

Abstract
The putative proteasome-associated proteins Mpa (Mycobaterium proteasomal ATPase) and PafA (proteasome accessory factor A) of the human pathogen Mycobacterium tuberculosis (Mtb) are essential for virulence and resistance to nitric oxide. However, a direct link between the proteasome protease and Mpa or PafA has never been demonstrated. Furthermore, protein degradation by bacterial proteasomes in vitro has not been accomplished, possibly due to the failure to find natural degradation substrates or other necessary proteasome co-factors. In this work, we identify the first bacterial proteasome substrates, malonyl Co-A acyl carrier protein transacylase and ketopantoate hydroxymethyltransferase, enzymes that are required for the biosynthesis of fatty acids and polyketides that are essential for the pathogenesis of Mtb. Maintenance of the physiological levels of these enzymes required Mpa and PafA in addition to proteasome protease activity. Mpa levels were also regulated in a proteasome-dependent manner. Finally, we found that a conserved tyrosine of Mpa was essential for function. Thus, these results suggest that Mpa, PafA, and the Mtb proteasome degrade bacterial proteins that are important for virulence in mice.
Keywords: Mpa, PafA, proteasome, substrates, tuberculosis
Introduction
Most individuals who are infected with Mycobacterium tuberculosis (Mtb) do not develop tuberculosis. In these cases, the immune system is able to prevent the bacteria from growing uncontrollably. However, despite effective control of Mtb growth in healthy individuals, Mtb is rarely sterilized from the body (Hernandez-Pando et al, 2000). Mtb resides primarily in macrophages, a cell type that normally controls bacterial growth. Although numerous aspects of the immune system are responsible for slowing the growth of Mtb, increasing data suggest that nitric oxide (NO) is essential for this process (Nozaki et al, 1997; MacMicking et al, 1997b; Bekker et al, 2001; Chan et al, 2001; Scanga et al, 2001; Jung et al, 2002). NO, which can form other reactive nitrogen intermediates (RNI), has antimicrobial activity and is produced by the inducible nitric oxide synthase (iNOS) in macrophages and other cell types (MacMicking et al, 1997a). Genetic inactivation of iNOS in mice results in severe susceptibility to Mtb; iNOS−/− mice succumb rapidly after infection with wild type (WT) Mtb (40–80 days) (MacMicking et al, 1997b; Mogues et al, 2001; Scanga et al, 2001; Darwin et al, 2003). In contrast, WT mice live more than a year after infection with WT Mtb.
In an effort to identify new targets for tuberculosis therapy, over 10 000 Mtb mutants were screened for hyper-susceptibility to the lethal effects of NO and other RNI (Darwin et al, 2003). This screen identified Mycobacterium proteasomal ATPase (Mpa), which forms hexameric ATPase rings, and proteasome accessory factor A (PafA), a protein of unknown function (Darwin et al, 2003, 2005). Because Mpa and PafA are encoded in proximity to the Mtb proteasome genes (prcA and prcB) and Mpa is homologous to ATPases found in the regulatory cap of the eukaryotic 26S proteasome, these proteins were presumed to be involved in proteasome function (Nagy et al, 1997).
Proteasomes are present in all eukaryotes and archaea, and in some bacteria of the class Actinomycetes, which includes Mtb (Butler et al, 2006). The proteasome is a multisubunit, ATP-dependent protease and is the major cytosolic protein degradation structure in eukaryotic cells (Baumeister et al, 1998). 20S proteasome core particles are composed of two rings of catalytic β-subunits sandwiched by rings of α-subunits that form a barrel-shaped structure. Access to the channel and the catalytic sites of the β-subunits is obstructed by a gate formed by the α-subunits (Groll et al, 1997; Unno et al, 2002; Benaroudj et al, 2003; Hu et al, 2006; Lin et al, 2006). The eukaryotic 26S proteasome is composed of the 20S core particle and one or two 19S regulatory caps. The cap is composed of numerous proteins, including six AAA (ATPases associated with various activities) ATPases, and is involved in the recognition, unfolding, and translocation of ubiquitinated proteins into the 20S proteasome core (Pickart and Cohen, 2004). It is not known how proteins are targeted for degradation in prokaryotes where 19S regulatory caps and ubiquitin appear to be absent. Attempts to demonstrate ATPase-dependent degradation of proteins by bacterial proteasome cores in vitro have not been successful (our unpublished data) (Wolf et al, 1998), most likely due to the lack of additional co-factors required for protein degradation.
Little is known about bacterial proteasome biology. Questions that remain to be answered include those regarding the role of proteasomes in bacteria, the requirement of cofactors for proteasome function, and the mechanism by which substrates are targeted for degradation by the proteasome. In this work, we identify the first natural substrates of a bacterial proteasome that will allow us to begin to address these questions.
Results
The C-terminus of Mpa is essential for Mtb virulence and resistance to RNI
Earlier studies described mpa607, a mutant with a transposon insertion in the penultimate codon, which caused the deletion of the last two amino acids: tyrosine and leucine (Darwin et al, 2003). It was previously determined that Mpa forms a hexameric AAA ATPase that is homologous to proteasomal ATPases (Darwin et al, 2005). Mpa607 retains the ability to hydrolyze ATP in vitro (Darwin et al, 2005). Notably, the mpa607 strain was as sensitive to RNI in vitro as the mpa null strain, suggesting that the terminal amino acids are essential for the function of Mpa in RNI resistance (Darwin et al, 2003, 2005).
It was reported that the mpa607 mutant was attenuated in mice, as determined by enumerating colony-forming units (CFU) from the lungs and spleens 8 weeks after infection (Darwin et al, 2003). Since that study, we have determined that WT mice and mice deficient for iNOS live significantly longer when infected with the mpa607 mutant compared to infection with WT Mtb (Figure 1A). WT mice infected with the mpa607 strain of Mtb had a 60% survival rate after one and a half years, whereas those infected with WT Mtb only had a 20% survival rate (Figure 1A). Importantly, WT mice that had been infected with any of the mutant strains of Mtb were killed around 600 days after infection for reasons other than tuberculosis; these mice exhibited symptoms of aging, including tumors and perturbed walking, but were otherwise maintaining robust body weight and active movement (not shown). In contrast, WT mice infected with WT Mtb were hunched, thin, ruffled, slow-moving and had labored breathing, which are typical symptoms of mice succumbing to Mtb infection (not shown). Although genetic inactivation of iNOS resulted in increased susceptibility of the mice to Mtb, the mpa607 mutant was still severely attenuated in these mice compared to the WT Mtb strain (Figure 1A). Similar effects were seen when mice were infected with the pafA null strain of Mtb (Figure 1A) or the mpa null strain (Darwin et al, 2005). This outcome demonstrates that, like the mpa null strain (Darwin et al, 2005), the mpa607 and the pafA null strains are highly attenuated in the mouse model of Mtb infection and support the hypothesis that Mpa and PafA operate in a common pathway. Additionally, these experiments indicate that the C-terminus of Mpa is essential for Mpa function.
Figure 1.
