

Abstract
Approximately 75% of eukaryotic proteins contain more than one so-called independently folding domain. However, there have been relatively few systematic studies to investigate the effect of interdomain interactions on protein stability and fewer still on folding kinetics. We present the folding of pairs of three-helix bundle spectrin domains as a paradigm to indicate how complex such an analysis can be. Equilibrium studies show an increase in denaturant concentration required to unfold the domains with only a single unfolding transition; however, in some cases, this is not accompanied by the increase in m value, which would be expected if the protein is a truly cooperative, all-or-none system. We analyze the complex kinetics of spectrin domain pairs, both wild-type and carefully selected mutants. By comparing these pairs, we are able to demonstrate that equilibrium data alone are insufficient to describe the folding of multidomain proteins and to quantify the effects that one domain can have on its neighbor.
Keywords: alpha-helix, protein folding, spectrin, m value, equilibrium denaturation
Protein domains are independent, evolutionary units that can form a single domain protein on their own or recombine with others to form part of a multidomain protein. The extent of recombination means that the vast majority of eukaryotic proteins contain more than one “independently folding” domain (1). Most protein folding studies consider these domains in isolation (2), but it may be important to consider these domains in their context; neighboring domains may be independent or there can be effects on both the stability and kinetic behavior in multidomain proteins (3–16). A complicating factor involves choice of domain boundaries (17). An early study of the two-domain fibronectin type III (fnIII) pair FNfn9 and FNfn10 (FNfn9–10) of fibronectin found that FNfn9 was much less stable alone than in FNfn9–10 (18). However, later studies showed that the original FNfn9 was “too short.” A longer form of FNfn9 had the same stability as in FNfn9–10 (13). Evidence suggests that any effects are not simply due to crowding; Ig domains from titin behave just as the isolated domain even when both N- and C-terminal neighboring domains are attached (12); other protein-specific factors must play a role. The domain interfaces in multidomain proteins vary considerably. There may be an extensive interaction surface between domain pairs, whereas in other systems there are many fewer contacts. Elements of secondary structure can extend from one domain to another. So little work has been done in this area that it is difficult to relate types of interfaces to cooperative behavior between domains. A few investigations suggest that the nature of this interface might be critical. β-Sandwich proteins are commonly found in multidomain proteins; a few have been studied in detail. Whereas the fnIII domains of fibronectin and the Ig domains of titin, with short linkers and few interdomain interactions, fold and unfold independently (12, 13, 15), similar domains with extensive interfaces (both Ig and γ-crystallin domains) show increases in stability (8, 16, 19) and changes in folding kinetics. Only one study has used protein engineering to investigate the molecular basis for interdomain stabilizing effects in detail (20). This field of investigation is important to understand protein folding in the context of multidomain proteins, which make up ≈75% of human proteins.
So few systems have been studied in detail (both kinetic and equilibrium studies on both single and multidomain proteins) that consistent mechanistic schemes of description, investigation and analysis have not been described, so it is difficult to compare different studies. In some cases, multidomain proteins have been investigated, but there are no data on the constituent domains alone (e.g., refs. 10, 19, and 21–23). An interesting paradigm is the three-helix bundle spectrin domains (24–29). These have an extended helix, so that helix C of one domain is contiguous with helix A of the next (Fig. 1). Pairs of spectrin domains unfold at significantly higher concentrations of denaturant, or at higher temperatures, than the domains alone, and only a single unfolding transition is observed (20, 30, 31). In one case, a folded domain has been shown to both increase the rate of folding and decrease the rate of unfolding of its neighbor (6). What is puzzling about the spectrin repeats is that, in some domain pairs, the increase in [denaturant]50% ([D]50%) is accompanied by an increase in unfolding m value, as expected if the two domains are unfolding as a single, larger cooperative unit, in other cases there is an increase in [D]50% but no concomitant increase in the m value. We have suggested that lower-than-expected m values are inconsistent with all-or-none “cooperative” folding behavior (20). Here we investigate this apparent anomaly by using the two-domain pairs R1516 and R1617 from α-spectrin. In both cases, the N-terminal domain folds first followed by the C-terminal domain. Whether apparent equilibrium “cooperativity” is observed (i.e., whether the m value is consistent with the unfolding of the protein as a single cooperative unit) depends on the relative rate constants for the folding of the constituent domains. That is, apparent equilibrium cooperativity depends on the kinetic behavior.
