


Abstract
There has been a long-standing interest in the discovery of unnatural nucleotides that can be incorporated into DNA by polymerases. However, it is difficult to predict which nucleotide analogs will prove to have biological relevance. Therefore, we have developed a new screening method to identify novel substrates for DNA polymerases. This technique uses the polymerase itself to select a dNTP from a pool of potential substrates via incorporation onto a short oligonucleotide. The unnatural nucleotide(s) is then identified by high-resolution mass spectrometry. By using a DNA polymerase as a selection tool, only the biologically relevant members of a small nucleotide library can be quickly determined. We have demonstrated that this method can be used to discover unnatural base pairs in DNA with a detection threshold of ≤10% incorporation.
INTRODUCTION
DNA polymerases are ubiquitous and necessary enzymes for all living organisms. During DNA replication, polymerases normally only use four different nucleotides; however, a great number of other nucleotides are able to act as substrates for various polymerases. Many of these nucleotides or nucleotide analogs have found therapeutic applications, whereas others are useful tools for biochemical and biological studies. It is, therefore, of great interest to develop new methodology to identify novel nucleotides that can act as substrates for DNA polymerases.
All of the major replicative polymerases share a similar overall conformation and appear to use a similar two metal catalytic center, implying a similar mechanism of nucleotide incorporation (1,2). However, the mechanism(s) by which different polymerases select between right and wrong (d)NTPs vary substantially, as can be seen by the differential incorporation of various analogs by different polymerases (3–6). For example, DNA pol α1, a B family polymerase, does not require the base pair formed between the incoming dNTP and the template base being replicated to have either a similar shape as a correct base pair or Watson–Crick hydrogen bonds. In contrast, both human and herpes primase choose whether or not to polymerize a NTP based on the formation of Watson–Crick hydrogen bonds between the incoming NTP and the template base.
A further complication in nucleotide design is the differential interaction of polymerases with a base analog present in a dNTP versus in the template. Although a given analog may act as an acceptable substrate when in the template strand, it may not be incorporated as a dNTP, or vice versa (3,7,8). In combination with the different mechanisms of nucleotide selectivity, this lack of consistency in polymerase behavior makes the de novo design of nucleotide analogs difficult. Moreover, it emphasizes the importance of examining nucleotide analogs in both the template and growing strands of DNA.
In addition to single nucleotide analogs, much effort has been focused on the search for novel base pairs that can function seamlessly in DNA (7). The addition of one or more new base pairs in DNA has great potential for applications as wide ranging as data storage, molecular computers and biochemical insight into the function of DNA polymerases themselves. However the discovery of a true third base pair remains elusive. A significant hindrance to progress in this area is the time necessary to synthesize and test potential novel base pairs. Thus far, research into nucleotide analogs has relied on one at a time trial and error approach. This results in a substantial amount of time being spent on failed base pairs. In order to circumvent this, we have developed a new approach to base pair discovery that allows for the selection of biologically acceptable base pairs from a pool of nucleotides (Figure 1). The key features of our approach are the use of a DNA polymerase as the selection tool and mass spectrometry as the identification tool. Mass spectrometry has been used previously to identify naturally modified nucleotides in RNA (9). Through this, it has been shown to be a capable method of analysis for identification of a single compound in a heterogeneous mixture. Furthermore, the sensitivity of LC-MS detection makes it a feasible tool for the analysis of very small quantities of material.
Figure 1.

Overview of nucleotide selection scheme. A template containing a nucleotide analog is incubated with a library of nucleoside triphosphate analogs and a DNA polymerase. The extended oligonucleotides are separated, enzymatically digested to nucleosides and analyzed by LC-MS.