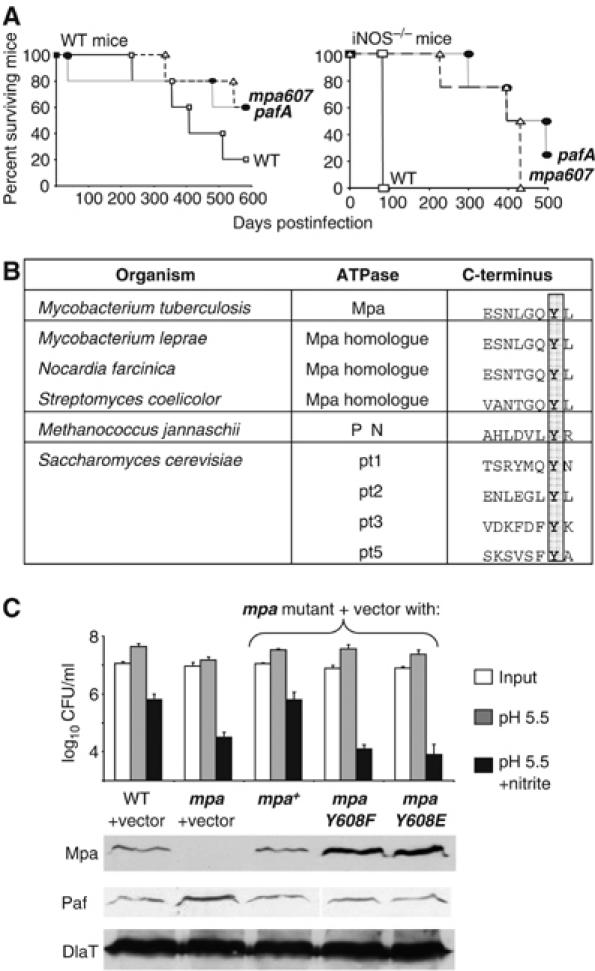
The C-terminus of Mpa is essential for Mtb virulence and resistance to RNI. (A) Survival of WT and iNOS−/− C57BL/6 mice infected with WT, mpa607 and pafA strains of Mtb. Each data point represents the percentage of mice that were alive at the time point shown on the x axis. (B) Alignment of the C-terminus of Mpa with the C-termini of proteasome-associated ATPases from representative actinomycetes, archaea and yeast. The conserved penultimate tyrosines are highlighted. Sequences were compiled from the NCBI server. (C) Substitution of the penultimate tyrosine of Mpa abolished protection against RNI in Mtb and increased the steady-state levels of Mpa protein. (Top) Each data point represents the average of triplicate samples. These results are representative of at least three independent experiments. Error bars represent +s.d. (Middle) (Bottom) Immunoblot analysis of Mpa, PafA and DlaT (dihydrolipoamide acyltransferase, loading control) in Mtb lysates from the same cultures harvested just prior to the nitrite survival assay.
Because the truncation in Mpa607 drastically impaired the virulence of Mtb, we sought to define the functional relevance of the missing residues. We aligned the WT Mpa sequence with the sequences of other proteasome-associated ATPases to determine if any of the C-terminal amino acids were conserved. We found that the penultimate tyrosine of Mpa was conserved across all proteasome-bearing bacteria, most archaea, and some of the ATPase subunits of the eukaryotic 19S cap (Figure 1B). The conservation of the penultimate tyrosine across all domains of life suggested that this residue is important for the proper functioning of these proteasome-associated molecules.
We mutagenized the conserved penultimate tyrosine to determine if it was essential for Mpa function. We substituted the tyrosine with phenylalanine (MpaY608F) to test if a closely related amino acid would affect Mpa function. Additionally, we mutated the tyrosine to glutamic acid (MpaY608E) to introduce a negative charge that can mimic constitutive tyrosine phosphorylation. This strategy was based on reports that the related eukaryotic ATPase Cdc48 (p97/valosin-containing protein) was determined to be phosphorylated on the penultimate tyrosine; mutagenesis of this tyrosine to glutamate resulted in Cdc48 that behaved as if it were constitutively phosphorylated (Egerton et al, 1992; Madeo et al, 1998).
The mpa point mutant plasmids were introduced individually into the mpa null strain of Mtb on an integrative plasmid. Both mpaY608 alleles failed to protect Mtb against RNI in vitro (Figure 1C). Notably, the mpaY608F Mtb strain was as vulnerable to RNI as the mpa null strain, despite replacement of the penultimate tyrosine with another aromatic amino acid (Figure 1C). The mpaY608E Mtb strain was also as susceptible to RNI as the mpa null strain (Figure 1C), suggesting that phosphorylation of the tyrosine is not important for Mpa-dependent protection against RNI. Consistent with this hypothesis, phosphotyrosine immunoblot analysis of Mtb lysates did not detect tyrosine-phosphorylated Mpa (data not shown).
Because the Mtb strains expressing the mutant mpa alleles were as sensitive to RNI as the mpa null strain, we checked for the presence of the MpaY608-mutant proteins with antibodies against Mpa. The mutant proteins were observed at higher steady-state levels than those seen in WT Mtb or the mpa null strain complemented with WT mpa (Figure 1C) (Darwin et al, 2005). We also examined PafA levels in these strains. PafA was present at slightly increased levels in the mpa null mutant but not in the penultimate tyrosine mutants or the mpa complemented strain (Figure 1C). It is not clear if the increased amount of PafA in the mpa null strain is significant in light of the observation that PafA levels appear WT in the other Mpa-defective strains.
Taken together, these data show that the extreme C-terminal residues of Mpa, particularly the highly conserved tyrosine, are indispensable for Mpa function. Furthermore, the results suggest that the same residues are important for the maintenance of WT steady-state protein levels of Mpa.
Mpa and PafA work with the Mtb proteasome to regulate Mpa protein levels
The accumulation of the MpaY608-mutant proteins was not entirely unexpected because previous analysis revealed elevated levels of the Mpa607 protein in the mpa607 mutant (Darwin et al, 2005). Additionally, studies of Mpa with mutations in the ATPase catalytic site demonstrated increased steady-state levels of Mpa (Darwin et al, 2005). These data suggested that the process that regulates Mpa protein levels requires the penultimate tyrosine and ATPase activity of Mpa.
Because both Mpa and PafA are thought to be involved in a common pathway, we also tested whether endogenous Mpa levels were affected in the pafA null strain. The pafA null strain showed increased abundance of Mpa (Figure 2A) comparable to that seen in the Mtb strains expressing the MpaY608 point mutants (Figure 1C) or the Mpa catalytic site mutants (Darwin et al, 2005). Taken together, these results further establish a link between Mpa and PafA and suggest that Mpa protein levels are regulated by a mechanism that requires at least Mpa and PafA.
Figure 2.
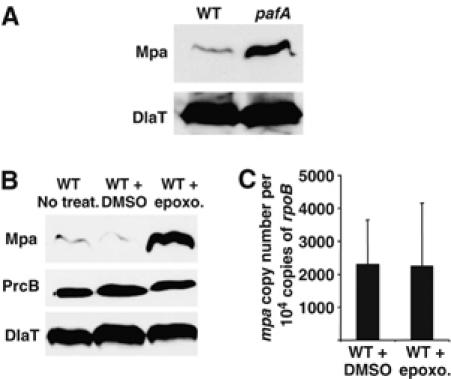
Mpa protein levels are regulated by the Mtb proteasome. (A) Anti-Mpa immunoblots of whole-cell lysates from a WT and the pafA null strain of Mtb. Both strains contain pMV306, the empty vector used for complementation analysis. DlaT shows equivalent loading. (B) (Top) Anti-Mpa immunoblots of whole-cell lysates from WT Mtb and WT Mtb that had been incubated with DMSO or 50 μM of the proteasome inhibitor epoxomicin in DMSO. (Middle) Anti-PrcB immunoblots of the same samples. (Bottom) Immunodetection of DlaT was used as a loading control for both experiments. (C) QRTPCR analysis of mpa transcript levels in WT Mtb that had been incubated with either DMSO or 50 μM epoxomicin in DMSO. The copy number of each transcript was normalized to a non-differentially regulated RNA, rpoB for each strain tested. Values represent two biologically replicate RNA samples.