Fig. 1.

Structure of the two-domain spectrin fragment R1516. The C helix of R15 forms a continuous helix with the A helix of R16 (Protein Data Bank ID 1U5P).
This study suggests that kinetic analysis is critical for the investigation of the effects that one domain may have on its neighbors; equilibrium data alone are not sufficient. It also shows that unexpected equilibrium m values or changes in m values may be diagnostic of complex changes in folding kinetics.
Results
Equilibrium Studies.
A comparison of the equilibrium denaturation curves of the individual domains of R15, R16, and R17 to those of R1516 and R1617 are shown in Fig. 2a. CD and fluorescence traces all overlay. In R1516 and R1617, only a single transition is observed, with a higher apparent [urea]50% than for the constituent domains. Importantly, however, whereas the m value of the transition increases for R1617 (consistent with the unfolding of the entire protein as a single cooperative unit), the m value of R1516 remains the same as that of the individual domains (Table 1). It has been shown that both mutation and change of conditions can induce noncooperative equilibrium behavior in R1617 (20), resulting in a decrease in m value and in some cases a loss of coincidence of CD and fluorescence data (Fig. 7, which is published as supporting information on the PNAS web site).
Fig. 2.

Equilibrium and kinetic data for R1516. (a) Equilibrium denaturation curves of spectrin R1516 (purple), R1617 (black), and their constituent domains, R15 (blue), R16 (red), and R17 (cyan). These data follow the change in fluorescence at 350 nm (filled circles) and change in CD signal at 222 nm (open circles), which all overlay well. Note that the apparent m values of all proteins are the same, within error, as judged by the slope of the transition, except for R1617, which has an increased m value (Table 1). Data for individual domains were taken from ref. 40; data for R1617 were taken from ref. 20. (b) A plot of the natural logarithm of the observed rate constants for R1516. Data from fluorescence (filled symbols) and CD (open symbols) measurements are shown. Closed squares show rate constants which could only be observed in double-jump, interrupted refolding experiments. Blue, data for the R15 domain in R1516 (with R16 unfolded); Red, data for the R16 domain in R1516 (with R15 folded). For details on the assignment of the phases, see Supporting Text. (Note that a third, proline isomerization-limited phase was also observed in refolding experiments, but this has been omitted for clarity; see Supporting Text).
Table 1.
The folding of spectrin domains alone and in the presence of a neighboring domain
| Protein | Equilibrium m value (kcal·mol−1·M−1) | kfH2O (s−1) | kfH2O (s−1) | ΔΔGD-N(kcal·mol−1)* |
|---|---|---|---|---|
| R15† | 1.8 (±0.1) | 30,000 (±6,000) | 1.7 (±0.3) | — |
| R16† | 1.9 (±0.1) | 125 (±3) | 2.6 × 10−3 (±0.3 × 10−3) | — |
| 17† | 2.0 (±0.1) | 30 (±2) | 4.0 × 10−4 (±0.3 × 10−4) | — |
| R1516 | 1.8 (±0.1) | — | — | — |
| R15 in presence of unfolded R16 | — | 26,000 (±4,000) | 6.0 × 10−2 (±2.0 × 10−2) | 1.9 (±0.4) |
| R16 in presence of folded R15 | — | 730 (±40) | 7.5 × 10−4 (±1.9 × 10−4) | 1.7 (±0.2) |
| R1617‡ | 3.0 (±0.1) | — | — | — |
| R16 in presence of unfolded R17‡ | — | 4.2 (±0.2) | 9.4 × 10−4 (±1.8 × 10−4) | 1.2§ (±0.4) |
| R17 in presence of folded R16‡ | — | 1,000 (±200) | 1.3 × 10−5 (±0.6 × 10−5) | 4.1 (±0.5) |
*ΔΔGD-N determined from changes in rate constants of folding and unfolding compared to the domain alone. Note the high uncertainty due largely to errors from extrapolating unfolding data over a significant range of urea concentrations to 0 M denaturant, and for R15 of the refolding rate constants (which are very fast) to 0 M denaturant.
