

Abstract
We report here a comparative analysis of the genome sequence of Methanosarcina barkeri with those of Methanosarcina acetivorans and Methanosarcina mazei. The genome of M. barkeri is distinguished by having an organization that is well conserved with respect to the other Methanosarcina spp. in the region proximal to the origin of replication, with interspecies gene similarities as high as 95%. However, it is disordered and marked by increased transposase frequency and decreased gene synteny and gene density in the distal semigenome. Of the 3,680 open reading frames (ORFs) in M. barkeri, 746 had homologs with better than 80% identity to both M. acetivorans and M. mazei, while 128 nonhypothetical ORFs were unique (nonorthologous) among these species, including a complete formate dehydrogenase operon, genes required for N-acetylmuramic acid synthesis, a 14-gene gas vesicle cluster, and a bacterial-like P450-specific ferredoxin reductase cluster not previously observed or characterized for this genus. A cryptic 36-kbp plasmid sequence that contains an orc1 gene flanked by a presumptive origin of replication consisting of 38 tandem repeats of a 143-nucleotide motif was detected in M. barkeri. Three-way comparison of these genomes reveals differing mechanisms for the accrual of changes. Elongation of the relatively large M. acetivorans genome is the result of uniformly distributed multiple gene scale insertions and duplications, while the M. barkeri genome is characterized by localized inversions associated with the loss of gene content. In contrast, the short M. mazei genome most closely approximates the putative ancestral organizational state of these species.
Biological methanogenesis by the methane-producing archaea has a significant role in the global carbon cycle. This process is one of several anaerobic degradative processes that complement aerobic degradation by utilizing alternative electron acceptors in habitats where O2 is not available (39). The efficiency of this microbial process is directly dependent upon the interaction of three metabolically distinct groups of microorganisms: the fermentative and acetogenic bacteria and the methanogenic archaea. The methanogenic archaea have two pivotal roles in methanogenic consortia (28). By consuming hydrogen for methanogenesis and effectively lowering its partial pressure by the process of interspecies hydrogen exchange, the methanogens provide a thermodynamically favorable environment for the fermentative and acetogenic species to utilize protons as electron acceptors. This interaction enables fermenters to conserve more energy by producing a more oxidized product, acetate, which is also a substrate for methanogenesis. The second role of the methanogens is the fermentation of acetate, which accounts for 70% of the global methane produced by biological methane production (28). The net effect of these microbial interactions is the diversion of protons to hydrogen and carbon to acetate, which ultimately yields methane and carbon dioxide via methanogenesis.
The genus Methanosarcina includes the most metabolically diverse species of methanogenic archaea. Whereas most methanogenic species grow by obligate CO2 reduction with H2, methyl reduction with H2, aceticlastic fermentation of acetate, or methylotrophic catabolism of methanol, methylated amines, and dimethylsulfide, most Methanosarcina spp. can grow by all four catabolic pathways (49). Methanosarcina acetivorans was recently reported to grow also nonmethanogenically with CO (35). In addition to their appetency for all known methanogenic substrates, most Methanosarcina spp. can grow in a minimal mineral medium and fix molecular nitrogen (6, 26). They can adapt to intracellular solute concentrations ranging from freshwater to three times that found in seawater (38) by osmoregulatory mechanisms that enable them to synthesize or accumulate osmoprotectants and modify their outer cell envelope (41). This metabolic diversity is reflected in the relatively large genome sizes of Methanosarcina acetivorans (5.8 Mb) and Methanosarcina mazei (4.1 Mb) and the relatively large number of putative coding sequences, 4,524 and 3,371, respectively, compared with those of other methanogenic archaea (13, 17). The adaptive success of these species is further evidenced by the occurrence of multiple paralogs in the genomes, including multiple catabolic methyltransferases and carbon monoxide dehydrogenases, all three known types of nitrogenases, and all four known chaperoning systems (8, 13, 17).
Methanosarcina barkeri Fusaro was isolated from sediment from Lago del Fusaro, a freshwater coastal lagoon west of Naples, Italy (22). In contrast, M. acetivorans was isolated from marine sediments (40) and M. mazei was isolated from sewage sludge (12). M. barkeri utilizes all four methanogenic pathways described above and exhibits a dichotomous morphology. When grown on freshwater medium, this species grows as large multicellular aggregates embedded in a heteropolysaccharide matrix (Fig. 1A) composed primarily of d-galactosamine and d-glucuronic acid, termed methanochondroitin (24), whereas in marine medium these species grow as individual cells surrounded only by a protein cell surface layer (S layer) (38). This isolate has been one of the methanosarcinal strains most frequently studied for the physiology, biochemistry, and bioenergetics of methanogenesis (39). The development of a tractable methanosarcinal gene transfer system has led to a number of recent reports on the mechanisms of methanogenesis using genetic approaches (36).
FIG. 1.

Thin-section electron micrographs of M. barkeri Fusaro. Cells cultured in low-saline medium (A) grow as multicellular aggregates embedded in a methanochondroitin matrix (mc). Cells cultured in marine medium (B) grow as single cells without the methanochondroitin outer layer. When grown with hydrogen, gas vesicles (gv) are observed in some cells. Bar, 1.0 μm.
Herein we describe the genome of M. barkeri, which represents the third methanosarcinal genome sequenced. In addition to comparison of the genome annotations, this is the first three-way analysis of the complete genomes of closely related species in the methanogenic Euryarchaeota. Results reveal extensive gene rearrangements in M. barkeri relative to M. acetivorans and M. mazei and a high degree of conservation within the fragments, providing insight into the mechanisms of structural modification and the functional organization of the methanosarcinal genome.
MATERIALS AND METHODS
Growth conditions.
The source for Methanosarcina barkeri Fusaro (DSM 804) was described previously (31). M. barkeri was grown in F medium (38) with 0.1 M trimethylamine. Where described, growth was tested with 0.1 M sodium formate or with a headspace of 200 kPa H2-CO2 (80:20) substituted for trimethylamine. Cultures were incubated statically at 35°C in the dark. Growth was monitored by measuring the optical density at 550 nm with a Spectronic 21 instrument and by measuring methanogenesis by gas chromatography as described previously (41, 42).
Genome sequencing, assembly, and finishing.
Genomic DNA was isolated from M. barkeri Fusaro as described previously (5). The genome of M. barkeri was sequenced at the Joint Genome Institute (JGI) by using a combination of 3-kb, 8-kb, and 40-kb (fosmid) DNA libraries. All general aspects of library construction and sequencing performed at the JGI can be found at http://www.jgi.doe.gov/. Draft assemblies were based on 89,216 total reads. All three libraries provided 13-fold sequence coverage of the genome. The Phred/Phrap/Consed software package (http://www.phrap.com) was used for sequence assembly and quality assessment (15, 16, 19). After the shotgun stage, reads were assembled with parallel Phrap (High Performance Software, LLC). Possible misassemblies were corrected with Dupfinisher (21) or transposon bombing of bridging clones (EZ-Tn5 <R6Kyori/KAN-2>Tnp transposome kit; Epicenter Biotechnologies, Madison, WI). Gaps between contigs were closed by editing in Consed, custom primer walk, or PCR amplification (Roche Applied Science, Indianapolis, IN). A total of 2,389 additional reactions were necessary to close gaps and to raise the quality of the finished sequence. The completed genome sequences of M. barkeri contain 85,812 reads, achieving an average of 12-fold sequence coverage per base, with an error rate of less than 1 in 100,000. The sequences of M. barkeri include a chromosome and plasmid and can be accessed by GenBank accession numbers CP000099 and CP000098, respectively, or from the JGI IMG site (http://img.jgi.doe.gov) as taxon identification no. 623520000.