Proteasome-associated ATPases mediate the entry of protein substrates into the catalytic chamber of the proteasome (Kohler et al, 2001a; Smith et al, 2005). It has been suggested that the C-termini of these ATPases are important for opening the gate of the proteasome core (Forster et al, 2005). These findings, in conjunction with the differences in Mpa protein levels observed in the various mpa mutant strains, led us to the hypothesis that Mpa stimulates gate opening of the Mtb proteasome core and promotes its own turnover. If Mpa were a substrate of the proteasome, then Mpa protein levels would increase in the absence of proteasome protease activity. We inhibited the Mtb proteasome in cultures treated with epoxomicin, a specific eukaryotic proteasome inhibitor that is effective against the Mtb proteasome (Kisselev and Goldberg, 2001; Darwin et al, 2003; Lin et al, 2006) and assessed Mpa levels in Mtb. Consistent with our hypothesis, Mpa protein was more abundant in a WT strain that had been treated with proteasome inhibitor than in a WT strain incubated with nothing or the vehicle control (Figure 2B). In contrast, the levels of the proteolytic β-subunit of the Mtb proteasome, PrcB, or the loading control protein dihydrolipoamide acyltransferase (DlaT) remained constant (Figure 2B). This finding demonstrates that the proteasome-dependent regulation of Mpa was specific. Quantitative real-time PCR (QRTPCR) showed that there were no changes in mpa transcript levels between the WT strain incubated with or without epoxomicin (Figure 2C), suggesting the regulation of Mpa protein levels occurs post-transcriptionally.
Taken together, the findings of these experiments are the first to link Mpa and PafA to proteasome protease activity. Moreover, this activity appears to regulate the steady-state levels of a specific protein, Mpa, the presumptive proteasome-associated ATPase. To our knowledge, this is also the first demonstration that proteasomal ATPase levels can be autoregulated by the proteasome pathway.
FabD and PanB levels are regulated by the Mtb proteasome in an Mpa- and PafA-dependent manner
As Mpa protein levels were regulated by the concerted efforts of Mpa, PafA and the Mtb proteasome, we hypothesized that Mpa is necessary for the proteasome-dependent degradation of other protein substrates in Mtb. To search for other natural proteasome substrates in Mtb, we compared the steady-state proteomes of WT and mpa null strains using two-dimensional (2D) gel electrophoresis. Global changes in protein levels were not observed between the WT and mutant strains; however, this could have been due to the limit of sensitivity of 2D gel analysis. Two proteins were reproducibly more abundant in gels prepared with lysates from the mpa null strain when compared to gels prepared from WT strains (Figure 3A). This was observed whether the strains were grown under standard conditions or in acidified media with or without nitrite (Figure 3A). The two proteins were identified by matrix assisted laser desorption/ionization-time of flight (MALDI-TOF) analysis as malonyl Co-A acyl carrier protein transacylase (FabD) (Kremer et al, 2001) and ketopantenoate hydroxymethyltransferase (PanB) (Chaudhuri et al, 2003; Sugantino et al, 2003).
Figure 3.
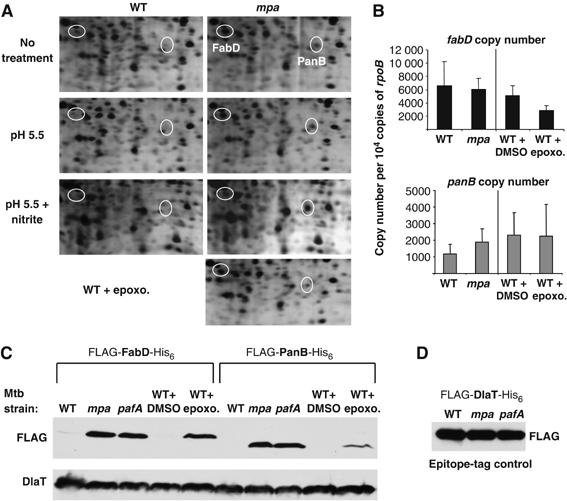
FabD and PanB are candidate substrates of the Mtb proteasome. (A) 2-D gel electrophoresis of total lysates from untreated WT Mtb compared to mpa mutant Mtb and WT Mtb incubated with the proteasome inhibitor epoxomicin. Spots corresponding to FabD and PanB are circled. The 2-D gels were prepared with filtered total cell lysates from the Mtb strains and separated by isoelectric focusing on a pH 4–8 gradient in the first dimension and 10% SDS–PAGE in the second dimension. Only the portion of the gels that showed reproducible differences is shown. Proteins were visualized by silver staining and the relevant spots were identified by MALDI-TOF. Spots corresponding to FabD and PanB were observed in at least four different paired samples by 2D gel analysis. Nine peptides representing 44% coverage of FabD and six peptides representing 37% coverage of PanB were identified. (B) QRTPCR analysis of fabD and panB transcript levels. The copy number of each transcript was normalized to non-differentially regulated rpoB. Values are shown as copy number per 10 000 copies of rpoB and represent two biologically replicate RNA samples. The differences in the transcript values for fabD and panB between the WT and mpa strains were not statistically significant as determined by unpaired t-tests (P>0.05). The two- to three-fold difference in fabD expression after epoxomicin treatment is statistically significant (P=0.0002). (C) Anti-FLAG immunoblots of whole-cell lysates to examine the steady-state protein levels of epitope-tagged versions of FabD and PanB in untreated WT, mpa null, pafA null and proteasome-inhibited WT Mtb strains. WT Mtb was incubated with 50 μM epoxomicin in DMSO to inhibit proteasome protease activity. Immunodetection of DlaT is shown as a loading control. (D) Anti-FLAG immunoblots of whole-cell lysates to examine the steady-state protein levels of recombinant DlaT in untreated WT, mpa null and pafA null Mtb strains. Equivalent cell numbers were analyzed.
To determine if the difference in protein levels was due to an increase in fabD or panB transcripts in the mpa mutant, we analyzed the transcript levels of both genes by QRTPCR. There were no statistically significant increases in fabD or panB transcripts in the mpa null strain compared to the WT strain, demonstrating that the increased abundance of FabD and PanB in the mpa mutant was not due to amplified gene expression (Figure 3B).
To test the hypothesis that FabD and PanB are Mtb proteasome substrates, we examined steady-state FabD and PanB protein levels in proteasome protease inhibited WT Mtb. Similar to the proteome of the mpa null strain, endogenous FabD (as confirmed by MALDI-TOF) and presumed PanB spots were increased in intensity in proteasome-inhibited samples compared to untreated WT Mtb samples (Figure 3A, far right). There was no statistically significant change in the panB transcript between the treated and untreated WT Mtb strains; however, this was not the case for fabD (Figure 3B). Expression of fabD was reduced 50–60% in epoxomicin treated cultures (P=0.0002; Student's unpaired t-test). One possible explanation is that the accumulation of FabD results in increased amounts of long-chain fatty acid intermediates in the cell that exert a negative effect on fabD expression. This form of gene control is well characterized for the fad/fab genes of Escherichia coli (Raman and DiRusso, 1995; Raman et al, 1997), but it is not known if a similar mechanism occurs in Mtb. Furthermore, it is not clear why this would occur in epoxomicin treated samples and not in the mpa mutant. Nonetheless, this result emphasizes the observation that FabD protein levels are not increased due to an increase in fabD transcription in the proteasome-defective strains.