†Data taken from ref. 40.
‡Data taken from ref. 20.
§R16 in ΔΔGD-N in R1617 calculated taking into account the presence of an intermediate with a stability of 2.6 kcal·mol−1.
Kinetics Studies of R1516 and R1617.
R1516.
The folding kinetics of R1516 were studied by using CD and fluorescence stopped-flow. Single- and double-jump experiments allowed the assignment of the R15 and R16 domains within the R1516 construct (for details of the assignment of the kinetic phases, see Supporting Text and Fig. 8, which are published as supporting information on the PNAS web site). The folding is simple and sequential: R15DR16D ⇆ R15NR16D ⇆ R15NR16N, where D and N represent denatured and native domains, respectively.
R15 folds rapidly then R16 folds more slowly; both folding phases are observed in single-jump experiments (Fig. 2b). The unfolding is more complex. R16 unfolds first and then R15 unfolds with a higher rate constant. As there is a slow unfolding phase (R16) followed by a faster phase (R15) only the unfolding of R16 can be observed in single jump unfolding experiments (unfolding of R15 can be seen in double-jump, interrupted refolding experiments) (Fig. 2b).
R1617.
The folding of R1617 has been described (6). R16 folds first, followed by R17. R17 folds faster than R16. As this fast phase follows the slower R16 folding phase only the folding of R16 can be seen in single jump experiments (double jump interrupted unfolding experiments were necessary to see the folding of R17 in R1617) (Fig. 3a). In the unfolding, R17 unfolds first then R16. Below 6 M urea, the unfolding of R16 is faster than R17 unfolding and so this phase is not observed in single-jump unfolding experiments. However, due to differences in the slope of the unfolding arms, the chevrons cross at ≈6 M urea. Above 6 M urea, both unfolding phases are observable in single jump experiments; R17 unfolds first (fast) and R16 unfolds second (more slowly).
Fig. 3.

Kinetics of R1617 wild-type and mutants. The data shown in red represent the folding and unfolding of the R16 domain in R1617, and the data shown in cyan represent the folding and unfolding data for the R17 domain in R1617. The data shown in filled symbols were determined by using single-jump stopped flow measurements. The data shown in open circles could only be observed in double-jump, interrupted unfolding experiments. (a) The kinetics of R1617 wild-type (data taken from ref. 6 where details of the full assignment of the kinetic phases can also be found). (b) The kinetics of R1617 S20A. As for wild-type at [urea] below ≈6 M, only a single unfolding phase can be detected. (c) The kinetics of R1617 L203A. Two unfolding phases are observed at all [urea]. Note that the rollover in the folding kinetics of R1617 reflects dead-time formation of a collapsed intermediate, which is marginally stable, has little secondary structure, and is not populated at equilibrium (6).
Two mutants of R1617 were studied to investigate the effects of the kinetics on apparent equilibrium cooperativity: S20A, which apparently unfolds cooperatively at equilibrium (high m value and coincidence of CD and fluorescence data), and L203A, which unfolds noncooperatively at equilibrium (low m value and noncoincidence of CD and fluorescence data). S20A has the same kinetic signature as wild type (Fig. 3b). However, L203A has two unfolding phases observable at all concentrations of urea (Fig. 3c).
Discussion
Equilibrium m Values and Cooperative Unfolding.