As a proof of principle, we first tested the selection scheme on a known unnatural base pair. For this, the bases 2,4-diaminopyrimidine (‘κ’) and xanthine (‘X’) (Figure 2) were chosen for their known ability to be moderately incorporated by some DNA polymerases (10,11). Using a template containing dκ and the Klenow Fragment of Escherichia coli DNA polymerase I (KF), we were able to both selectively identify dXTP from a pool of nucleotides and detect dXTP at low levels of incorporation (∼10%).
Figure 2.

The positive control unnatural base pair: 2,4-diaminopyrimidine (κ) and xanthine (X).
MATERIALS AND METHODS
All reagents were purchased from Sigma-Aldrich unless otherwise noted. Enzymes were purchased from New England BioLabs, unless otherwise noted. Solvents were distilled immediately before use and synthetic reactions were carried out under N2 (g). The synthesis of dκ required significant alteration from the published procedure (Supplemental Data). dXTP was synthesized as reported previously (12). DNA templates were synthesized on an ABI 394 DNA synthesizer.
5′ End labeling and annealing of DNA
DNA was 5′-[32P]-labeled using polynucleotide kinase and [γ-32P]ATP, and the reaction was stopped by heating to 65°C for 15 min. Slow cooling of this mixture allowed for annealing of the self-priming template. DNA was desalted before use on a P2 BioGel column.
Polymerization assays with pol α and KF
Assays contained 1 µM to 1 mM 5′-[32P]DNA, 50 mM Tris–HCl (pH 7.6), 10 mM MgCl2, 1 mM DTT, 0.05 mg/ml BSA and various concentrations of dXTP and enzyme. For the selection method, polymerization reactions were initiated by the addition of KF (0.5 U/µl), incubated at 37°C for 60 min and quenched by adding an equal volume of gel loading buffer (90% formamide in 1× TBE). Quenched reaction mixtures were loaded directly onto a polyacrylamide gel (20% acrylamide, 1× TBE, 0.8 mm × 300 mm × 375 mm).
DNA recovery
The primer and primer +1 bands in the polyacrylamide gel were visualized by autoradiography. After localization, the primer +1 band was carefully excised and the DNA was extracted. The extraction procedure used was a modification of published procedures (13). Briefly, the sample was thoroughly crushed in a small tube, then subjected to three consecutive extractions with ∼0.5 ml buffer each [5 M NH4AcO, 1 mM EDTA and 10 mM Mg(AcO)2]. The extractions, all on a rotator, were of the following durations: (i) 3 h at 37°C; (ii) overnight at 4°C; (iii) 3 h at 37°C. After each extraction, the mixture was filtered and the filtrate saved; after the final extraction, the gel was rinsed with a further 0.5 ml buffer. The combined filtrates were then dried to a solid, which was desalted on a P2 Bio-Gel column.
Oligonucleotide digestion
DNA was incubated with calf intestinal phosphodiesterase I (Sigma, 7 mU per 1 nmol reaction; this preparation also contains alkaline phosphatase as a contaminant, and thus it gives nucleosides as the product) at 37°C for 2 h. Following this, the proteins were removed using a Microcon-10 filtration tube. The samples were washed twice to ensure complete recovery of the nucleosides. Nucleoside mixtures were desalted by flash chromatography using C18-derivitized silica. A column (60 mm × 5 mm) was pretreated with methanol and equilibrated with water. After a wash of one column volume water, the nucleosides were eluted with 100% methanol. The methanol was removed by evaporation before LC-MS analysis.
LC-MS
Liquid chromatography was performed on an Agilent C18 microbore column (50 µM diameter) at a flow rate of 10 µl/min. The LC eluent flowed directly into a PE Sciex Q-Star Pulsar mass spectrometer. Mass standards consisted of a solution of PEG-200 enriched in the lower mass region by passing the solution through a reverse phase cartridge. Because the longer chains are retained slightly on reverse phase media, this effectively skews the molecular weight standard solution to a lower mass region, which is more useful for nucleosides. This solution was concurrently introduced into the spectrometer using a t-joint fitted between the LC outlet and the MS inlet.