Annotation and analysis.
Genes were predicted with a combination of GLIMMER and CRITICA (2, 11). These gene predictions were then run through a pipeline that identifies gene overlaps, missed genes, and incorrect start sites (29). The gene predictions were then manually curated. Functional predictions were generated automatically based on the presence of hits to COG (47), Pfam (3), and Interpro (32) families.
Whole-genome alignment and analysis.
Chromosome sequences (Table 1) in FASTA format were used to build single-sequence BLAST databases, which served as the subject sequences for comprehensive WuBlast (W. Gish, 1996-2004, http://BLAST.wustl.edu), BLASTN, and TBLASTX paired comparisons both as whole sequences and as segmented comparisons using the following parameters: span2, noseqs, filter = none, hspmax = 10,000, gspmax = 10,000. Similarly, all coding sequence features were built into databases and a BLAST search was done to generate a comprehensive set of pairwise comparisons. BLASTN outputs were captured into a database of high-scoring segment pair (HSP) features cross-referenced to a sequence and sequence feature database. Outputs were also directly parsed by Cross (D. Maeder, 1998-2006, http://bigm.umbi.umd.edu/materials/software/Cross.pub/) for display and interactive examination of comparative features.
TABLE 1.
Comparison of genome features among Methanosarcina spp.
| Organism | Taxonomic IDa no. | GenBank accession no. | Designation | Genome length (bp) | G+C content (%) | No. of features | Length of genome portion with features (bp) | % of genome with features | % of genome featureless | Mean no. of nt/feature |
|---|---|---|---|---|---|---|---|---|---|---|
| M. acetivorans | 188937 | NC_003552 | Chromosome | 5,751,494 | 42.7 | 4,540 | 4,262,934 | 74 | 26 | 939 |
| M. mazei | 192952 | NC_003901 | Chromosome | 4,096,345 | 41.5 | 3,371 | 3,074,712 | 75 | 25 | 912 |
| M. barkeri Fusaro | 269797 | NC_007355 | Chromosome | 4,837,408 | 39.2 | 3,680 | 3,390,164 | 70 | 30 | 921 |
| 269797 | NC_007349 | Plasmid | 36,358 | 33.6 | 18 | 21,099 | 58 | 42 | 1,172 | |
| 269797 | Genome | 4,873,766 | 39.2 | 3,698 | 3,411,263 | 69 | 31 | 922 |
ID, identification.
The paired-comparison database GRIT runs under the database manager MySQL (MySQL AB) and consists of source, feature, fragment, and link tables. The feature table was populated with predicted gene product features derived from GenBank or JGI (Table 1) with a foreign key pointing to a source table. The link table contains BLASTN HSP scores and identities with foreign keys pointing to entries in the fragment table, which contains positional information about HSPs with a foreign key pointing to the feature from which it was derived. This schema allows the construction of a structured query language (SQL) query that directly and rapidly retrieves sets of features which are either unique within a set of source chromosomes or describe a set of genes common at an arbitrary level of similarity between two or more sources. BLASTN comparisons facilitate measurement of significant similarity in homologs of closely related organisms and manage noncoding sequences; TBLASTX or BLASTX comparisons are similarly applicable for comparison of less closely related sequences. Washu BLASTN was used with the parameters span2, filter = none, hspmax = 10,000, gspmax = 10,000 and was wrapped in a Perl script for automatic iteration through multiple pairwise BLAST searches. Output data were then parsed and HSPs stored in GRIT. A web interface to the queries and databases is available at http://bigm.umbi.umd.edu/dat/genome/ and is elaborated below.
Cumulative skew analysis was performed using skew (D. Maeder, 2001, http://bigm.umbi.umd.edu/materials/software/skew/), which implements the algorithm of Grigoriev (20). Repeat analysis emerged directly from unfiltered BLAST and was confirmed using MUMmer (11). Putative origins of replication were explored by examining regions with locally separated inverted repeats in close upstream proximity to the orc1 and cdc6 genes.
Chromosomal sequence similarity was calculated as a distance derived from BLASTN comparisons in the GRIT database by using Perl script cross match.pl, which generates distance matrices in MEGA2 format based on the following equation: 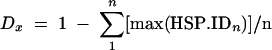 , where n is the length of the genome and HSP.ID is the maximal fractional identity at position n of sequence x and where HSP.ID exceeds a threshold of, e.g., 0.67. The mean distance, D, is calculated independently for each axis. This measure of distance is comparable with hybridization techniques, as it yields a fractional nucleotide similarity between organisms that considers stringency.
, where n is the length of the genome and HSP.ID is the maximal fractional identity at position n of sequence x and where HSP.ID exceeds a threshold of, e.g., 0.67. The mean distance, D, is calculated independently for each axis. This measure of distance is comparable with hybridization techniques, as it yields a fractional nucleotide similarity between organisms that considers stringency.
Synteny of any gene was measured by comparing the order of the gene's left and right neighbors with those of their best matched homologous genes in the comparable genome. Downstream synteny (SI) is expressed as the ratio of the ordinal distance between a gene, G, and its downstream neighbor, R (which is always 1), and the distance between a corresponding orthologous gene, G′, and the ortholog of R, R′. This may be calculated as follows: 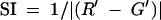 , with 0 < SI ≤ 1. Cumulative deviations from the mean of SI were calculated for intelligible display. Intergenic interval was calculated in the same manner.
, with 0 < SI ≤ 1. Cumulative deviations from the mean of SI were calculated for intelligible display. Intergenic interval was calculated in the same manner.
Microscopy.
For thin-section electron micrographs, cells were fixed with 2% glutaraldehyde and 2% osmium tetroxide and dehydrated in a graded series of ethanol mixtures. Cells were embedded and sectioned in Epon resin and then poststained with uranyl acetate and lead citrate as described previously (42). A Joel JEM-1200 EX II transmission electron microscope at 80 kV was used to generate thin-section micrographs.
RESULTS AND DISCUSSION
The genome of Methanosarcina barkeri was sequenced using a combination of whole-genome shotgun and directed finishing as described in Materials and Methods. The genome consists of a circular chromosome of 4,837,408 bp and a circular 36,358-bp extrachromosomal element (Table 1). The M. barkeri genome, which is intermediate in size between that of Methanosarcina acetivorans (5.8 Mb) and that of Methanosarcina mazei (4.1 Mb), is the second-largest genome among the archaea. The extrachromosomal element is 6.7 times larger than the only other methanosarcinal extrachromosomal element, plasmid pC2A from M. acetivorans (30).