We validated the results seen on the 2-D gels by using immunoblotting to follow changes in the levels of epitope-tagged versions of FabD and PanB in recombinant strains of Mtb. WT, mpa and pafA strains of Mtb were transformed with plasmids encoding FLAG-fabD-His6 and FLAG-panB-His6 cloned downstream of a heterologous Mycobacterium bovis Bacille Calmette-Guerin hsp60 promoter (Scholz et al, 2000) and an E. coli ribosome binding site. Anti-FLAG immunoblots of total cell lysates revealed a dramatic increase in the steady-state levels of the FLAG-tagged proteins in the mpa and pafA mutant strains compared to WT Mtb (Figure 3C). This further strengthened the functional association between Mpa and PafA. We also observed increased levels of the recombinant proteins in a WT strain that had been incubated with proteasome inhibitor (Figure 3C). In contrast, the recombinant proteins were barely detectable in the untreated WT strain, consistent with the results of the endogenous proteins observed by 2D gel analysis (Figure 3C).
In contrast to FabD and PanB, epitope-tagged DlaT protein was equally abundant in the WT and mutant Mtb strains (Figure 3D). These data suggest that the accumulation of recombinant FabD and PanB in the mutant strains was due to intrinsic features of these proteins, rather than experimental artifact. Furthermore, the design of the heterologous expression system controls for possible changes in transcription and translation initiation rates, supporting the hypothesis that the recombinant protein levels were increased because the proteins were not efficiently degraded. Therefore, FabD and PanB steady-state levels, like those of Mpa, are regulated in Mtb by a pathway that requires Mpa, PafA and proteasome protease activity.
We assessed the potential effects the accumulation of FabD and PanB might have on several important fatty acids. Mycolic acid methyl esters, fatty acid methyl esters, methylated lipids, triacyl glycerols, and wax esters in WT, mpa, mpa-complemented and pafA strains were examined. All of these molecules appeared to be normal in mpa and pafA mutant strains (Supplementary Figure S1). However, we cannot rule out other effects that are not detectable by even these extensive assays. We also observed that the mpa mutant consistently appeared to have reduced uptake of palmitic acid (Supplementary Figure S1C and D), whereas uptake looked WT in the complemented and pafA strains. We do not yet understand the significance of this phenomenon. Despite this observation, our analysis suggests that there are no dramatic changes in the fatty acid and lipid profiles of mpa and pafA mutants.
The C-terminus of Mpa is required for maintaining wild-type steady-state levels of FabD and PanB
The C-terminal Mpa mutants were unable to maintain WT Mpa protein levels, suggesting a loss of function of Mpa. Therefore, we determined whether or not the two amino acid truncated form of Mpa was able to regulate the protein levels of the newly discovered candidate substrates. We examined the steady-state protein levels of FLAG-FabD-His6 and FLAG-PanB-His6 in the mpa607 strain. Anti-FLAG immunoblots revealed increased levels of FLAG-FabD-His6 and FLAG-PanB-His6 in the mpa607 strain compared to WT Mtb (Figure 4), similar to the increases seen in the mpa null and pafA null strains of Mtb (Figure 3C). The inability of Mpa607 to regulate the recombinant protein levels further suggests that the C-terminal residues are essential for Mpa to function in the proteasome degradation pathway.
Figure 4.
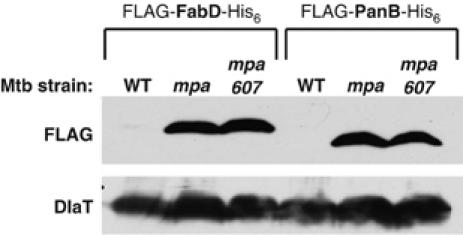
The C-terminus of Mpa is necessary to promote the apparent proteasome-dependent degradation of FabD and PanB. Anti-FLAG immunoblots of whole-cell lysates to examine the steady-state protein levels of epitope-tagged versions of FabD and PanB in WT, mpa null, mpa607 Mtb strains. Immunodetection of DlaT is shown as a loading control.
Discussion
Mpa, PafA and the Mtb proteasome were previously linked to each other because of their ability to provide resistance to RNI in Mtb (Darwin et al, 2003). In addition, both mpa and pafA mutant strains of Mtb are similarly attenuated in mice, suggesting that both Mpa and PafA participate in a common pathway needed for in vivo survival. We further substantiate this association with the discovery that Mpa, PafA and the proteasome are involved in the regulation of FabD, PanB and Mpa protein levels. Mpa and PafA are equally important in maintaining physiologic levels of FabD and PanB because the amounts of these proteins are similarly affected in mpa and pafA mutant strains. The inability to regulate Mpa protein levels in the absence of pafA or in proteasome protease-inhibited WT Mtb further provides evidence that Mpa, PafA and the proteasome core are functionally linked. Thus, we propose a model that suggests Mpa interacts with the proteasome core in a PafA-dependent manner to promote the degradation of proteins such as FabD, PanB and Mpa itself (Figure 5).
Figure 5.
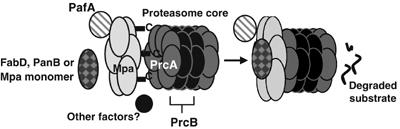
Proposed model of the Mtb proteasome. The C-termini of Mpa hexamers may interact with the PrcA subunits, thereby promoting opening of the gate formed by the PrcA N-termini and allowing access into the proteolytic core. Interactions of Mpa with the protease core (β-subunit=PrcB; α-subunit=PrcA) appear to require the presence of PafA. Direct interactions between substrates, Mpa, PafA and the proteasome core have not yet been detected, possibly due to the requirement of additional factors.
In order to conclude that the Mtb proteasome degrades FabD, PanB and Mpa, it will be necessary to reconstitute the proteasomal system in vitro. Proteolysis of peptide substrates by the Mtb 20S proteasome has been demonstrated in vitro (Lin et al, 2006). However, this process is not comparable to proteolysis of a globular substrate; accessory factors, such as an ATPase, are not required to promote peptide degradation. Previous attempts to reconstitute degradation of a folded protein by the homologous Rhodococcus erythropolis proteasome system in vitro with 20S proteasome cores and the ATPase ARC have been unsuccessful (Wolf et al, 1998). This is not surprising because we found that the Mtb proteasome system requires at least two co-factors, Mpa and PafA, in addition to the proteasome core to regulate the levels of candidate substrates. So far, direct interactions between Mpa, PafA and the proteasome core have not been detected, most likely due to the lack of other co-factors required for protein–protein contacts (MJ Pearce and KH Darwin, unpublished results). We are currently working to determine if proteasomal degradation of endogenous substrates requires additional co-factors in Mtb.
