dagenais GR, Yusuf S. Long-term effects of ramipril on cardiovascular events and on diabetes: results of the HOPE study extension. Circulation. 2005;112:1339–1346. doi: 10.1161/CIRCULATIONAHA.105.548461. [DOI] [PubMed] [Google Scholar]
- 13.Barbe F, Su JB, Guyene TT, Crozatier B, Menard J, Hittinger L. Bradykinin pathway is involved in acute hemodynamic effects of enalaprilat in dogs with heart failure. Am J Physiol. 1996;270:H1985–1992. doi: 10.1152/ajpheart.1996.270.6.H1985. [DOI] [PubMed] [Google Scholar]
- 14.Hornig B, Kohler C, Drexler H. Role of bradykinin in mediating vascular effects of angiotensin-converting enzyme inhibitors in humans. Circulation. 1997;95:1115–1118. doi: 10.1161/01.cir.95.5.1115. [DOI] [PubMed] [Google Scholar]
- 15.Erdös EG, Deddish PA, Marcic BM. Potentiation of bradykinin actions by ACE inhibitors. Trends Endocrinol Metab. 1999;10:223–229. doi: 10.1016/s1043-2760(99)00156-3. [DOI] [PubMed] [Google Scholar]
- 16.Minshall RD, Tan F, Nakamura F, Rabito SF, Becker RP, Marcic B, Erdös EG. Potentiation of the actions of bradykinin by angiotensin I converting enzyme (ACE) inhibitors. The role of expressed human bradykinin B2 receptors and ACE in CHO cells. Circul Res. 1997;81:848–856. doi: 10.1161/01.res.81.5.848. [DOI] [PubMed] [Google Scholar]
- 17.Marcic B, Deddish PA, Skidgel RA, Erdös EG, Minshall RD, Tan F. Replacement of the transmembrane anchor in angiotensin I-converting enzyme (ACE) with a glycosylphosphatidylinositol tail affects activation of the B2 bradykinin receptor by ACE inhibitors. J Biol Chem. 2000;275:16110–16118. doi: 10.1074/jbc.M909490199. [DOI] [PubMed] [Google Scholar]
- 18.Marcic B, Deddish PA, Jackman HL, Erdös EG. Enhancement of bradykinin and resensitization of its B2 receptor. Hypertension. 1999;33:835–843. doi: 10.1161/01.hyp.33.3.835. [DOI] [PubMed] [Google Scholar]
- 19.Chen Z, Tan F, Erdös EG, Deddish PA. Hydrolysis of angiotensin peptides by human angiotensin I-converting enzyme and the resensitization of B2 kinin receptors. Hypertension. 2005;46:1368–1373. doi: 10.1161/01.HYP.0000188905.20884.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Changeux JP, Edelstein SJ. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 21.Siegel RM, Chan FK, Zacharias DA, Swofford R, Holmes KL, Tsien RY, Lenardo MJ. Measurement of molecular interactions in living cells by fluorescence resonance energy transfer between variants of the green fluorescent protein. Sci STKE. 2000;2000:PL1. doi: 10.1126/stke.2000.38.pl1. [DOI] [PubMed] [Google Scholar]
- 22.Bastiaens PI, Majoul IV, Verveer PJ, Soling HD, Jovin TM. Imaging the intracellular trafficking and state of the AB5 quaternary structure of cholera toxin. Embo J. 1996;15:4246–4253. [PMC free article] [PubMed] [Google Scholar]
- 23.Deddish PA, Jackman HL, Skidgel RA, Erdös EG. Differences in the hydrolysis of enkephalin congeners by the two domains of angiotensin converting enzyme. Biochem Pharmacol. 1997;53:1459–1463. doi: 10.1016/s0006-2952(97)00087-7. [DOI] [PubMed] [Google Scholar]
- 24.Herrick-Davis K, Grinde E, Mazurkiewicz JE. Biochemical and biophysical characterization of serotonin 5-HT2C receptor homodimers on the plasma membrane of living cells. Biochemistry. 2004;43:13963–13971. doi: 10.1021/bi048398p. [DOI] [PubMed] [Google Scholar]