Methanosarcina barkeri chromosome structure and content.
A total of 3,680 putative protein-coding genes longer than 200 bp, which together cover 70% of the genome, were identified (Table 1). The average protein-coding region of M. barkeri, at 921 bp, is within 2% of M. acetivorans and M. mazei coding regions, while its average intergenic region, at 393 bp, is considerably larger than those of M. acetivorans (328 bp) and M. mazei (303 bp). A further 71 RNA features were identified, including three sets of ribosomal RNAs (5S, 16S, and 23S) and 62 tRNAs covering all amino acids and pyrrolysine, which is encoded by the UAG codon in methylamine methyltransferase genes. One thousand seven hundred eighty hypothetical protein open reading frames (ORFs) accounted for nearly half of all protein features, with 1,837 putative functional protein assignments based on similarity to identified protein sequences in public databases. Of hypothetical protein genes conserved at the 80% nucleotide level, 289 were shared with M. acetivorans and 249 with M. mazei, of which 105 were common to both and should be considered highly conserved unidentified genes.
Gene annotation.
There were 128 ORFs with sequence identities greater than 67% to genes in the NCBI sequence database but without sequence identity to other methanosarcinal genomes (http://bigm.umbi.umd.edu/materials/Methanosarcina/) (also see the supplemental material). Some of these features are highlighted below.
The M. barkeri genome included the full complement of genes encoding enzymes in the CO2 and methyl reduction with H2, methylotrophic, and aceticlastic pathways (13, 17). In addition to these, a complete formate dehydrogenase operon (MbarA 1561 to 1562), fdhAB, with high sequence identity to catabolic formate dehydrogenase from several formate-utilizing methanogens, was detected. Methanosarcina spp. have never been reported to utilize formate for growth, and fdhAB has not been detected previously in this genus (7). Attempts to grow M. barkeri on 50 mM formate in this study were unsuccessful, and the addition of sodium formate to cultures containing trimethylamine or hydrogen did not enhance growth, which suggests that either the operon is not expressed under the conditions tested or it does not have a catabolic role. M. barkeri lacks genes encoding a two-subunit nucleoside diphosphate-forming acetyl-coenzyme A (CoA) synthetase (acdAB) that is found in M. acetivorans (MA3168 and MA3602) and M. mazei (MM0358 and MM0493) but has a remnant of this enzyme, pseudogene MbarA 3662. The sequence adjacent to the 5′ end includes the same order of gene orthologs found in M. acetivorans and M. mazei, but the 3′ end is adjacent to a sequence inversion, which further suggests that it is a truncated acdA sequence. This enzyme catalyzes one of two pathways for generating acetyl-CoA; the other is the CO dehydrogenase/acetyl-coenzyme A synthase that catalyzes aceticlastic catabolism in Methanosarcina spp. There are no ORFs encoding nucleoside diphosphate-forming acetyl-CoA synthetases close to characterized acetyl-CoA synthetases, but there are nucleoside monophosphate-forming acyl-CoA synthetases with unknown functions (MbarA 267, 2172, and 2821) that could potentially function as acetyl-CoA synthetases. Alternatively, it is also possible that the CO dehydrogenase/acetyl-coenzyme A synthase fulfills the function of both enzymes in M. barkeri.
Among genes encoding biosynthetic functions, a group of 14 sequential ORFs encode predicted gas vesicles with highest identity to gvpANOFGJKLM (MbarA 326 to 339) in the haloarchaea, which includes the minimal gene set for expression of vesicle in Haloferax volcanii (34). Although there are no prior reports of gas vesicles in M. barkeri Fusaro, gas vesicles have been reported in another strain of M. barkeri, FR-1, and in Methanosarcina vacuolata, which has a DNA-DNA reassociation value of 61% with the type strain of M. barkeri (1, 51, 52). Interestingly, M. barkeri has three sequential copies of gvpA that encode the ribs of the vesicle wall and influence the strength and width of the vesicles (4). The 33.5-kb region that includes the gvp operon may have been acquired from vesicle-synthesizing strains, as it is flanked by transposons. Gas vesicles have been proposed to be an early organelle of prokaryote motility, and they are often regulated by light and oxygen partial pressure (45, 48). In contrast to the other methanosarcinal strains that express gas vesicles with methanol and acetate, M. barkeri gas vesicles were observed only in cells grown with H2-CO2 in liquid and on solidified medium (Fig. 1B), which suggests that they might be expressed as part of a chemotactic mechanism in response to hydrogen gradients. M. barkeri possesses a full complement of chemotaxis genes, but unlike M. acetivorans and M. mazei it has only a single copy of the chemotaxis genes (with the exception of cheY) instead of two and lacks a cheC homolog. The functional role of these chemotaxis genes in Methanosarcina spp. is currently unknown, and additional types of motility, such as flagellar motility, have not been observed for these species. Osmoregulatory genes detected in the M. barkeri genome, including kefC (MbarA 671) for potassium uptake at low solute concentrations and ablAB (MbarA 669 to 670) for Nɛ-acetyl-β-lysine at high solute concentrations, indicate that this strain adapts to changes in extracellular solute concentrations (43) by mechanisms similar to those for other methanosarcinal species. Interestingly, M. barkeri also has ORFs (MbarA 22 to 23) with high identity to two enzymes required for N-acetylmuramic acid synthesis, which is unique among the sequenced archaea. Prior analysis of the cell wall composition of M. barkeri Fusaro failed to detect muramic acid (22). In addition, the ORFs and flanking ORFs MbarA 20 to 21 and MbarA 24 to 26 encode proteins with high sequence identity to the proteobacteria, which suggests that this DNA fragment was acquired by lateral gene transfer.
Another unique feature of the M. barkeri genome is the detection of a putative operon encoding a bacterial P450-specific ferredoxin reductase (MbarA 1947 to 1945). The family of heme protein monooxygenases known as cytochrome P450 plays a critical role in the synthesis and degradation of many xenobiotics and physiologically important compounds (37, 50). All known P450s are multicenter enzymes consisting of a heme, or P450, component with associated reductase components. The gene encoding the putative cytochrome P450 in M. barkeri is flanked immediately upstream by genes encoding a ferredoxin and ferredoxin reductase, which is typical of bacterial class I three-component systems. For catalytic activity, cytochrome P450 must be associated with the electron donor partner proteins ferredoxin/ferredoxin reductase complex (46). Cytochrome P450 has not been detected previously in the archaea. All three predicted proteins encoded by the putative operon have 54 to 62% sequence identity with cytochrome P450 from Myxococcus spp., and proteins encoded by genes immediately flanking the operon have high sequence identity to methanosarcinal genes. This suggests that this operon was acquired by M. barkeri through a lateral gene transfer event. Another putative operon encoding oxygen-dependent cytochrome d oxidase cydAB was also identified in the genome of M. barkeri and the other two methanosarcinal genomes. The presence of these oxygen-dependent genes along with one catalase and two superoxide dismutase genes suggests that these proteins protect methanosarcinal species from oxygen or that they may support microaerophilic growth by a currently undescribed mechanism. As cytochrome P450 catalyzes an oxygen-requiring reaction and has not been detected previously in an anaerobe, the detection of this gene in M. barkeri raises intriguing questions about the function of this gene product in this obligately anaerobic methanogen.