RESULTS AND DISCUSSION
The selection strategy outlined in Figure 1 was developed using a known unnatural base pair. A short, self-pairing oligonucleotide containing dκ was incubated with KF and a pool of dNTPs. This allows the polymerase to select for a novel base pair through covalent linkage of the unnatural dNTP (Y) to the same DNA strand as it partner. Any base pair thus selected will be de facto biologically relevant because of the method of its selection. PAGE was used to separate the positive hits by virtue of the difference in mobility of the original DNA strand and one that is extended by one additional nucleotide. Subsequent enzymatic digestion of the extracted oligonucleotides and mass spectral analysis of the resultant nucleosides then provides the molecular formula of the positive hits. From this, the identity of the incorporated nucleotide is easily determined.
Several DNA polymerases and template sequences were tested for use in the primer extension selection (Figure 3). In designing the DNA strand, we sought to use the shortest possible sequence for two reasons: (i) a smaller number of natural nucleosides in the final LC-MS sample increases the ability to detect the lower abundance unnatural nucleoside; and (ii) baseline separation of the primer and primer +1 bands by PAGE is necessary for accurate identification of only extended products, and is more easily achieved for smaller pieces of DNA. Additionally, the short loop sequence was designed to give maximum duplex stability (14,15). We found that the 23mer shown in Figure 4 provided an optimum primer:template for balancing a short sequence with enzymatic activity (Figure 4). In this sequence context, both KF and pol α incorporated dXTP into DNA across from template dκ (Figure 5). However, Moloney murine leukemia virus reverse transcriptase and T4 DNA polymerase did not detectably incorporate the analog (<1%; data not shown). As KF incorporates dXTP opposite dκ much more effectively than does pol α, all further studies were performed with this enzyme. Manipulation of concentrations of dXTP and KF allowed us to control the amount of primer extension, and thereby assess the threshold for nucleoside detection by this method.
Figure 3.

Sequences of oligonucleotides studied.
Figure 4.

Testing the DNA sequences in Figure 3 for activity as substrates for pol α and KF. The polymerase used and the oligonucleotide length are indicated in the figure. For each length template, the 5 lanes are the: 1, no enzyme control; 2, no dNTP control; 3, +1 extension; 4, +2 extension; and 5, full extension.
Figure 5.
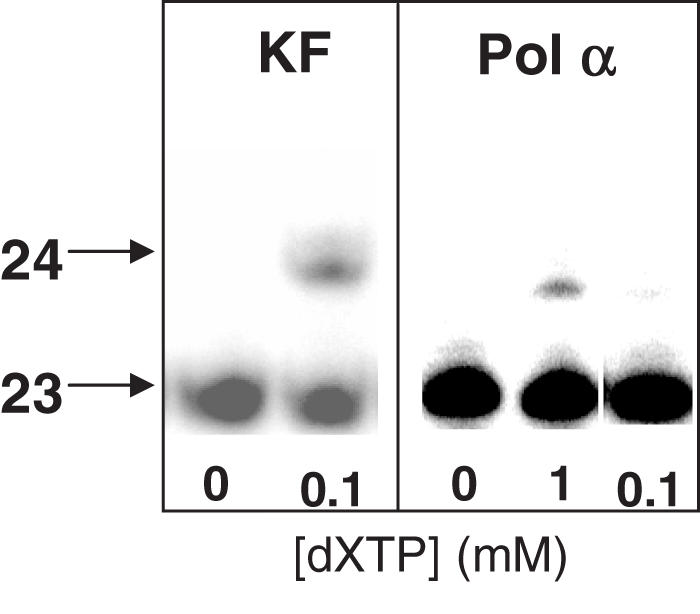
Incorporation of dXTP opposite dκ by KF and pol α. Polymerase and amount of dXTP used are indicated. The lengths of the DNA bands are indicated with arrows.