It is well established that the equilibrium m value reflects the total change in accessible surface area on unfolding (32). Large proteins have higher m values than small proteins. For a protein with two domains with the same structure (and thus the same m value, as for R1516) there are several possible scenarios, all of which have been observed experimentally.
The domains fold and unfold entirely independently.
In this case, what is observed depends on the [D]50% of the two domains. (i) If the two domains have widely separated [D]50%, then two transitions will be observed and each will have the m value of the domain alone. This has been seen in natural two- and three-domain fragments of titin (12, 15) and in artificial two-domain constructs (e.g., ref. 33). (ii) If the two domains have the same [D]50%, as is the case for two identical domains cloned in tandem then only a single transition will be observed. This has been observed in tandem repeats of identical domains made for AFM experiments (34) and in a two-domain repeat of the B domain of protein A (35). (iii) However, if the two domains have different, but close [D]50% values then a single transition will be observed but the apparent m value will be lower than for either domain alone (e.g., mutants of spectrin domain pairs, ref. 20).
The two domains unfold as a single cooperative unit in an all-or-none fashion: No intermediate states between fully unfolded and completely folded proteins are populated at equilibrium.
In this case, only a single transition will be observed, and the m value will be approximately double that of the single domains; doubling the size of CI2 by inserting a second domain into a loop resulted in almost double the m value (36). These scenarios are all modeled in Fig. 9, which is published as supporting information on the PNAS web site.
For R1617, a single transition is seen at equilibrium and the m value is ≈1.6 times that of R16 or R17. Thus, R1617 displays apparently cooperative all-or-none behavior, and there is no evidence for any accumulation of partly folded intermediate species.
Equilibrium Behavior of R1516.
Only a single transition is observed for R1516 and the CD and fluorescence data overlay (Fig. 2a), usually taken to be an indication of cooperative folding (secondary and tertiary structure are forming concomitantly). However, the m value is the same as R15 and R16 alone; it does not appear to be folding as a single cooperative unit, unlike R1617 (Table 1).
Folding Pathways of R1516 and R1617 Are the Same.
In both R1516 and R1617 the N-terminal domain folds first, at a rate similar to the domain alone. This domain (R15 in R1516 and R16 in R1617) is stabilized by its unfolded neighbor by decreasing the unfolding rate constant. The C-terminal domain folds second, and unfolds first, and is stabilized by its folded neighbor through both an increase in its folding rate constant and a decrease in its unfolding rate constant (Fig. 4).
Fig. 4.

The folding pathways of R1516 (a) and R1617 (b). The rate constants shown are the folding and unfolding rates extrapolated to 0 M denaturant. In both cases, the N-terminal domain folds first, followed by the C-terminal domain. In R1617, there is also a low stability partly folded early intermediate (I1) that has little secondary structure but can be detected by a dead-time change in fluorescence (6).
The Effect of Neighboring Domains in R1516 on Stability and Kinetics.
We can compare R15 alone and R15 in R1516 (Fig. 5 and Table 1). The folding rate constant of R15 is unaffected by unfolded R16. However, the unfolding rate constant of R15 is significantly reduced in the presence of unfolded R16. R15 is stabilized by ≈1.9 kcal·mol−1 by unfolded R16. We never observe the folding or unfolding of R15 in the presence of folded R16. However, the unfolding of R15 in fully folded R1516 must be slower than the unfolding of the R16 domain; i.e., R15 must be stabilized to a greater extent by folded R16 than by unfolded R16.
Fig. 5.

Chevron plots for R15 and R16 alone and in R1516. The dependence of the observed rate constants for folding and unfolding against [urea]. Blue filled circles, R15 domain alone; blue open circles, R15 in R1516; the solid lines are fits to a two-state equation. In R1516, R15 is folding and unfolding in the presence of an unfolded R16 domain. The folding rate constants are the same as for R15 alone, but R15 unfolds more slowly in R1516. Red filled circles, R16 domain alone; red open circles, R16 in R1516; the solid lines are fits to a sequential transition state model (41, 42). In R1516, R16 is folding and unfolding in the presence of a folded R15 domain. The R16 domain folds more rapidly and unfolds more slowly in R1516.