A comparison of gene role categories among the three species is shown in Table 2. The genomes were analyzed also for classes underrepresented or missing in the M. barkeri genome compared with the 1-Mb-larger M. acetivorans genome. Most of the genes absent from M. barkeri were unidentified ORFs, but identified genes included primarily ORFs encoding transporter proteins, sensory proteins, cell surface proteins, and polysaccharide synthesis proteins. All essential biosynthetic and catabolic genes were conserved in M. barkeri, including multiple copies of confirmed methyltransferases, but several hypothetical methyltransferases of unknown function were not present (17). As reported for M. mazei, which has a genome 1.7 Mb smaller than that of M. acetivorans, M. barkeri lacks also two multigene operons proposed to be linked to energy conservation in M. acetivorans during growth on acetate. The lack of mrpABCDEFG operon (MA4572 to MA4566) H+/Na+ antiporter and rnfABCDGE Na+ transporting NADH oxidoreductase in both M. barkeri and M. mazei supports the hypothesis that these gene products replace the function of the Ech hydrogenase, which is absent in M. acetivorans, by generating a transmembrane ion gradient for ATP synthesis during growth on acetate (25). Genes glnP and glnQ, encoding glutamine transporter proteins, were absent from M. barkeri but present in the other two methanosarcinal species. Finally, M. barkeri also lacks a low-affinity phosphate transporter (MA2935), suggesting it originated in a phosphate-poor environment. Two other transporters are missing in M. barkeri, a gluconate transporter (MA0021) and a dicarboxylate transporter (MA2961). This suggests that M. barkeri has less ability to take up organic compounds than the other two Methanosarcina spp.
TABLE 2.
Gene role categories and numbers of genes for three Methanosarcina spp.a
| Role | No. of genes
|
||
|---|---|---|---|
| M. barkerib | M. acetivorans | M. mazei | |
| Amino acid biosynthesis | 126 | 152 | 129 |
| Biosynthesis of cofactors, prosthetic groups, and carriers | 143 | 174 | 141 |
| Cell envelope | 293† | 277 | 235 |
| Cellular processes | 118 | 131 | 141 |
| Central intermediary metabolism | 604† | 259 | 140 |
| DNA metabolism | 133 | 179 | 150 |
| Energy metabolism | 821† | 431 | 325 |
| Fatty acid and phospholipid metabolism | 21 | 31 | 31 |
| Hypothetical proteins | 908* | 1,022 | 1,527 |
| Hypothetical proteins, conserved | 133* | 253 | 261 |
| Mobile and extrachromosomal element functions | 167 | 201 | 106 |
| Protein fate | 135 | 132 | 130 |
| Protein synthesis | 158 | 157 | 155 |
| Purines, pyrimidines, nucleosides, and nucleotides | 62 | 71 | 75 |
| Regulatory functions | 160 | 209 | 117 |
| Signal transduction | 4 | 0 | 1 |
| Transcription | 50 | 55 | 43 |
| Transport and binding proteins | 341 | 360 | 205 |
| Unclassified | 515* | 2,089 | 1,058 |
| Unknown function | 285 | 111 | 128 |
Derived from TIGR CMR (http://cmr.tigr.org/tigr-scripts/CMR/shared/GetNumAndPercentGenesInARole.cgi?Porg=ntmb02).
For M. barkeri, the lower relative numbers for hypothetical and unclassified genes (*) are compensated for by increased prediction frequency (†).
Plasmid structure and content.
The 36.4-kb plasmid in M. barkeri has not been detected previously. In contrast to the smaller, 5.4-kb plasmid pC2A in M. acetivorans, which appears to replicate by a rolling-circle mechanism (30), the M. barkeri extrachromosomal element lacks a putative repA gene. Instead, it has a cdc6 homolog in a region of highly repetitive sequence (discussed below), which suggests a novel mechanism of synchronous replication. Interestingly, one of the extrachromosomal ORFs (MbarB 3749) has 44% sequence similarity to an ATPase associated with chromosomal partitioning ParA, but the nucleotide recognition component ParB was not detected (9). These combined characteristics suggest that the extrachromosomal element replicates with cell division. In addition to the putative cdc6 and partitioning protein, the ORFs include four genes possibly associated with methanochondroitin synthesis, seven hypothetical genes of unknown function, and five putative transposases. The ORFs in the plasmid did not have identities equivalent to ORFs found in the M. acetivorans and M. mazei genomes, which might have suggested a critical function for the extrachromosomal element.
Features revealed by whole-genome comparison.
Sets of gene features shared between genomes were determined (see the supplemental material) and organized as sets of paralogous genes. This approach was pursued at several different levels of identity. The data for the 80% identity level are presented in Fig. 2. When excess paralogs (the difference between the number of paralog clusters and the genes they contain) are expressed as a fraction of total features in the paralog set, M. acetivorans has the highest fraction, at 14% to 15%, and M. mazei the lowest, at 10% to 11%, with M. barkeri intermediate. This correlation between genome size and paralogy suggests a model of genome growth driven by gene duplication and is consistent with our previous observation of high levels of paralogy in the heat shock proteins of M. acetivorans (8).
FIG. 2.

Venn diagram for three Methanosarcina sp. genomes, indicating the numbers of genome features with at least 80% nucleotide identity. Numbers in parentheses are the numbers of common paralog groups, and adjacent numbers are the gene counts for the contributing organisms. Values for M. barkeri are given in bold type, values for M. mazei are given in italics, and values for M. acetivorans are underlined.
For the purpose of comparing whole-genome gene feature sets, we have analyzed BLAST comparisons of genome pairs and assigned related paralog clusters in each genome which are similar to those of comparable genomes. These data may be freely examined using arbitrary similarity thresholds at http://bigm.umbi.umd.edu/dat/genome/venn.php and for fixed thresholds of 90%, 85%, 80%, and 75% with cluster counts of 26, 192, 785, and 1,573, respectively, for three-way similarity in the tables in the supplemental material or at http://bigm.umbi.umd.edu/materials/Methanosarcina/. Eighty percent was selected as a representative threshold, as more than half the genes are represented and those that are clustered have significant similarity. The “unique” method, on the other hand, allows sequences with similarity no greater than an arbitrary threshold with respect to other genomes to be rapidly identified (http://bigm.umbi.umd.edu/dat/genome/unique.php). M. acetivorans has the highest number of uncommon features, with 656 and 499 at 60% and 50%, respectively. By comparison, M. barkeri has 350 and 340 and M. mazei has 236 and 156 under the same conditions consonant with numbers proportional to genome size.