Our genetic analysis suggests that a truncation at the C-terminus of Mpa prevents the proteasome-dependent degradation of FabD, PanB and Mpa. A possible explanation for these results is that the C-terminus of Mpa is essential for opening the gate of the Mtb 20S proteasome. Studies in other organisms employing proteasomal ATPase and proteasome gate mutants suggest that the ATPases are responsible for activating 20S proteasomes by opening the gate to allow access to the site of proteolysis in the core particle (Groll et al, 1997; Kohler et al, 2001b; Benaroudj et al, 2003; Smith et al, 2005). The importance of the ATPase C-terminus in proteasome gate opening is supported by a finding that C-terminal truncation of the archaebacterial ATPase proteasome activating nucleotidase prevented it from degrading casein in vitro (Forster et al, 2005). In an analogous bacterial degradation system, the C-terminus of the ATPase HslU (ClpY) is required for the activation of the HslV (ClpQ) protease. The binding of ATP to HslU induces a conformational change in its C-terminus that allows the C-terminus to interact with pockets inside the HslV protease causing an opening of the HslV entry pore (Wang et al, 2001; Seong et al, 2002). Together, these studies establish a theme that the C-termini of protease-associated ATPases are critical for the interaction with and the activation of their partner proteases, and our data indicate that the same might be true for the activation of the Mtb proteasome by Mpa. Interestingly, the amino-acid substitution in MpaY608F leaves an aromatic ring missing a hydroxyl group that is present in the usual tyrosine side chain. This missing hydroxyl group may be important for forming hydrogen bonds with residues in the Mtb proteasome α-subunits that stabilize the interaction of Mpa with the proteasome.
Eukaryotic proteasomes contribute to the regulation of many cellular processes by degrading proteins (Coux et al, 1996), but there is little data regarding how the 26S proteasome components are themselves turned over (Chen and Hochstrasser, 1996; Schmidtke et al, 1996; Hirano et al, 2005). Our results indicate that Mpa, the proposed proteasome-associated ATPase, is itself degraded by the Mtb proteasome. Studies of the Clp protease in Gram-negative bacteria have revealed that the ClpA ATPase is a substrate of the ClpP protease, a situation that resembles our observation with Mpa (Maurizi et al, 1990). In the Clp system, either the ClpA or ClpX ATPase associates with ClpP to direct the degradation of particular substrates (Gottesman, 2003). It is possible that the degradation of ClpA by ClpP quickly resets the protease system to allow interactions with ClpX. It is tempting to speculate that a similar situation might apply to Mtb because Mpa is not essential for normal growth in vitro, while the proteasome protease genes are predicted to be essential or required for optimal growth (Darwin et al, 2003; Sassetti et al, 2003). Perhaps, there are additional ATPase regulators of the proteasome in Mtb that function in protein turnover.
FabD and PanB are involved in fatty acid metabolism and FabD is also required for polyketide synthesis. About 6% of the Mtb genome (∼250 genes) is devoted to the metabolism of fatty acids, suggesting that these macromolecules are important for the mycobacterial lifestyle (Kinsella et al, 2003). So far, the fatty acid profiles of the mutant strains appear to be normal; however, we cannot rule out that changes are occurring in the mutant strains. Importantly, we do not believe that the accumulation of these enzymes is necessarily responsible for the attenuation of the mpa and pafA mutants.
Although our proteomic analyses identified two putative substrates, it is likely that more exist. We are currently exploring other methods to identify additional proteins that are regulated by the Mtb proteasome with the hope that their discovery will help us understand how proteins are targeted for proteolysis. Furthermore, identification of these substrates may reveal why the mpa and pafA mutants are attenuated in vivo.
This work has identified the first putative natural substrates of a prokaryotic proteasome. These proteins will be useful tools as we continue to study Mpa, PafA and the Mtb proteasome in their involvement in cellular protein degradation and virulence. Furthermore, we have described the essentiality of the extreme C-terminus of Mpa for proteasome-dependent regulation of protein levels, and we have elucidated an auto-regulatory circuit involving proteasome-associated factors. Most importantly, we have provided the first compelling evidence that Mpa, PafA and the Mtb proteasome core protease function to regulate protein levels in Mtb. We favor the hypothesis that Mpa and PafA work directly with the proteasome to promote the degradation of target proteins; however, other mechanisms of regulation may exist. Regardless of the underlying mechanism, a better understanding of the biology of the Mtb proteasome and the characterization of its substrates may provide new targets for the treatment of tuberculosis.
Materials and methods
Bacterial strains, growth conditions and primers
The bacterial strains, plasmids and primers used in this study are listed in Table I. All primers were purchased from Invitrogen. Mtb strains were grown in Middlebrook 7H9 broth (Difco) supplemented with 0.2% glycerol, 0.05% Tween-80, 0.5% bovine serum albumin, 0.2% dextrose and 0.085% sodium chloride (ADN). Cultures were grown without shaking in either 25 or 75 cm2 vented flasks (Corning) in humidified incubators with 5% CO2 at 37°C. 7H11 agar (Difco) supplemented with oleic acid, albumin, dextrose and catalase (BBL) was used for growth on solid medium. The antibiotics kanamycin and hygromycin were each used at a concentration of 50 μg ml−1 as needed. For epoxomicin experiments, cultures were grown in 7H9+ADN to an OD580 of ∼0.5 after which either a final concentration of 50 μM epoxomicin in DMSO (Boston Biochem) or an equal volume of DMSO was added. Cultures were harvested 4 days after addition of the compound as described below.
Table 1.
Bacterial strains, plasmids and primers used in this work
| Strain | Genotype/sequence | Source or reference |
|---|---|---|
| Plasmids | ||
| pET24b(+) | Kanr; for production of epitope-tagged protein | Novagen |
| pMV306 | Hygr; integrates at attB site on the Mtb chromosome | Stover et al (1991) |
| pMV-mpa | Hygr; pMV306 with 2.6 kb ClaI fragment containing mpa | Darwin et al (2003) |
| pMN402 | Hygr; replicating mycobacterial plasmid with gfp under the control of the BCG hsp60 promoter | Scholz et al (2000) |
| pMV-mpaY608F | Hygr; pMV-mpa but with a Y608 → F point mutation | This work |
| pMV-mpaY608E | Hygr; pMV-mpa but with a Y608 → E point mutation | This work |
| pMN-FLAG-fabD-His6 | Hygr; pMN402 with gfp replaced by FLAG and His6-tagged fabD | This work |
| pMN-FLAG-panB-His6 | Hygr; pMN402 with gfp replaced by FLAG and His6-tagged panB | This work |
| pMN-FLAG-dlaT-His6 | Hygr; pMN402 with gfp replaced by FLAG and His6-tagged dlaT | This work |
| E. coli | ||
| DH5α | supE44 ΔlacU169 (ϕ80 lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | Gibco, BRL |
| Mtb | ||
| H37Rv | wild type (WT) American Type Culture Collection 25618 | ATCC |
| MHD4 | Kanr; mpa608∷ΦMycoMarT7 | Darwin et al (2003) |
| MHD5 | Kanr; mpa77∷ΦMycoMarT7 (Kanr) | Darwin et al (2003) |
| MHD18 | Hygr; H37Rv pMV306 | Darwin et al (2003) |
| MHD22 | Hygr, Kanr; MHD5 pMV306 | Darwin et al (2003) |
| MHD23 | Hygr, Kanr; MHD5 pMV-mpa | Darwin et al (2003) |
| MHD60 | Hygr, Kanr; MHD5 pMV-mpaY608F | This work |
| MHD61 | Hygr, Kanr; MHD5 pMV-mpaY608E | This work |
| MHD62 | Hygr, Kanr; pafA282∷ΦMycoMarT7 pMV306 | This work |
| Primers | ||
| mpaY608Ffa | GAGTCCAACCTCGGCCAGTTCCTGTAGGGCTCAGGCGGTCAC | |
| mpaY608Fra | GTGACCGCCTGAGCCCTACAGGAACTGGCCGAGGTTGGACTC | |
| mpaY608Efa | GATACCGAGTCCAACCTCGGCCAGGAGCTGTAGGGCTCAGGCGGTCACC | |
| mpaY608Era | GGTGACCGCCTGAGCCCTACAGCTCCTGGCCGAGGTTGGACTCGGTATC | |
| FLAGpanBNdef2 | GGAATTCCATATGGATTACAAGGATGACGACGATAAGATGTCTGAGCAGACTATCTATGGG GCC | |
| panBNotr2 | ATAAGAATGCGGCCGCGAAACTGTGTTCGTCAGCGGGGAA | |
| FLAGfabDNdef2 | GGAATTCCATATGGATTACAAGGATGACGACGATAAGATGATTGCGTTGCTCGCACCCGGA | |
| fabDHindIIIr2 | CCCAAGCTTTAGGTTTGCCAGCTCGTCCAGGTC | |
| PET24NheIf | CTAGCTAGCCCTCTAGAAATAATTTTGTTTAAC | |
| PET24PstIr | AACTGCAGTCAGTGGTGGTGGTGGTG | |
| RBSFLAGdlaTPacI | CCTTAATTAAGAAGGAGATATACATATGGATTACAAGGATGACGACGATAAGATGGCCTTC TCCGTCCAGATGCCG | |
| His6dlaTPstI | AACTGCAGTCAGTGGTGGTGGTGGTGGTGCAGTCCTAAATCGGCCTCGAACGC | |
| rpoBf1 | TCGTTCTCTGACCCTCGTTTC | |
| rpoBr1 | ACGTGCCCTTCTCGGTCATCA | |
| mpaf1 | CGAGAATGTCATCGTGATCG | |
| mpar1 | GGCAAGAACTCGGTCAGGTA | |
| fabDf | CAAACCGAGGGAATGTTGTC | |
| fabDr | GCTATCACACCGGCGATT | |
| panBf | GACCAAGATCCGCACCC | |
| panBr | CCTCGTAGCTGCCGAAC | |
| aUnderlined nucleotides indicate those changed from the WT sequence. | ||
Plasmids and site-directed mutagenesis
WT and mutant forms of mpa were expressed from pMV306 (HygR), which integrates into the attB site on the Mtb chromosome. For site-directed mutagenesis of pMV-mpa, the Stratagene QuikChange XL system was used as described elsewhere (Darwin et al, 2005).