After quenching the reactions and separating the primer and primer +1 products by PAGE, the products were located by autoradiography and the primer +1 product extracted from the gel. Optimization of extraction conditions resulted in a maximum recovery of ∼70% of the DNA from the gel. This was then enzymatically digested to yield the free nucleosides. The crude mixture of nucleosides was passed through a C18 column before analysis by LC-MS in order to reduce the amount of salt, which contaminates the detector.
Initial studies identified LC conditions that gave efficient separation of nucleosides and MS parameters for the sensitive detection of the six nucleosides present in the mixture (Table 1). In order to obtain a highly accurate mass of the nucleoside samples, they must contain an internal mass standard. This was accomplished using a post-column t-joint to concurrently infuse both the sample and a solution of molecular weight standards into the MS. Detection and mass analysis parameters were then optimized using mixtures of purified nucleosides in similar concentrations to that of predicted samples from the selection scheme.
Table 1.
LC method for nucleoside separation
| Time | %B |
|---|---|
| 0 | 0 |
| 5 | 0 |
| 20 | 15 |
| 22 | 50 |
| 23 | 50 |
| 25 | 0 |
Solvent A, 0.1% formic acid; Solvent B, 80% CH3CN in A.
In order to fully test the scheme, we subjected 1 nmol of DNA containing dκ (Figure 4, DNAκ) to a round of selection with dXTP, under several different conditions. In one set of experiments, the template DNA was incubated with KF and a range of concentrations of dXTP to assess the lower limit of incorporation needed for detection of a positive hit. In another set, DNAκ and KF were incubated with dXTP in the presence of a variety of other nucleotides, both natural and unnatural (Figure 6). This latter experiment served two purposes: (i) to test if the presence of inactive dNTPs will interfere with the detection of the active ones; and (ii) to verify that the nucleotides which should not be incorporated are not detected as false positives.
Figure 6.

Composition of the dNTP mixtures used in the full run-through of the selection scheme. The concentration of each nucleotide was 100 µM. dRTP, deoxyribose 5′triphosphate.
In all samples, dκ was easily detected, even at <1% predicted extension of the primer (i.e. <10 pmol product). Additionally, the accuracy of the mass assigned was quite good, 3.2 mmu or less from the exact mass (most samples <1 mmu from the exact mass). This level of accuracy allows for the deduction of the molecular formula of the molecule detected, providing an unambiguous identification of the nucleoside. The results for the incoming nucleotide dXTP were also positive, though less straightforward than for dκ. In the absence of other dNTPs, dX was detected (as its free base, xanthine) in all samples, save the one containing the lowest concentration of dXTP, implying that incorporation must be 10% to detect a hit (∼100 pmol). When it was detected, its experimental mass was <4 mmu from the exact mass of xanthine. On average, the accuracy was slightly less than was seen for dκ, but was still within the range needed for the assignment of the molecular formula of the base (Table 2). A detailed example of the data analysis is included in Supplementary Data.
Table 2.
Detection of the unnatural base pair by MS
| [dXTP] (µM) | Expected incorporation | Δmmu dκ | Δmmu dX |
|---|---|---|---|
| 1 | <1% (<10 pmol) | 3.2 | nd |
| 10 | 10% (100 pmol) | 0.2 | 0.5 |
| 50 | 25% (250 pmol) | 0.3 | 3.9 |
| 100 | 35% (350 pmol) | 0.6 | 0.2 |
| 500 | >50% (>500 pmol) | 0.7 | 1.0 |
| MIXTURE A | <35% (<350 pmol) | 1.5 | 0.7 |
| MIXTURE B | <35% (<350 pmol) | 0.1 | 2.5 |
| MIXTURE C | <35% (<350 pmol) | 0.7 | 0.6 |
dκ was detected as the [MH]+ ion and dX as the [BH]+ ion (the free base form of dX). [dXTP] refers to the amount used in the first step of the selection scheme. The experiments were performed with 1 nmol of DNAκ, and the expected amount of extended DNA was estimated from prior kinetic studies; for the mixtures, the incorporation of dXTP is expected to be significantly lower due to competition by the inactive dNTPs. Although the data were not quantified, visual inspection of the autoradiograms used to locate the products indicated that the amount of extension was similar to the predicted amount. Δmmu refers to the difference between the detected mass and the known mass in millimass units. nd, not detected.