At the 80% three-way identity level, M. acetivorans, M. barkeri, and M. mazei have 924, 893, and 881 genes falling into 785 paralogous clusters with similarity to the other Methanosarcina spp. However, when their respective transposase contributions of 68, 50, and 49 are discounted the residual differences in relative paralog counts are small. With about 50% of all paralogs being transposase, it is difficult to identify gene duplication events that may not have been driven by transposition-mediated duplication. Chromosome extension in all three organisms must be affected by transposition, but such effects are not uniformly distributed in M. barkeri (Fig. 3). Whole-genome distances (Table 3) based on maximal local alignments indicate that the genomes are quite similar in overall content but that M. acetivorans and M. mazei are marginally more closely related. This is in qualitative agreement with DNA-DNA hybridization experiments (44), which showed reassociation values of 28% between M. acetivorans and M. mazei and 18% between these species and M. barkeri. This result underscores the comparability of these sequences, with the exception of the plasmid sequence.
FIG. 3.

Asymmetric fragmentation in M. barkeri. The top panel shows cumulative deviations from the mean in the M. barkeri genome for synteny with respect to M. acetivorans (a), M. mazei (b), or intergenic interval (c). The cumulative transposon count is superimposed (d). The bottom panel shows uniformly scaled BLASTN cross plots of the M. barkeri chromosome with those of M. mazei and M. acetivorans, with the origin regions circled.
TABLE 3.
Intergenomic distancesa
| Organism or plasmid | Distance
|
|||
|---|---|---|---|---|
| M. acetivorans | M. mazei | M. barkeri | M. barkeri plasmid | |
| M. acetivorans | 0 | 0.489 | 0.487 | 0.570 |
| M. mazei | 0.517 | 0 | 0.480 | 0.591 |
| M. barkeri | 0.552 | 0.569 | 0 | 0.512 |
| M. barkeri plasmid | 0.881 | 0.883 | 0.836 | 0 |
Calculated using equation 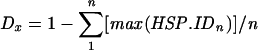 , based on whole-genome maximal local nucleotide sequence identity considering only HSPs with identities of >67% (from left) or 55% (from top).
, based on whole-genome maximal local nucleotide sequence identity considering only HSPs with identities of >67% (from left) or 55% (from top).
Location of origins of replication.
In archaea, origins of replication are invariably found in close proximity to the origin recognition complex gene (orc1), sometimes also referred to as cell division control protein 6 (cdc6) (27). When genes are densely packed, searches for putative origins of replication are directed at proximal intergenic regions. In the three Methanosarcina spp., there are two highly conserved paralogous copies of these genes in relatively close mutual proximity (about 100 kb or 300 kb apart in M. barkeri) situated on opposite strands and directed away from each other, a finding consistent with observations of other archaea with multiple functional origins of replication (reviewed in reference 23). Flanking downstream ORFs are conserved (Table 4). The putative origins of replication are located in the upstream intergenic regions of approximately 1,600 nucleotides (nt) (ORI A) or 800 nt (ORI B) in extent and are somewhat conserved at the nucleotide level. In the chromosomal origin of replication region, gene products are approximately 95% identical across all species. Noncoding origin features (ORI A and ORI B) are not as well conserved (E ≤ 1e−44 in ORI A and E ≤ 1e−8 in ORI B) and show only weak similarity between ORI A and ORI B. They are extremely AT rich (∼70%) and may show unconserved inverted-repeat structures.
TABLE 4.
Identification of conserved features for chromosomal origins of replication in Methanosarcina spp.a
| Description or designation | Strand | GenInfo identifier sequence identification no.
|
Location (nt)
|
||||
|---|---|---|---|---|---|---|---|
| M. acetivorans | M. barkeri | M. mazei | M. acetivorans | M. barkeri | M. mazei | ||
| Conserved hypothetical protein ORF | Plus | 20093437 | 73668510 | 21227413 | |||
| Conserved hypothetical protein ORF | Plus | 20093438 | 73668511 | 21227414 | |||
| Conserved hypothetical protein ORF | Plus | 20093439 | 73668512 | 21227415 | |||
| orc1 A | Minus | 20088900 | 73668513 | 21227416 | |||
| ORI A | 1529-3357 | 1189197-1191361 | 1564667-1566241 | ||||
| Interorigin regionb | Variable | ||||||
| Conserved hypothetical protein ORF | Minus | 20088983 | 73668731 | 21227479 | |||
| ORI B | 104571-105272 | 1481179-1482170 | 1654267-1655075 | ||||
| orc1 B | Plus | 20088984 | 73668732 | 21227480 | |||
| hsp60 | Minus | 20088985 | 73668733 | 21227481 | |||
Highly conserved genes and features, which are presented as an aligned feature set, are found in the immediate proximity of the dual origins of replication of all three Methanosarcina spp. ORI A and ORI B are putative origins of replication, orc1 A and orc1 B encode putative replication initiation proteins, and hsp60 encodes a putative heat-shock induced chaperone. The first origin of M. acetivorans spans the annotation origin.
The interorigin region was ∼100 kbp for M. acetivorans, ∼300 kbp for M. barkeri, and ∼100 kbp for M. mazei.
A replication complex is initiated when the Orc1 (Cdc6) protein binds to cognate DNA at the origin and allows the recruitment of Mcm and the rest of the replication machinery. An approximate inverted repeat (Fig. 4) could allow a pseudosymmetrical double hairpin to form a crucifix motif similar to a Holliday junction, thereby initiating bidirectional replication from a point, with complexes bypassing each other to replicate the origin at the beginning of replication. The concurrent presence of more than one active origin would cause contention for DNA, so there must be an implicit mechanism to control which origin and which origin recognition complex protein are dominant. It is notable that the downstream neighbor of the secondary Orc1 B protein is a highly conserved Hsp60 class heat shock protein, as this suggests a possible stress-associated switching mechanism. The putative origins of replication are located centrally within the most highly conserved and syntenous regions of the respective genomes (Fig. 4), consistent with the observation of Eisen et al. (14) of symmetrical inversion about the origin of replication. GC skew analysis (results not shown) is not useful in this case as there is a high level of strand inversion and rearrangement.
FIG. 4.

Sequence alignment of the M. barkeri ORI A self-complementary region (1190360 to 1191059) using BLASTN. Numbers at ends of the bottom sequences are genomic locations.