- 25.Ferreira SH. Bradykinin-potentiating factor. In: Erdös EG, Back N, Sicuteri F, editors. Hypotensive Peptides. Proceedings of the International Symposium Firenze, Italy, 1965. Springer-Verlag; New York: 1966. pp. 356–367. [Google Scholar]
- 26.Vogel R, Werle E, Zickgraf-Rüdel G. Neure Aspekte der Kininforschung. I Potenzierung und Blockierung der biologischen Kininwirkung. Z klin Chem u klin Biochem. 1970;8:177–185. [PubMed] [Google Scholar]
- 27.Mueller S, Gothe R, Siems WD, Vietinghoff G, Paegelow I, Reissmann S. Potentiation of bradykinin actions by analogues of the bradykinin potentiating nonapeptide BPP9alpha. Peptides. 2005;26:1235–1247. doi: 10.1016/j.peptides.2005.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rubin B, Laffan RJ, Kotler DG, O'Keefe EH, Demaio DA, Goldberg ME. SQ14,225 (D-3-mercapto-2-methylpropanoyl-L-proline), a novel orally active inhibitor of angiotensin I-converting enzyme. J Pharmacol Exp Ther. 1978;204:271–280. [PubMed] [Google Scholar]
- 29.Erdös EG, Skidgel RA. Metabolism of bradykinin by peptidases in health and disease. In: Farmer SG, editor. The Kinin System. Academic Press; San Diego, CA: 1997. pp. 111–141. [Google Scholar]
- 30.Marcic BM, Erdös EG. Protein kinase C and phosphatase inhibitors block the ability of angiotensin I-converting enzyme inhibitors to resensitize the receptor to bradykinin without altering the primary effects of bradykinin. J Pharmacol Exp Ther. 2000;294:605–612. [PubMed] [Google Scholar]
- 31.Ignjatovic T, Stanisavljevic S, Brovkovych V, Skidgel RA, Erdös EG. Kinin B1 receptors stimulate nitric oxide production in endothelial cells: signaling pathways activated by angiotensin I-converting enzyme inhibitors and peptide ligands. Mol Pharmacol. 2004;66:1310–1316. doi: 10.1124/mol.104.001990. [DOI] [PubMed] [Google Scholar]
- 32.Ignjatovic T, Tan F, Brovkovych V, Skidgel RA, Erdös EG. Novel mode of action of angiotensin I converting enzyme inhibitors. Direct activation of bradykinin B1 receptor. J Biol Chem. 2002;277:16847–16852. doi: 10.1074/jbc.M200355200. [DOI] [PubMed] [Google Scholar]
- 33.Stanisavljevic S, Ignjatovic T, Deddish PA, Brovkovych V, Zhang K, Erdös EG, Skidgel RA. Angiotensin I-converting enzyme inhibitors block PKCɛ by activating bradykinin B1 receptors in human endothelial cells. J Pharmacol Exp Ther. 2006;316:1153–1158. doi: 10.1124/jpet.105.093849. [DOI] [PubMed] [Google Scholar]
- 34.Marianayagam NJ, Sunde M, Matthews JM. The power of two: protein dimerization in biology. Trends in Biochemical Sciences. 2004;29:618–625. doi: 10.1016/j.tibs.2004.09.006. [DOI] [PubMed] [Google Scholar]
- 35.Salahpour A, Angers S, Mercier JF, Lagace M, Marullo S, Bouvier M. Homodimerization of the β2-adrenergic receptor as a prerequisite for cell surface targeting. J Biol Chem. 2004;279:33390–33397. doi: 10.1074/jbc.M403363200. [DOI] [PubMed] [Google Scholar]
- 36.Pfeiffer M, Kirscht S, Stumm R, Koch T, Wu D, Laugsch M, Schroder H, Hollt V, Schulz S. Heterodimerization of substance P and μ-opioid receptors regulates receptor trafficking and resensitization. J Biol Chem. 2003;278:51630–51637. doi: 10.1074/jbc.M307095200. [DOI] [PubMed] [Google Scholar]
- 37.Deddish PA, Marcic BM, Tan F, Jackman HL, Chen Z, Erdös EG. Neprilysin inhibitors potentiate effects of bradykinin on B2 receptor. Hypertension. 2002;39:619–623. doi: 10.1161/hy0202.103298. [DOI] [PubMed] [Google Scholar]