A comparison of the kinetics of R16 alone to that of R16 in R1516 (Fig. 5 and Table 1) shows that both the folding and unfolding rate constants are affected by the neighboring folded R15 domain. R16 is stabilized by ≈1.7 kcal·mol−1 by folded R15. We never observe R16 in the presence of unfolded R15; R15 folds much more rapidly than R16 and can only be observed to unfold after the R16 domain has unfolded.
Equilibrium Cooperativity Can Be Explained by the Kinetics.
From the folding and unfolding rate constants, the equilibrium populations of different species at all urea concentrations can be determined (Fig. 6): the native state, N (both domains folded), the denatured state, D (both domains unfolded), and the intermediate species, I (for R1516, I has a folded R15 and an unfolded R16 domain; for R1617 I has a folded R16 and an unfolded R17 domain). In the transition region, R1516 has a significant population of intermediate (≈40%), but in R1617, I never accumulates to >10%. Modeling of the data to predict equilibrium curves (Fig. 6 c and d) demonstrates that population of I at equilibrium is the direct cause of the lower than expected m value in R1516.
Fig. 6.

The equilibrium populations of the native (N), intermediate (I), and denatured (D) species at different denaturant concentrations (determined from the kinetic rate constants). (a) R1516. (b) R1617 wild-type. (e) R1617 with the mutation S20A. (d) R1617 with the mutation L203A. The filled circles represent the population of N, the open circles represent the population of I (folded R15 in R1516 and folded R16 in R1617 wild type and mutants), and the filled triangles represent the population of D. The population of I is negligible at all denaturant concentrations for wild-type R1617 and the apparently cooperative mutant S20A. I is significantly populated in R1516 and the “noncooperative” mutant of R1617, L203A. These data were used to model the expected equilibrium data for R1516 (c) and wild-type R1617 (d). It was assumed that the two domains had the same CD signal (as they have approximately the same number of helical residues) and that R16 (two Trp residues) has double the fluorescence change of R15 or R17 (one Trp each). The modeled and experimental data overlay. Thus, it is apparent that it is the population of an intermediate at equilibrium that leads to a lower equilibrium m value in R1516 (and the L203A mutant of R1617) than in wild-type R1617.
I accumulates in the transition region of R1516 and not R1617 because of the relative rates of the formation and degradation. In R1617 I (with only R16 folded) folds to N faster than it is formed (Fig. 4) (the rapid folding of I to N cannot be observed except in double-jump experiments). Thus, for R1617, there is a single rate-determining step for the formation of N, and I does not accumulate. In R1516, I, with only R15 folded, forms more rapidly than it folds to N, thus I accumulates and both refolding phases are observed: the folding of R16 is slower than R15 even in the presence of folded R15. In unfolding conditions, in the transition region, in both R1516 and R1617, I unfolds more rapidly than it is formed.
This finding suggests that, for a two-domain protein where the domains fold sequentially, an apparently cooperative transition with a high m value will be observed only where there is a single observable rate constant for both folding and unfolding. The results from the mutants of R1617 are consistent with this hypothesis. Apparent equilibrium cooperativity is lost in the L203A mutant, which has more than one observable unfolding rate constant in the transition region. Analysis of the kinetic data shows that an intermediate accumulates to ≈80% at the apparent [D]50% (Fig. 6d). R1617 S20A maintains apparent equilibrium cooperativity (high m value and coincidence of CD and fluorescence signals). There is only one observable rate constant for both folding and unfolding in the transition region for this mutant. Only above 6.5 M urea are two unfolding rate constants observed (as in wild type). Further analysis shows that, as for wild type, I is never populated to >10% in the transition region (Fig. 6c).