The plasmid of M. barkeri presents a unique and distinct origin of replication characterized by an orc1 homolog (orc1 C) that is relatively weakly related to orc1 A and orc1 B (37%/66% with orc1 A and 21%/44% with orc1 B) (Table 4). The immediately adjacent upstream region of the plasmid DNA contains a 5.6-kb noncoding region (15.3% of the plasmid sequence) characterized by a highly repetitive sequence consisting of over 38 direct repeats of a 143-nt sequence, with few variations between them (see Fig. S1 in the supplemental material). The consensus of the AT-rich repeat sequence is ATCCCATTTCCTAAGCAGAGAATTAGTTTCCTAAGCAAAAAAAAaG ATTTCTGgcttagACCATTTCCTAAGCAAAACGATATCA GAAGACATAACAAGTTAGAAGAaAAAtAAgTTAAAA TTAGATATTAATCTGTATATAT, with internal repeats underlined and variable regions in lowercase. This sort of arrangement (ORI C) is quite unlike that of chromosomal ORI A and ORI B but has the capacity to present slidable bubbled-out complete repeat motifs and retain a quasi-stable structure (Fig. 5).
FIG. 5.

Proposed mechanism for conserved repetitive sequence to provide bubbled-out repeat motifs for initiation of replication. Pairs of quasi-stable bubbles might occur in pairs at arbitrary locations on opposite strands. All motifs are essentially identical.
Overall genomic organization.
A significant observation in the three-way comparison of the Methanosarcina genomes is the overall colinearity of M. mazei and M. acetivorans (Fig. 3, lower left panel). This attests to a history of conserved gene order and resistance to large-scale mosaicity. However, closer examination (Fig. 6) reveals that there is considerable deviation from the expected 45° slope for a line of identity. This is maintained between M. barkeri and M. mazei, indicating that M. acetivorans has been subject to uniformly distributed local elongation, which may arise from gene duplication, elongation of intergenic regions, or insertion of sequence by transposition. This may explain the large size of the M. acetivorans genome relative to those of the other Methanosarcina spp. Comparison of M. acetivorans and M. mazei genomes shows limited transposition and inversion, with nonorthogonal colinear tracts, which indicates that M. mazei has lost or M. acetivorans has gained material in a uniformly distributed fashion. M. mazei and M. barkeri genomes are orthogonal and share common extended tracts characterized by inversions and transpositions in either or both genomes. The fact that M. acetivorans and M. mazei are colinear indicates that M. barkeri has undergone numerous inversions and transpositions. Finally, the genomes of M. mazei and M. barkeri are orthogonal, but M. acetivorans has undergone distributed insertions. The overall results indicate that M. mazei most closely represents the ancestral state, with the least number of insertion and inversion events.
FIG. 6.

M. acetivorans is elongated due to distributed duplication events. Uniformly scaled, comparable 2-Mbp BLASTN cross plot comparisons reveal deviation from the dotted 45° slope of identity in M. acetivorans, with respect to M. barkeri (upper left) and M. mazei (lower right). The anticipated orthogonal relationship is observed in the M. barkeri/M. mazei comparison (upper right). The self-comparison of M. acetivorans (lower left) reveals multiple nonidentical repeat sequences characteristic of the multiple transposase genes found in all three genomes. M. barkeri comparisons show multiple strand inversions and transpositions relative to the other two genomes.
The M. barkeri genome is distinguished by having an organization that is well conserved with respect to the other Methanosarcina spp. in the region proximal to the origin of replication, where interspecies gene similarities are as high as 95% (Table 4). However, there is little apparent conservation of gene organization in the region most distal to the origin, where large-scale colinearity appears rare. The putative terminus of replication is observed to be a hot spot for reorganization (33). Two properties of M. barkeri were measured: synteny, which measures conservation of the local neighborhood with respect to comparable genomes, and intergenic interval, or the separation between successive genes, which measures relative content density. In the distal semigenome, the rate of change of synteny is negative in accord with the macroscopic observation of decreased colinearity, and the negatively correlated intergenic interval is greater than average, indicating a loss of gene content in this region (Table 3).
What might cause this wasteland effect? One possibility, given the symmetry with respect to the origin, is an accumulation of strand exchange failures in the replication process and subsequent “gene rot” of broken genes. The cross effect of random strand inversion noted by Eisen et al. (14) gives way to a shotgun effect. Another possibility is infiltration by transposons with transposase-mediated damage. Certainly there is an increased frequency of transposon genes in this area (Fig. 3, trace d), but this may be either causative or opportunistic, with the organism tolerating infiltration of already-dysfunctional sections of the chromosome. The possibility that CRISPRs (clustered regularly interspaced short palindromic repeats) might be involved in large-scale rearrangements was also investigated. All four known CRISPR-associated, or cas, genes typically found in association with the DNA repeats have been detected previously in the M. barkeri genome (18). In M. barkeri, CRISPR sequences are found in six distinct localities. None of these coincide with the margins of rearrangement with respect to the other Methanosarcina spp. but are found in intergenic regions in structurally unconserved regions. This contrasts with the genomes of Thermotoga spp., where large-scale DNA rearrangements appear to be associated with CRISPR DNA repeats and/or tRNA genes (10). Although CRISPRs might be involved in homologous recombination, their immediate environment is not strongly conserved and so it is impossible to say on that basis whether they are better accepted in nondeleterious locations or whether there is a localized deterioration of the immediate environment. In two locations, CRISPR elements are adjoined by colinear sequences, implying an insertion event in M. barkeri.
Conclusions.
Of the 3,680 open reading frames in M. barkeri, 746 had orthologs with better than 80% similarity to both M. acetivorans and M. mazei while 240 were unique (nonorthologous) among these species. An etiology for genome rearrangement is revealed by whole-genome comparison of three species of the genus Methanosarcina. The inverse correlation of intergenic size and synteny demonstrates a mechanism for the development of genome plasticity, which involves replication-associated inversion with concomitant gene damage and colonization by transposable elements. Gene duplication is also observed as a mechanism for genome extension. The organization of M. barkeri is well conserved with respect to the other Methanosarcina spp. in the region proximal to the origin of replication, with interspecies gene similarities as high as 95%. In the half genome most distant from the origin, it is however disordered and marked by increased transposase frequency and decreased gene synteny and gene density. Furthermore, we have observed a highly conserved double origin of replication, which suggests a mechanism for replication which allows a double start with pass-through, enabling the origin itself to be replicated. The apparent genome plasticity likely contributed to these species' ability to adapt to a broad range of environments as a result of genome elongation and enrichment for favorable phenotypes.
Supplementary Material
Acknowledgments
This work was performed under the auspices of the U.S. Department of Energy's Office of Science, Biological and Environmental Research Program and the by the University of California Lawrence Livermore National Laboratory under contract no. W-7405-Eng-48, Lawrence Berkeley National Laboratory under contract no. DE-AC03-76SF00098, and Los Alamos National Laboratory under contract no. W-7405-Eng-36.
K.R.S. was supported in part by NSF MCB Division of Cellular and Bioscience grant no. MCB0110762 and by DOE Energy Biosciences Program grant no. DE-FG02-93-ER20106. W.W.M. was supported in part by NSF MCB Division of Cellular and Biosciences grant no. MCB12466 and by DOE Energy Biosciences Program grant no. DE-FG02-02-ER15296.
Footnotes
Published ahead of print on 15 September 2006.
Supplemental material for this article may be found at http://jb.asm.org/.