In those samples containing dNTP mixtures during the polymerization reaction, xanthine was readily detected. Under these conditions, the amount of elongated product was reduced ∼6-fold as compared to the identical assay (100 µM dXTP) that lacked any other natural or analog dNTPs. Thus, other dNTPs inhibit dXTP polymerization but are not incorporated in place of dXTP, as predicted by the selectivity of KF for polymerization of dXTP opposite dκ. Furthermore, we were unable to detect incorporation of any of the other analog dNTPs included in the mixtures, thereby providing a direct demonstration of the utility of using a DNA polymerase to identify a novel base pair from a library of dNTP analogs.
The enhanced sensitivity towards dκ relative to dX may have resulted from several causes. Primarily, in the LC-MS analysis of pure nucleoside solutions, dX reproducibly gave a less intense signal than the dκ. This likely results from a difference in the case of protonation of the two molecular structures. Diaminopyrimidine has two free amino groups, which are relatively easy to protonate, whereas xanthine has none. The general trend of free amino groups leading to a more intense MS signal holds true for the four natural nucleosides as well. One additional possibility is that there was actually a higher concentration of dκ due to contamination of unextended template in the samples. However, this seems unlikely because analytical scale reactions showed that overlap between the primer and primer +1 bands separated by gel electrophoresis is well under 1%.
A complicating factor with regard to the mass accuracy of the dX peak is contamination by other species. First, dX and deoxyguanosine slightly overlap during elution from the LC. Because the 13C isotope peak of [guanine·H]+ has the same mass as the protonated molecular peak of xanthosine, this leads to a slight skewing of the mass peak. Second, small amounts of non-nucleoside contaminants in the final sample are an inevitable result of the manipulations that the sample goes through before LC-MS analysis. Unfortunately, one of these contaminants gives a small peak that overlaps with xanthosine. This leads to a greater variability in the accuracy of the detection of dX. In the least concentrated sample (extension <1%), the contaminant completely obscures any dX that may be present. However, despite the above issues, the mass accuracy and sensitivity for detection of dX, though noticeably worse than for dκ, is still quite reasonable and allows for ready identification of the incorporation of dXTP.
In looking at the broader implications of these results, this selection method clearly works. The incorporation of dXTP was readily detected at levels as low as 10% extension (100 pmol). Given that xanthine ionizes quite poorly, and that the template nucleoside, dκ, was detected with even better mass accuracy and sensitivity, we feel that 10% incorporation represents a true lower limit of detection using this system. Importantly, incorporation of dX was detected in mixtures of several natural and unnatural nucleotides, and as predicted based on the studies using KF and individual analogs (data not shown), incorporation of no other nucleoside analogs was detected in these samples (i.e. no false positives). This gives direct proof that this method can be used to detect a novel base pair from a pool of nucleotide analogs. Although current methods of synthesizing dNTPs one at a time limits the utility of a secelection-based method, we have developed recently a solid phase method that allows one to synthesize libraries of nucleosides and nucleoside triphosphates (16).
A major advantage of this method is that the chromatographic separation of the final nucleoside mixture allows for the use of multiple analogs with similar or even idenitcal nominal masses. Given the large chromatographic differences often observed between nucleotides with extremely similar structures, this allows for fine-tuning of the library to be screened. By customizing the LC protocol, one could maximize the number of nucleotides in the pool, though one is restricted to compounds that are either separable by chromatography or are of different molecular weight (e.g. some regioisomers may co-elute, preventing distinction between them). Importantly, one can avoid customization of the LC method if the different nucleotide analogs have sufficiently different masses, while still retaining a detection method quite sensitive to small molecular weight changes (i.e. 1 a.m.u.). This offers an advantage over using a method such as MALDI on unfragmented DNA containing unnatural nucleotides, which requires much larger differences in nominal molecular weight (∼5 a.m.u.), significantly restricting the nucleotide library composition.