Conclusion
Although ≈75% of all human proteins contain more than one domain, there have been few investigations into the effect that the folding of one domain can have on the folding of a neighbor. Interdomain interactions may have important biological significance. During protein synthesis, cotranslational folding of domains, one at a time will protect against misfolding and/or aggregation (37). Catalysis of the folding of one domain by a preformed neighbor might also enhance the efficiency of folding after synthesis (6). Furthermore, in the cell, long-lived proteins will undergo a number of individual domain unfolding events. An unfolded domain is vulnerable to proteolysis or aggregation. Stabilizing interdomain interactions may protect the protein by both increasing the unfolding half-life (decreasing the likelihood of unfolding) and speed up refolding (recovery of the native fold).
How far cooperative folding, if observed, depends on the nature of the interface between neighboring domains is unknown. Little work has been done to assess whether folding pathways themselves are affected by interdomain interactions, and in the case of one protein, phosphoglycerate kinase, the data are somewhat contradictory (11, 38, 39). For progress to be made in this field, common, systematic methods of analysis are vital. Direct comparison must be made between domains in isolation and the multidomain protein. Importantly, this analysis of spectrin domain pairs demonstrates that equilibrium data alone are insufficient to quantify the effects of one domain on another. Moreover, m value differences can reflect complex kinetics that can only be dissected by detailed experimental analysis, and unexpectedly low m values can indicate the presence of an unfolding intermediate, even where CD and fluorescence data coincide.
Materials and Methods
Protein Mutagenesis and Purification.
The sequences of R15, R16, R17, and R1516 and R1617 are described in Fig. 10, which is published as supporting information on the PNAS web site. The proteins were mutated, expressed, and purified as described (40).
Reagents.
All experiments were carried out in sodium phosphate buffer, pH 7.0, at 25 ± 0.1°C. Concentrations of urea solutions were determined by refractive index. R1617 wild type and mutants were studied in 5 mM DTT.
Thermodynamic Measurements.
The thermodynamic properties of R1516 were determined as described (20). Samples were left to equilibrate for at least 4 h.
Kinetic Studies.
Kinetic studies were carried out by using an Applied Photophysics SX.18MX and an Applied Photophysics π*-180 instrument. Protein concentration was ≈1 μM for fluorescence measurements and ≈5 μM for CD measurements. An excitation wavelength of 280 nm was used with emission monitored at wavelengths >320 nm. CD data were collected at 222 nm. Between 10 and 15 kinetic traces were obtained at all [urea]. The data for single-jump experiments were fitted by using Kaleidagraph (Synergy Software, Reading, PA); double-jump experiments were fitted globally using Prism (GraphPad, San Diego, CA). All rate constants were independent of protein concentration.
Supplementary Material
Acknowledgments
We thank Chris Waudby for help with the kinetic modeling of the populations of species and Terrence Oas and Kathryn Scott for helpful discussion. This work was funded by the Wellcome Trust. J.C. is a Wellcome Trust Senior Research Fellow.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS direct submission.