REFERENCES
- 1.Archer, D. B., and N. R. King. 1984. Isolation of gas vesicles from Methanosarcina barkeri. J. Gen. Microbiol. 130:167-172. [Google Scholar]
- 2.Badger, J. H., and G. J. Olsen. 1999. CRITICA: coding region identification tool invoking comparative analysis. Mol. Biol. Evol. 16:512-524. [DOI] [PubMed] [Google Scholar]
- 3.Bateman, A., E. Birney, R. Durbin, S. R. Eddy, K. L. Howe, and E. L. Sonnhammer. 2000. The Pfam protein families database. Nucleic Acids Res. 28:263-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beard, S. J., P. K. Hayes, F. Pfeifer, and A. E. Walsby. 2002. The sequence of the major gas vesicle protein, GvpA, influences the width and strength of halobacterial gas vesicles. FEMS Microbiol. Lett. 213:149-157. [DOI] [PubMed] [Google Scholar]
- 5.Boccazzi, P., K. J. Zhang, and W. W. Metcalf. 2000. Generation of dominant selectable markers for resistance to pseudomonic acid by cloning and mutagenesis of the ileS gene from the archaeon Methanosarcina barkeri Fusaro. J. Bacteriol. 182:2611-2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bomar, M., K. Knoll, and F. Widdel. 1985. Fixation of molecular nitrogen by Methanosarcina barkeri. FEMS Microbiol. Lett. 31:47-55. [Google Scholar]
- 7.Boone, D. R., W. B. Whitman, and Y. Koga. 2001. Genus Methanosarcina, p. 268-276. In D. R. Boone and R. W. Castenholz (ed.), Bergey's manual of systematic bacteriology. Springer, New York, N.Y.
- 8.Conway de Macario, E., D. L. Maeder, and A. J. L. Macario. 2003. Breaking the mould: archaea with all four chaperoning systems. Biochem. Biophys. Res. Commun. 301:811-812. [DOI] [PubMed] [Google Scholar]
- 9.Davis, M. A., K. A. Martin, and S. Austin. 1992. Biochemical activities of the ParA partition protein of the P1 plasmid. Mol. Microbiol. 6:1141-1147. [DOI] [PubMed] [Google Scholar]
- 10.DeBoy, R. T., E. F. Mongodin, J. B. Emerson, and K. E. Nelson. 2006. Chromosome evolution in the Thermotogales: large-scale inversions and strain diversification of CRISPR sequences. J. Bacteriol. 188:2364-2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Delcher, A. L., D. Harmon, S. Kasif, O. White, and S. L. Salzberg. 1999. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 27:4636-4641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deppenmeier, U., M. Blaut, A. Jussofie, and G. Gottschalk. 1988. A methyl-CoM methylreductase system from methanogenic bacterium strain Gö1 not requiring ATP for activity. FEBS Lett. 241:60-64. [DOI] [PubMed] [Google Scholar]
- 13.Deppenmeier, U., A. Johann, T. Hartsch, R. Merkl, R. A. Schmitz, R. Martinez-Arias, A. Henne, A. Wiezer, S. Bäumer, C. Jacobi, H. Brüggemann, T. Lienard, A. Christmann, M. Bömeke, S. Steckel, A. Bhattacharyya, A. Lykidis, R. Overbeek, H.-P. Klenk, R. P. Gunsalus, H. J. Fritz, and G. Gottschalk. 2002. The genome of Methanosarcina mazei: evidence for lateral gene transfer between bacteria and archaea. J. Mol. Microbiol. Biotechnol. 4:453-461. [PubMed] [Google Scholar]
- 14.Eisen, J., J. Heidelberg, O. White, and S. Salzberg. 2000. Evidence for symmetric chromosomal inversions around the replication origin in bacteria. Genome Biol. 1:RESEARCH0011. [DOI] [PMC free article] [PubMed]
- 15.Ewing, B., and P. Green. 1998. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8:186-194. [PubMed] [Google Scholar]
- 16.Ewing, B., L. Hillier, M. C. Wendl, and P. Green. 1998. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 8:175-185. [DOI] [PubMed] [Google Scholar]
- 17.Galagan, J. E., C. Nusbaum, A. Roy, M. G. Endrizzi, P. Macdonald, W. FitzHugh, S. Calvo, R. Engels, S. Smirnov, D. Atnoor, A. Brown, N. Allen, J. Naylor, N. Stange-Thomann, K. DeArellano, R. Johnson, L. Linton, P. McEwan, K. McKernan, J. Talamas, A. Tirrell, W. J. Ye, A. Zimmer, R. D. Barber, I. Cann, D. E. Graham, D. A. Grahame, A. M. Guss, R. Hedderich, C. Ingram-Smith, H. C. Kuettner, J. A. Krzycki, J. A. Leigh, W. X. Li, J. F. Liu, B. Mukhopadhyay, J. N. Reeve, K. Smith, T. A. Springer, L. A. Umayam, O. White, R. H. White, E. C. de Macario, J. G. Ferry, K. F. Jarrell, H. Jing, A. J. L. Macario, I. Paulsen, M. Pritchett, K. R. Sowers, R. V. Swanson, S. H. Zinder, E. Lander, W. W. Metcalf, and B. Birren. 2002. The genome of Methanosarcina acetivorans reveals extensive metabolic and physiological diversity. Genome Res. 12:532-542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Godde, J. S., and A. Bickerton. 2006. The repetitive DNA elements called CRISPRs and their associated genes: evidence of horizontal transfer among prokaryotes. J. Mol. Evol. 62:718-729. [DOI] [PubMed] [Google Scholar]
- 19.Gordon, D., C. Abajian, and P. Green. 1998. Consed: a graphical tool for sequence finishing. Genome Res. 8:195-202. [DOI] [PubMed] [Google Scholar]
- 20.Grigoriev, A. 1998. Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res. 26:2286-2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Han, C. S., and P. Chain. 2006. Finishing repeat regions automatically with Dupfinisher, p. 141-146. In H. R. A. H. Valafar (ed.), Proceedings of the 2006 International Conference on Bioinformatics & Computational Biology. CSREA Press, Las Vegas, Nev.