To make epitope-tagged proteins, panB and fabD were amplified by PCR from Mtb genomic DNA using primers encoding the FLAG epitope (MDYKDDDDKI; Table I). The resulting PCR products were cloned into pET24b(+), which adds a His6 tag to the cloned gene. The pET24b(+)-cloned inserts, including the consensus E. coli ribosome-binding site from pET24b(+) and both epitope tags, were PCR amplified and cloned into pMN402 that has the Mtb hsp60 promoter that allows expression of the cloned gene. To make the epitope-tagged control plasmid, dlaT was PCR amplified from Mtb genomic DNA using a 5′ primer that encoded the FLAG epitope and a 3′ primer that encoded the His6 epitope with sequences exactly as found in the fabD and panB plasmids (Table I). The amplified product was cloned directly into pMN402.
Pfu polymerase (Stratagene) was used for all PCR-product cloning. Restriction enzymes were purchased from New England Biolabs. All clones were sequenced by the NYU DNA Sequencing Core Facility to confirm the correct DNA sequence. DNA was introduced into Mtb by electroporation as described previously (Hatfull and Jacobs, 2000).
Nitrite survival assay and mouse infections
Nitrite survival assays and mouse infections were carried out as described in detail elsewhere (Darwin et al, 2003, 2005). For mouse infections, groups of mice purchased from Jackson Laboratories were infected with aerosolized Mtb using a Glas-Col Inhalation Exposure System (Terre Haute, IN). Each mouse received about 100–200 CFU as determined by enumerating bacteria from the lungs of three mice per Mtb strain, 24 h after infection. Mice were humanely killed upon observation of any symptoms of illness (hunched posture, labored breathing, thinning and ruffled fur).
Immunoblotting
Total protein lysates were prepared from equivalent cell numbers, determined by culture optical density at wavelength 580 (OD580). Either 5 or 10 OD equivalents were harvested. Bacteria were collected by centrifugation and washed in 5 ml of 0.05% Tween-80 in phosphate buffered saline, resuspended in 300 μl of lysis buffer (100 mM Tris–Cl, 100 mM KCl, 1 mM EDTA, 5 mM MgCl2, pH 8) and transferred to bead beating tubes with 200 μl of zirconia silica beads (BioSpec Products). Cells were lysed by bead beating 2–3 times in a BioSpec Mini Bead Beater for 30 s. Total cell lysate (150 μl) was mixed with 50 μl of protein sample buffer. The samples were boiled at 100°C for 10 min and equal volumes representing equivalent cell numbers were separated by SDS–PAGE in a 10% gel.
For immunodetection, FLAG antibodies were purchased from Sigma (St Louis, MO) and were used according to the manufacturer's instructions. Polyclonal antibodies to Mpa-His6, PrcB-His6 and DlaT-His6 were used as described elsewhere (Darwin et al, 2005; Tian et al, 2005; Lin et al, 2006). For all immunoblots, experimental membranes were stripped and incubated with anti-DlaT to check equivalent loading of samples (Darwin et al, 2005; Tian et al, 2005). Horseradish peroxidase (HRP) coupled anti-rabbit secondary antibodies were used according to the manufacturer's instructions (Amersham). Detection of HRP was performed using either SuperSignal West Pico or West Femto Chemiluminescent Substrate (Pierce).
QRTPCR
Mtb strains were grown to late logarithmic phase (OD580∼1.0) in 7H9+ADN broth in the presence of the appropriate antibiotics. Cultures were added to an equal volume of 5 M guanidium isothiocyanate, 0.5% sodium-N-lauryl sarcosine, 25 mM tri-sodium citrate, 0.1 M β-mercaptoethanol to stop transcription and cells were harvested by centrifugation. RNA was extracted from the bacterial pellets using TRIzol Reagent (Invitrogen). The pellets were resuspended in 1.2 ml TRIzol Reagent and the samples were lysed by bead beating as described above. The remaining RNA purification steps were carried out as described in the TRIzol protocol and repeated to ensure removal of genomic DNA. The Reverse Transcription System (Promega) was used to synthesize cDNA from 100 ng of Mtb RNA with 4 ng of random hexamers (Amersham) to prime synthesis. The cDNA equivalent of ∼1.9 ng of total Mtb RNA was analyzed by quantitative PCR using Platinum SYBR Green qPCR SuperMix UDG (Invitrogen) in a DNA Engine Opticon 2 Continuous Fluorescence Detection System (MJ Research) (for primers, see Table I). For quantification, transcript copy numbers were determined by comparison to a standard curve of Mtb genomic DNA. The relative amounts of each transcript were normalized to that of rpoB, a control gene that is not differentially regulated in any of our Mtb strains (unpublished data). cDNA were synthesized from at least two biologically replicate RNA samples and QRTPCR was performed twice, in triplicate, for each cDNA.
Proteomics analysis
Two-dimensional SDS–PAGE analysis of filtered (0.22 μm) soluble Mtb lysates was performed at Kendrick Labs (www.kendricklabs.com). Gels were silver stained and protein spots were excised and shipped by Kendrick Labs to the Columbia University Protein Core Facility for MALDI-TOF analysis.