References
- 1.Teichmann SA, Chothia C, Gerstein M. Curr Opin Struct Biol. 1999;9:390–399. doi: 10.1016/S0959-440X(99)80053-0. [DOI] [PubMed] [Google Scholar]
- 2.Jackson SE. Fold Des. 1998;3:R81–R91. doi: 10.1016/S1359-0278(98)00033-9. [DOI] [PubMed] [Google Scholar]
- 3.Zarnt T, Tradler T, Stoller G, Scholz C, Schmid FX, Fischer G. J Mol Biol. 1997;271:827–837. doi: 10.1006/jmbi.1997.1206. [DOI] [PubMed] [Google Scholar]
- 4.Kirkitadze MD, Dryden DT, Kelly SM, Price NC, Wang X, Krych M, Atkinson JP, Barlow PN. FEBS Lett. 1999;459:133–138. doi: 10.1016/s0014-5793(99)01205-3. [DOI] [PubMed] [Google Scholar]
- 5.Kirkitadze MD, Krych M, Uhrin D, Dryden DT, Smith BO, Cooper A, Wang X, Hauhart R, Atkinson JP, Barlow PN. Biochemistry. 1999;38:7019–7031. doi: 10.1021/bi982453a. [DOI] [PubMed] [Google Scholar]
- 6.Batey S, Scott KA, Clarke J. Biophys J. 2006;90:2120–2130. doi: 10.1529/biophysj.105.072710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cordier-Ochsenbein F, Guerois R, Baleux F, Huynh-Dinh T, Lirsac PN, Russo-Marie F, Neumann JM, Sanson A. J Mol Biol. 1998;279:1163–1175. doi: 10.1006/jmbi.1998.1829. [DOI] [PubMed] [Google Scholar]
- 8.Rothlisberger D, Honegger A, Pluckthun A. J Mol Biol. 2005;347:773–789. doi: 10.1016/j.jmb.2005.01.053. [DOI] [PubMed] [Google Scholar]
- 9.Robertsson J, Petzold K, Lofvenberg L, Backman L. Cell Mol Biol Lett. 2005;10:595–612. [PubMed] [Google Scholar]
- 10.Head JG, Houmeida A, Knight PJ, Clarke AR, Trinick J, Brady RL. Biophys J. 2001;81:1570–1579. doi: 10.1016/s0006-3495(01)75811-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osvath S, Kohler G, Zavodszky P, Fidy J. Protein Sci. 2005;14:1609–1616. doi: 10.1110/ps.051359905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scott KA, Steward A, Fowler SB, Clarke J. J Mol Biol. 2002;315:819–829. doi: 10.1006/jmbi.2001.5260. [DOI] [PubMed] [Google Scholar]
- 13.Steward A, Adhya S, Clarke J. J Mol Biol. 2002;318:935–940. doi: 10.1016/S0022-2836(02)00184-5. [DOI] [PubMed] [Google Scholar]
- 14.Wenk M, Jaenicke R, Mayr EM. FEBS Lett. 1998;438:127–130. doi: 10.1016/s0014-5793(98)01287-3. [DOI] [PubMed] [Google Scholar]
- 15.Politou AS, Gautel M, Improta S, Vangelista L, Pastore A. J Mol Biol. 1996;255:604–616. doi: 10.1006/jmbi.1996.0050. [DOI] [PubMed] [Google Scholar]
- 16.Jager M, Gehrig P, Pluckthun A. J Mol Biol. 2001;305:1111–1129. doi: 10.1006/jmbi.2000.4342. [DOI] [PubMed] [Google Scholar]
- 17.Politou AS, Gautel M, Joseph C, Pastore A. FEBS Lett. 1994;352:27–31. doi: 10.1016/0014-5793(94)00911-2. [DOI] [PubMed] [Google Scholar]
- 18.Spitzfaden C, Grant RP, Mardon HJ, Campbell ID. J Mol Biol. 1997;265:565–579. doi: 10.1006/jmbi.1996.0736. [DOI] [PubMed] [Google Scholar]
- 19.Rudolph R, Siebendritt R, Nesslauer G, Sharma AK, Jaenicke R. Proc Natl Acad Sci USA. 1990;87:4625–4629. doi: 10.1073/pnas.87.12.4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Batey S, Randles LG, Steward A, Clarke J. J Mol Biol. 2005;349:1045–1059. doi: 10.1016/j.jmb.2005.04.028. [DOI] [PubMed] [Google Scholar]
- 21.Santra MK, Banerjee A, Krishnakumar SS, Rahaman O, Panda D. Eur J Biochem. 2004;271:1789–1797. doi: 10.1111/j.1432-1033.2004.04096.x. [DOI] [PubMed] [Google Scholar]