- 22.Kandler, O., and H. Hippe. 1977. Lack of peptidoglycan in the cell walls of Methanosarcina barkeri. Arch. Microbiol. 113:57-60. [DOI] [PubMed] [Google Scholar]
- 23.Kelman, L. M., and Z. Kelman. 2004. Multiple origins of replication in archaea. Trends Microbiol. 12:399-430. [DOI] [PubMed] [Google Scholar]
- 24.Kreisl, P., and O. Kandler. 1986. Chemical structure of the cell wall polymer of Methanosarcina. Syst. Appl. Microbiol. 7:293-299. [Google Scholar]
- 25.Li, Q., L. Li, T. Rejtar, D. J. Lessner, B. L. Karger, and J. G. Ferry. 2006. Electron transport in the pathway of acetate conversion to methane in the marine archaeon Methanosarcina acetivorans. J. Bacteriol. 188:702-710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lobo, A. L., and S. H. Zinder. 1988. Diazotrophy and nitrogenase activity in the archaebacterium Methanosarcina barkeri 227. Appl. Environ. Microbiol. 54:1656-1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopez, P., H. Philippe, H. Myllykallio, and P. Forterre. 1999. Identification of putative chromosomal origins of replication in archaea. Mol. Microbiol. 32:883-886. [DOI] [PubMed] [Google Scholar]
- 28.Lovley, D. R., and M. J. Klug. 1982. Intermediary metabolism of organic matter in the sediments of a eutrophic lake. Appl. Environ. Microbiol. 43:552-560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Markowitz, V., F. Korzeniewski, K. Palaniappan, E. Szeto, G. Werner, A. Padki, X. Zhao, I. Dubchak, P. Hugenholtz, I. Anderson, A. Lykidis, K. Mavromatis, N. Ivanova, and N. C. Kyrpides. 2006. The integrated microbial genomes (IMG) system. Nucleic Acids Res. 34:D344-D348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Metcalf, W. W., J. K. Zhang, E. Apolinario, K. R. Sowers, and R. S. Wolfe. 1997. A genetic system for archaea of the genus Methanosarcina: liposome-mediated transformation and construction of shuttle vectors. Proc. Natl. Acad. Sci. USA 94:2626-2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Metcalf, W. W., J.-K. Zhang, X. Shi, and R. S. Wolfe. 1996. Molecular, genetic, and biochemical characterization of the serC gene of Methanosarcina barkeri Fusaro. J. Bacteriol. 178:5797-5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mulder, N. J., R. Apweiler, T. K. Attwood, A. Bairoch, A. Bateman, D. Binns, P. Bradley, P. Bork, P. Bucher, L. Cerutti, et al. 2005. InterPro, progress and status in 2005. Nucleic Acids Res. 33:D201-D205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Myllykallio, H., P. Lopez, P. Lopez-Garcia, R. Heilig, W. Saurin, Y. Zivanovic, H. Philippe, and P. Forterre. 2000. Bacterial mode of replication with eukaryotic-like machinery in a hyperthermophilic archaeon. Science 288:2212-2215. [DOI] [PubMed] [Google Scholar]
- 34.Offner, S., and F. Pfeifer. 1995. Complementation studies with the gas vesicle-encoding p-vac region of Halobacterium salinarium PHH1 reveal a regulatory role for the p-gvpDE genes. Mol. Microbiol. 16:9-19. [DOI] [PubMed] [Google Scholar]
- 35.Rother, M., and W. W. Metcalf. 2004. Anaerobic growth of Methanosarcina acetivorans C2A on carbon monoxide: an unusual way of life for a methanogenic archaeon. Proc. Natl. Acad. Sci. USA 101:16929-16934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rother, M., and W. W. Metcalf. 2005. Genetic technologies for archaea. Curr. Opin. Microbiol. 8:745-751. [DOI] [PubMed] [Google Scholar]
- 37.Sono, M., M. P. Roach, E. D. Coulter, and J. H. Dawson. 1996. Heme-containing oxygenases. Chem. Rev. 96:2841-2887. [DOI] [PubMed] [Google Scholar]
- 38.Sowers, K. R. 1995. Growth of Methanosarcina spp. as single cells, p. 61-62. In F. T. Robb, K. R. Sowers, S. DasSharma, A. R. Place, H. J. Schreier, and E. M. Fleischmann (ed.), Archaea: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 39.Sowers, K. R. 2004. Methanogenesis, p. 659-679. In M. E. Schaechter (ed.), The desk encyclopedia of microbiology. Elsevier Academic Press, San Diego, Calif.
- 40.Sowers, K. R., S. F. Baron, and J. G. Ferry. 1984. Methanosarcina acetivorans sp. nov., an acetotrophic methane-producing bacterium isolated from marine sediments. Appl. Environ. Microbiol. 47:971-978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sowers, K. R., J. E. Boone, and R. P. Gunsalus. 1993. Disaggregation of Methanosarcina spp. and growth as single cells at elevated osmolarity. Appl. Environ. Microbiol. 59:3832-3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sowers, K. R., and J. G. Ferry. 1983. Isolation and characterization of a methylotrophic marine methanogen, Methanococcoides methylutens gen. nov., sp. nov. Appl. Environ. Microbiol. 45:684-690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sowers, K. R., and R. P. Gunsalus. 1995. Halotolerance in Methanosarcina spp.: role of Nɛ-acetyl-β-lysine, α-glutamate, glycine betaine, and K+ as compatible solutes for osmotic adaptation. Appl. Environ. Microbiol. 61:4382-4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sowers, K. R., J. L. Johnson, and J. G. Ferry. 1984. Phylogenetic relationships among the methylotrophic methane-producing bacteria and emendation of the family Methanosarcinaceae. Int. J. Syst. Bacteriol. 34:444-450. [Google Scholar]
- 45.Staley, J. T. 1980. The gas vacuole: an early organelle of prokaryote motility? Origins Life Evol. Biosph. 10:111-116. [Google Scholar]
- 46.Takemori, S., T. Yamazaki, and S. Ikushiro. 1993. Evolution and differentiation of P450 genes, p. 255-272. In T. Omura, Y. Ishimura, and Y. Fujii-Kuriyama (ed.), Cytochrome P450, 2nd ed. VCH Publishers, Inc., New York, N.Y.
- 47.Tatusov, R. L., N. D. Fedorova, J. D. Jackson, A. R. Jacobs, B. Kiryutin, E. V. Koonin, D. M. Krylov, R. Mazumder, S. L. Mekhedov, A. N. Nikolskaya, B. S. Rao, S. Smirnov, A. V. Sverdlov, S. Vasudevan, Y. I. Wolf, J. J. Yin, and D. A. Natale. 2003. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walsby, A. E. 1994. Gas vesicles. Microbiol. Rev. 58:94-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Welander, P. V., and W. W. Metcalf. 2005. Loss of the mtr operon in Methanosarcina blocks growth on methanol, but not methanogenesis, and reveals an unknown methanogenic pathway. Proc. Natl. Acad. Sci. USA 102:10664-10669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Whitlock, J. P., Jr., and M. S. Denison. 1995. Induction of cytochrome P450 enzymes that metabolize xenobiotics, p. 367-390. In P. R. Ortiz de Montellano (ed.), Cytochrome P450: structure, mechanism, and biochemistry. Plenum Press, New York, N.Y.
- 51.Zhilina, T. N., and G. A. Zavarzin. 1979. Comparative cytology of methanosarcinae and description of Methanosarcina vacuolata sp. nova. Microbiologiia 48:279-285. (In Russian.) [PubMed] [Google Scholar]
- 52.Zhilina, T. N., and G. A. Zavarzin. 1987. Methanosarcina vacuolata sp. nov., a vacuolated methanosarcina. Int. J. Syst. Bacteriol. 37:281-283. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.