Mycolic acid biosynthesis
Mtb cultures were grown to mid-log phase. In total, 5 μCi of 14C-acetate or 14C-propionate were added to 10 ml cultures and incubated for 16 h at 37°C. Mycolic acids were extracted from acetate or propionate fed culture pellets using the protocols described in the previous study (Phetsuksiri et al, 1999). In brief, the cells were harvested by centrifugation at 3000 g. The cell pellets were treated with 2 ml of 20% tetrabutyl-ammonium hydroxide at 100°C. After 12–14 h, 2 ml of dichloromethane and 300 μl of methyl iodide was added. All the tubes were mixed by gentle vortexing and incubated for 1 h at room temperature. After centrifugation at 2000 r.p.m. for 10 min, the aqueous layer was removed. The organic phase was washed twice with acidified water before drying under vacuum. The pellet was dissolved in 4 ml diethylether and liquid phase was removed by aspiration. The dried residue was dissolved in 200 μl of dichloromethane and quantified by scintillation counting. Approximately 10 000 c.p.m. per sample was spotted on silica gel 60 F254 TLC plates (Merck) and developed thrice in hexane:ethyl acetate (95:5, v/v).
Methylated (sulpholipid) lipid biosynthesis
Mtb cultures were grown to mid-log phase. 14C-propionate (5 μCi) was added to 10 ml cultures and incubation continued for 16 h at 37°C. Extractable lipids were isolated from cell pellets of propionate fed cultures using previously described methodologies (Converse et al, 2003). The harvested cell pellet was treated with 0.5 ml of chloroform:methanol (1:2, v/v) and vortexed. Acidified water (0.5 ml) was then added and organic and aqueous layers were separated by centrifugation for 30 s. The lower layer (organic chloroform layer) was transferred to a fresh glass tube and dried in a vacuum concentrator. The residue was resuspended in chloroform and approximately 25 000 c.p.m. per sample was spotted per sample. Lipids resolved on silica gel 60 Å with fluorescent indicator (Catalogue No. 4802-400, Whatman) or silica gel 60 Å F254(Catalogue No. 1.05554.0007, Merck), developed in chloroform:methanol:water (60:30:6, v/v) at ambient temperature.
Extraction of apolar lipids
Mtb cultures were grown to mid-log phase. 14C-palmitate (5 μCi) was added to 10 ml cultures and incubation continued for 16 h at 37°C. The 14C-palmitate-labelled apolar lipids were extracted by adding 2 ml of chloroform/0.3% NaCl (100/10; v/v) and 2 ml of petroleum ether to the cell pellet followed by stirring for 30 min. After centrifugation, the upper petroleum ether layer was removed, and 2 ml of petroleum ether was added to the lower phase, and the process was repeated. The combined petroleum ether extracts were then evaporated under nitrogen to yield apolar lipids that were resuspended in CH2Cl2 and analyzed by toluene/acetone (99/1; v/v) (Kremer et al, 2005). The wax esters were resolved on silica gel 60 Å F254 (Catalogue No. 1.05554.0007, Merck) and developed in hexanes:diethyl ether:acetic acid (80:20:1, v/v) (Kalscheuer and Steinbuchel, 2003).
Supplementary Material
Supplementary Figure S1
{kind=link}
Supplementary Figure S1 Legend
Acknowledgments
Antibodies to PrcB and DlaT were generous gifts from Gang Lin and Ruslana Bryk, respectively (Carl Nathan lab). Results presented in Figures 1A and 3A were obtained in part in Carl Nathan's lab with the support of Grant PO1-AI06293 (C Nathan, PI). We thank Joel Ernst for use of his QRTPCR equipment and Niaz Banaiee for QRTPCR advice. We thank Sabine Ehrt, Michael Garabedian, Alfred Goldberg and Carl Nathan for suggestions and advice. We thank Andrew Darwin, Ian Mohr and Carl Nathan for critically reviewing this manuscript. MJP was supported by 5T32 AI07189-25. RSG is a Howard Hughes Medical Institute International Fellow.
References
- Baumeister W, Walz J, Zühl F, Seemüller E (1998) The proteasome: paradigm of a self-compartmentalizing protease. Cell 92: 367–380 [DOI] [PubMed] [Google Scholar]
- Bekker L-G, Freeman S, Murray PJ, Ryffel B, Kaplan G (2001) TNF-α controls intracellular mycobacterial growth by both inducible nitric oxide synthase-dependent and inducible nitric oxide synthase-independent pathways. J Immunol 166: 6728–6734 [DOI] [PubMed] [Google Scholar]
- Benaroudj N, Zwickl P, Seemuller E, Baumeister W, Goldberg AL (2003) ATP hydrolysis by the proteasome regulatory complex PAN serves multiple functions in protein degradation. Mol Cell 11: 69–78 [DOI] [PubMed] [Google Scholar]
- Butler SM, Festa RF, Pearce MJ, Darwin KH (2006) Self-compartmentalized bacteria proteases and pathogenesis. Mol Microbiol 60: 553–562 [DOI] [PubMed] [Google Scholar]
- Chan ED, Chan J, Schluger NW (2001) What is the role of nitric oxide in murine and human host defense against tuberculosis? Am J Respir Cell Mol Biol 25: 606–612 [DOI] [PubMed] [Google Scholar]
- Chaudhuri BN, Sawaya MR, Kim CY, Waldo GS, Park MS, Terwilliger TC, Yeates TO (2003) The crystal structure of the first enzyme in the pantothenate biosynthetic pathway, ketopantoate hydroxymethyltransferase, from M. tuberculosis. Structure 11: 753–764 [DOI] [PubMed] [Google Scholar]
- Chen P, Hochstrasser M (1996) Autocatalytic subunit processing couples active site formation in the 20S proteasome to completion of assembly. Cell 86: 961–972 [DOI] [PubMed] [Google Scholar]
- Converse SE, Mougous JD, Leavell MD, Leary JA, Bertozzi CR, Cox JS (2003) MmpL8 is required for sulfolipid-1 biosynthesis and Mycobacterium tuberculosis virulence. Proc Natl Acad Sci USA 100: 6121–6126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coux O, Tanaka K, Goldberg AL (1996) Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem 65: 801–847 [DOI] [PubMed] [Google Scholar]
- Darwin KH, Ehrt S, Weich N, Gutierrez-Ramos J-C, Nathan CF (2003) The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302: 1963–1966 [DOI] [PubMed] [Google Scholar]
- Darwin KH, Lin G, Chen Z, Li H, Nathan C (2005) Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol Microbiol 55: 561–571 [DOI] [PubMed] [Google Scholar]
- Egerton M, Ashe OR, Chen D, Druker BJ, Burgess WH, Samelson LE (1992) VCP, the mammalian homolog of cdc48, is tyrosine phosphorylated in response to T cell antigen receptor activation. EMBO J 11: 3533–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster A, Masters EI, Whitby FG, Robinson H, Hill CP (2005) The 1.9 A structure of a proteasome-11S activator complex and implications for proteasome–PAN/PA700 interactions. Mol Cell 18: 589–599 [DOI] [PubMed] [Google Scholar]
- Gottesman S (2003) Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol 19: 565–587 [DOI] [PubMed] [Google Scholar]
- Groll M, Ditzel L, Lowe J, Stock D, Bochtler M, Bartunik HD, Huber R (1997) Structure of 20S proteasome from yeast at 2.4 A resolution. Nature 386: 463–471 [DOI] [PubMed] [Google Scholar]