- 22.Martin A, Schmid FX. J Mol Biol. 2003;329:599–610. doi: 10.1016/s0022-2836(03)00433-9. [DOI] [PubMed] [Google Scholar]
- 23.Sanchez IE, Morillas M, Zobeley E, Kiefhaber T, Glockshuber R. J Mol Biol. 2004;338:159–167. doi: 10.1016/j.jmb.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 24.Speicher DW, Marchesi VT. Nature. 1984;311:177–180. doi: 10.1038/311177a0. [DOI] [PubMed] [Google Scholar]
- 25.Winograd E, Hume D, Branton D. Proc Natl Acad Sci USA. 1991;88:10788–10791. doi: 10.1073/pnas.88.23.10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan Y, Winograd E, Viel A, Cronin T, Harrison SC, Branton D. Science. 1993;262:2027–2030. doi: 10.1126/science.8266097. [DOI] [PubMed] [Google Scholar]
- 27.Pascual J, Pfuhl M, Rivas G, Pastore A, Saraste M. FEBS Lett. 1996;383:201–207. doi: 10.1016/0014-5793(96)00251-7. [DOI] [PubMed] [Google Scholar]
- 28.Pascual J, Pfuhl M, Walther D, Saraste M, Nilges M. J Mol Biol. 1997;273:740–751. doi: 10.1006/jmbi.1997.1344. [DOI] [PubMed] [Google Scholar]
- 29.Kusunoki H, Minasov G, Macdonald RI, Mondragon A. J Mol Biol. 2004;344:495–511. doi: 10.1016/j.jmb.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 30.MacDonald RI, Cummings JA. Proc Natl Acad Sci USA. 2004;101:1502–1507. doi: 10.1073/pnas.0308059100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Macdonald RI, Pozharski EV. Biochemistry. 2001;40:3974–3984. doi: 10.1021/bi0025159. [DOI] [PubMed] [Google Scholar]
- 32.Myers JK, Pace N, Scholtz JM. Protein Sci. 1995;4:2138–2148. doi: 10.1002/pro.5560041020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Best RB, Li B, Steward A, Daggett V, Clarke J. Biophys J. 2001;81:2344–2356. doi: 10.1016/S0006-3495(01)75881-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rounsevell RW, Steward A, Clarke J. Biophys J. 2005;88:2022–2029. doi: 10.1529/biophysj.104.053744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arora P, Hammes GG, Oas TG. Biochemistry. 2006 doi: 10.1021/bi060923s. in press. [DOI] [PubMed] [Google Scholar]
- 36.Inaba K, Kobayashi N, Fersht AR. J Mol Biol. 2000;302:219–233. doi: 10.1006/jmbi.2000.4024. [DOI] [PubMed] [Google Scholar]
- 37.Netzer WJ, Hartl FU. Nature. 1997;388:343–349. doi: 10.1038/41024. [DOI] [PubMed] [Google Scholar]
- 38.Hosszu LL, Craven CJ, Spencer J, Parker MJ, Clarke AR, Kelly M, Waltho JP. Biochemistry. 1997;36:333–340. doi: 10.1021/bi961784p. [DOI] [PubMed] [Google Scholar]
- 39.Reed MA, Hounslow AM, Sze KH, Barsukov IG, Hosszu LL, Clarke AR, Craven CJ, Waltho JP. J Mol Biol. 2003;330:1189–1201. doi: 10.1016/s0022-2836(03)00625-9. [DOI] [PubMed] [Google Scholar]
- 40.Scott KA, Batey S, Hooton KA, Clarke J. J Mol Biol. 2004;344:195–205. doi: 10.1016/j.jmb.2004.09.037. [DOI] [PubMed] [Google Scholar]
- 41.Scott KA, Clarke J. Protein Sci. 2005;14:1617–1629. doi: 10.1110/ps.051377105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanchez IE, Kiefhaber T. J Mol Biol. 2003;325:367–376. doi: 10.1016/s0022-2836(02)01230-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
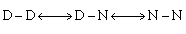{kind=link}
{kind=link}