One limitation of this method is the size of nucleotide libraries that can be screened. The final MS data must be analyzed manually, and the labor involved in this analysis ultimately limits the size of the starting pool. Nonetheless, small, targeted libraries can be quickly analyzed with multiple polymerases and the efficiency of library evaluation allows for the parallel screening of many pools at once. Thus, the library size limitation is not an insurmountable problem. For our method development studies, it was useful to have only one analog in the template. However, the pool of nucleotide analogs to be screened can also be incorporated into the template DNA. The number of possible base pairs would then be the square of the library size, substantially increasing diversity with minimal effort. Additionally, this method can also be used to detect the incorporation of nucleotide analogs across from both natural template bases and unnatural ones.
The potential applications for use of this method with natural DNA templates are manifold. For example, many nucleotide analogs that are currently used as drugs function by acting as substrates for a target DNA polymerase (17–19). A rapid screen that can be used with any polymerase, or multiple polymerases in parallel, allows for the discovery of new potential therapeutics from pools of candidates. This method can also monitor incorporation of nucleotide analogs into DNA to ensure the identity of the incorporated analog. Although such studies can usually be followed by analytical gel electrophoresis, if there is any question about the identity of the incorporated analog (owing to substrate contamination or decomposition, for example), the MS provides a way to positively identify only that compound which is enzymatically active.
We have demonstrated that our new selection strategy is a feasible one for use in the discovery of novel base pairs. This, coupled with the potential applications for the discovery of novel substrates for DNA polymerases using natural templates, makes this method a useful new tool for nucleotide analog research.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant GM54194 to R.D.K. and a Howard Hughes Medical Institute predoctoral fellowship to K.K. Funding to pay the Open Access publication charges for this article was provided by GM54194.
Conflict of interest statement. None declared.
REFERENCES
- 1.Doublie S., Sawaya M.R., Ellenberger T. An open and closed case for all polymerases. Structure. 1999;7:R31–R35. doi: 10.1016/S0969-2126(99)80017-3. [DOI] [PubMed] [Google Scholar]
- 2.Jager J., Pata J.D. Getting a grip: polymerases and their substrate complexes. Curr. Opin. Struct. Biol. 1999;9:21–28. doi: 10.1016/s0959-440x(99)80004-9. [DOI] [PubMed] [Google Scholar]
- 3.Horlacher J., Hottiger M., Podust V.N., Hubscher U., Benner S.A. Recognition by viral and cellular DNA polymerases of nucleosides bearing bases with nonstandard hydrogen bonding patterns. Proc. Natl Acad. Sci. USA. 1995;92:6329–6333. doi: 10.1073/pnas.92.14.6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morales J.C., Kool E.T. Varied molecular interactions at the active sites of several DNA polymerases: nonpolar nucleoside isosteres as probes. J. Am. Chem. Soc. 2000;122:1001–1007. doi: 10.1021/ja993464+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiaramonte M., Moore C.L., Kincaid K., Kuchta R.D. Facile polymerization of dNTPs bearing unnatural base analogues by DNA polymerase alpha and Klenow fragment (DNA polymerase I) Biochemistry. 2003;42:10472–10481. doi: 10.1021/bi034763l. [DOI] [PubMed] [Google Scholar]