- 38.Valentin G, Verheggen C, Piolot T, Neel H, Coppey-Moisan M, Bertrand E. Photoconversion of YFP into a CFP-like species during acceptor photobleaching FRET experiments. Nature Methods. 2005;2:801. doi: 10.1038/nmeth1105-801. [DOI] [PubMed] [Google Scholar]
- 39.Erdös EG, Gafford JT. Human converting enzyme. Clin Exper Hypertension. 1983;A5:1251–1262. doi: 10.3109/10641968309048855. [DOI] [PubMed] [Google Scholar]
- 40.Corvol P, Eyries M, Soubrier F. Peptidyl-dipeptidase A/angiotensin I-converting enzyme. In: Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of Proteolytic Enzymes. Vol. 1. Academic Press; San Diego, CA: 2004. pp. 332–346. [Google Scholar]
- 41.Skidgel RA, Erdös, EG. Biochemistry of angiotensin converting enzyme. In: Robertson JIS, Nicholls MG, editors. The Renin-Angiotensin System. Vol. 1. Gower Medical Publishers; London, England: 1993. pp. 10.11–10.10. [Google Scholar]
- 42.Jaspard E, Wei L, Alhenc-Gelas F. Differences in the properties and enzymatic specificities of the two active sites of angiotensin I-converting enzyme (kininase II). Studies with bradykinin and other natural peptides. J Biol Chem. 1993;268:9496–9503. [PubMed] [Google Scholar]
- 43.Soubrier F, Alhenc-Gelas F, Hubert C, Allegrini J, John M, Tregear G, Corvol P. Two putative active centers in human angiotensin I-converting enzyme revealed by molecular cloning. Proc Natl Acad Sci USA. 1988;85:9386–9390. doi: 10.1073/pnas.85.24.9386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kohlstedt K, Busse R, Fleming I. Signaling via the angiotensin-converting enzyme enhances the expression of cyclooxygenase-2 in endothelial cells. Hypertension. 2005;45:126–132. doi: 10.1161/01.HYP.0000150159.48992.11. [DOI] [PubMed] [Google Scholar]
- 45.Frohlich ED, Re RN, editors. The Local Cardiac Renin Angiotensin-Aldosterone System. Springer Science + Business Media, Inc.; New York: 2006. pp. 1–224. [Google Scholar]
- 46.Cruden NLM, Witherow FN, Webb DJ, Fox KAA, Newby DE. Bradykinin contributes to the systemic hemodynamic effects of chronic angiotensin-converting enzyme inhibition in patients with heart failure. Arterioscler Thromb Vasc Biol. 2004;24:1043–1048. doi: 10.1161/01.ATV.0000129331.21092.1d. [DOI] [PubMed] [Google Scholar]
- 47.Bhoola KD, Figueroa CD, Worthy K. Bioregulation of kinins: kallikreins, kininogens, and kininases. Pharmacol Rev. 1992;44:1–80. [PubMed] [Google Scholar]
- 48.Witherow FN, Helmy A, Webb DJ, Fox KAA, Newby DE. Bradykinin contributes to the vasodilator effects of chronic angiotensin-converting enzyme inhibition in patients with heart failure. Circulation. 2001;104:2177–2181. doi: 10.1161/hc4301.098252. [DOI] [PubMed] [Google Scholar]
- 49.Peng H, Carretero OA, Vuljaj N, Liao TD, Motivala A, Peterson EL, Rhaleb NE. Angiotensin-converting enzyme inhibitors: a new mechanism of action. Circulation. 2005;112:2436–2445. doi: 10.1161/CIRCULATIONAHA.104.528695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tschöpe C, Schultheiss HP, Walther T. Multiple interactions between the renin-angiotensin and the kallikrein-kinin systems: role of ACE inhibition and the AT1 receptor blockade. J Cardiovasc Pharmacol. 2002;39:478–487. doi: 10.1097/00005344-200204000-00003. [DOI] [PubMed] [Google Scholar]
- 51.Auch-Schwelk W, Bossaller C, Claus M, Graf K, Gräfe M, Fleck E. ACE inhibitors are endothelium dependent vasodilators of coronary arteries during submaximal stimulation with bradykinin. Cardiovasc Res. 1993;27:312–317. doi: 10.1093/cvr/27.2.312. [DOI] [PubMed] [Google Scholar]