- 6.Mitsui T., Kitamura A., Kimoto M., To T., Sato A., Hirao I., Yokoyama S. An unnatural hydrophobic base pair with shape complementarity between pyrrole-2-carbaldehyde and 9-methylimidazo[(4,5)-b]pyridine. J. Am. Chem. Soc. 2003;125:5298–5307. doi: 10.1021/ja028806h. [DOI] [PubMed] [Google Scholar]
- 7.Henry A.A., Romesberg F.E. Beyond A, C, G and T: augmenting nature's alphabet. Curr. Opin. Chem. Biol. 2003;7:727–733. doi: 10.1016/j.cbpa.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 8.Henry A.A., Yu C., Romesberg F.E. Determinants of unnatural nucleobase stability and polymerase recognition. J. Am. Chem. Soc. 2003;125:9638–9646. doi: 10.1021/ja035398o. [DOI] [PubMed] [Google Scholar]
- 9.Bangs J.D., Crain P.F., Hashizume T., McCloskey J.A., Boothroyd J.C. Mass spectrometry of mRNA cap 4 from trypanosomatids reveals two novel nucleosides. J. Biol. Chem. 1992;267:9805–9815. [PubMed] [Google Scholar]
- 10.Piccirilli J.A., Krauch T., Moroney S.E., Benner S.A. Enzymatic incorporation of a new base pair into DNA and RNA extends the genetic alphabet. Nature. 1990;343:33–37. doi: 10.1038/343033a0. [DOI] [PubMed] [Google Scholar]
- 11.Lutz M.J., Held H.A., Hottiger M., Hubscher U., Benner S.A. Differential discrimination of DNA polymerase for variants of the non-standard nucleobase pair between xanthosine and 2,4-diaminopyrimidine, two components of an expanded genetic alphabet. Nucleic Acids Res. 1996;24:1308–1313. doi: 10.1093/nar/24.7.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ludwig W., Follmann H. The specificity of ribonucleoside triphosphate reductase. Multiple induced activity changes and implications for deoxyribonucleotide formation. Eur. J. Biochem. 1978;82:393–403. doi: 10.1111/j.1432-1033.1978.tb12034.x. [DOI] [PubMed] [Google Scholar]
- 13.Sambrook J., Russell D.W. Molecular Cloning: A Laboratory Manual. 3 edn. Vol. 1. NY: Cold Spring Harbor Laboratory Press, Cold Spring Harbor; 2001. [Google Scholar]
- 14.Senior M.M., Jones R.A., Breslauer K.J. Influence of loop residues on the relative stabilities of DNA hairpin structures. Proc. Natl Acad. Sci. USA. 1988;85:6242–6246. doi: 10.1073/pnas.85.17.6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chou S.H., Zhu L., Reid B.R. On the relative ability of centromeric GNA triplets to form hairpins versus self-paired duplexes. J. Mol. Biol. 1996;259:445–457. doi: 10.1006/jmbi.1996.0331. [DOI] [PubMed] [Google Scholar]
- 16.Kincaid K. Ph.D. Thesis. Boulder, CO: University of Colorado; 2006. Studies of Nucleotide Analogues: DNA Polymerase Fidelity and the Solid Phase Synthesis of Nucleosides and Nucleotides. [Google Scholar]
- 17.Anderson K.S. Perspectives on the molecular mechanism of inhibition and toxicity of nucleoside analogs that target HIV-1 reverse transcriptase. Biochim. Biophys. Acta. 2002;1587:296–299. doi: 10.1016/s0925-4439(02)00092-3. [DOI] [PubMed] [Google Scholar]
- 18.Balzarini J., McGuigan C. Chemotherapy of varicella-zoster virus by a novel class of highly specific anti-VZV bicyclic pyrimidine nucleosides. Biochim. Biophys. Acta. 2002;1587:287–295. doi: 10.1016/s0925-4439(02)00091-1. [DOI] [PubMed] [Google Scholar]
- 19.Miura S., Izuta S. DNA polymerases as targets of anticancer nucleosides. Curr. Drug Targets. 2004;5:191–195. doi: 10.2174/1389450043490578. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.