- Hatfull GF, WR Jacobs J (2000) Molecular Genetics of Mycobacteria. Washington, DC: ASM Press [Google Scholar]
- Hernandez-Pando R, Jeyanathan M, Mengistu G, Aguilar D, Orozco H, Harboe M, Rook GA, Bjune G (2000) Persistence of DNA from Mycobacterium tuberculosis in superficially normal lung tissue during latent infection. Lancet 356: 2133–2138 [DOI] [PubMed] [Google Scholar]
- Hirano Y, Hendil KB, Yashiroda H, Iemura S, Nagane R, Hioki Y, Natsume T, Tanaka K, Murata S (2005) A heterodimeric complex that promotes the assembly of mammalian 20S proteasomes. Nature 437: 1381–1385 [DOI] [PubMed] [Google Scholar]
- Hu G, Lin G, Wang M, Dick L, Xu R-M, Nathan C, Li H (2006) Structure of the Mycobacterium tuberculosis proteasome and mechanism of inhibition by a peptidyl boronate. Mol Microbiol 59: 1417–1428 [DOI] [PubMed] [Google Scholar]
- Jung YJ, LaCourse R, Ryan L, North RJ (2002) Virulent but not avirulent Mycobacterium tuberculosis can evade the growth inhibitory action of a T helper 1-dependent, nitric oxide synthase 2-independent defense in mice. J Exp Med 196: 991–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalscheuer R, Steinbuchel A (2003) A novel bifunctional wax ester synthase/acyl-CoA:diacylglycerol acyltransferase mediates wax ester and triacylglycerol biosynthesis in Acinetobacter calcoaceticus ADP1. J Biol Chem 278: 8075–8082 [DOI] [PubMed] [Google Scholar]
- Kinsella RJ, Fitzpatrick DA, Creevey CJ, McInerney JO (2003) Fatty acid biosynthesis in Mycobacterium tuberculosis: lateral gene transfer, adaptive evolution, and gene duplication. Proc Natl Acad Sci USA 100: 10320–10325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisselev AF, Goldberg AL (2001) Proteasome inhibitors: from research tools to drug candidates. Chem Biol 8: 739–758 [DOI] [PubMed] [Google Scholar]
- Kohler A, Bajorek M, Groll M, Moroder L, Rubin DM, Huber R, Glickman MH, Finley D (2001a) The substrate translocation channel of the proteasome. Biochimie 83: 325–332 [DOI] [PubMed] [Google Scholar]
- Kohler A, Cascio P, Leggett DS, Woo KM, Goldberg AL, Finley D (2001b) The axial channel of the proteasome core particle is gated by the Rpt2 ATPase and controls both substrate entry and product release. Mol Cell 7: 1143–1152 [DOI] [PubMed] [Google Scholar]
- Kremer L, de Chastellier C, Dobson G, Gibson KJ, Bifani P, Balor S, Gorvel JP, Locht C, Minnikin DE, Besra GS (2005) Identification and structural characterization of an unusual mycobacterial monomeromycolyl-diacylglycerol. Mol Microbiol 57: 1113–1126 [DOI] [PubMed] [Google Scholar]
- Kremer L, Nampoothiri KM, Lesjean S, Dover LG, Graham S, Betts J, Brennan PJ, Minnikin DE, Locht C, Besra GS (2001) Biochemical characterization of acyl carrier protein (AcpM) and malonyl-CoA:AcpM transacylase (mtFabD), two major components of Mycobacterium tuberculosis fatty acid synthase II. J Biol Chem 276: 27967–27974 [DOI] [PubMed] [Google Scholar]
- Lin G, Hu G, Tsu C, Kunes YZ, Li H, Dick L, Parsons T, Li P, Chen Z, Zwickl P, Weich N, Nathan C (2006) Mycobacterium tuberculosis prcBA genes encode a gated proteasome. Mol Microbiol 59: 1405–1416 [DOI] [PubMed] [Google Scholar]
- MacMicking J, Xie QW, Nathan C (1997a) Nitric oxide and macrophage function. Annu Rev Immunol 15: 323–350 [DOI] [PubMed] [Google Scholar]
- MacMicking JD, North RJ, LaCourse R, Mudgett JS, Shah SK, Nathan CF (1997b) Identification of nitric oxide synthase as a protective locus against tuberculosis. Proc Natl Acad Sci USA 94: 5243–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo F, Schlauer J, Zischka H, Mecke D, Frohlich KU (1998) Tyrosine phosphorylation regulates cell cycle-dependent nuclear localization of Cdc48p. Mol Biol Cell 9: 131–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurizi MR, Clark WP, Kim SH, Gottesman S (1990) Clp P represents a unique family of serine proteases. J Biol Chem 265: 12546–12552 [PubMed] [Google Scholar]
- Mogues T, Goodrich ME, Ryan L, LaCourse R, North RJ (2001) The relative importance of T cell subsets in immunity and immunopathology of airborne Mycobacterium tuberculosis infection in mice. J Exp Med 193: 271–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy I, Geert S, Vanderleyden J, De Mot R (1997) Further sequence analysis of the DNA regions with the Rhodococcus 20S proteasome structural genes reveals extensive homolgy with Mycobacterium leprae. DNA Seq 7: 225–228 [DOI] [PubMed] [Google Scholar]
- Nozaki Y, Hasegawa Y, Ichiyama S, Nakashima I, Shimokata K (1997) Mechanism of nitric oxide-dependent killing of Mycobacterium bovis BCG in human alveolar macrophages. Infect Immun 65: 3644–3647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phetsuksiri B, Baulard AR, Cooper AM, Minnikin DE, Douglas JD, Besra GS, Brennan PJ (1999) Antimycobacterial activities of isoxyl and new derivatives through the inhibition of mycolic acid synthesis. Antimicrob Agents Chemother 43: 1042–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM, Cohen RE (2004) Proteasomes and their kin: proteases in the machine age. Nat Rev Mol Cell Biol 5: 177–187 [DOI] [PubMed] [Google Scholar]
- Raman N, Black PN, DiRusso CC (1997) Characterization of the fatty acid-responsive transcription factor FadR. Biochemical and genetic analyses of the native conformation and functional domains. J Biol Chem 272: 30645–30650 [DOI] [PubMed] [Google Scholar]
- Raman N, DiRusso CC (1995) Analysis of acyl coenzyme A binding to the transcription factor FadR and identification of amino acid residues in the carboxyl terminus required for ligand binding. J Biol Chem 270: 1092–1097 [DOI] [PubMed] [Google Scholar]
- Sassetti CM, Boyd DH, Rubin EJ (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol Microbiol 48: 77–84 [DOI] [PubMed] [Google Scholar]
- Scanga CA, Mohan VP, Tanaka K, Alland D, Flynn JL, Chan J (2001) The inducible nitric oxide synthase locus confers protection against aerogenic challenge of both clinical and laboratory strains of Mycobacterium tuberculosis in mice. Infect Immun 69: 7711–7717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidtke G, Kraft R, Kostka S, Henklein P, Frommel C, Lowe J, Huber R, Kloetzel PM, Schmidt M (1996) Analysis of mammalian 20S proteasome biogenesis: the maturation of beta-subunits is an ordered two-step mechanism involving autocatalysis. EMBO J 15: 6887–6898 [PMC free article] [PubMed] [Google Scholar]
- Scholz O, Thiel A, Hillen W, Niederweis M (2000) Quantitative analysis of gene expression with an improved green fluorescent protein. p6. Eur J Biochem 267: 1565–1570 [DOI] [PubMed] [Google Scholar]
- Seong IS, Kang MS, Choi MK, Lee JW, Koh OJ, Wang J, Eom SH, Chung CH (2002) The C-terminal tails of HslU ATPase act as a molecular switch for activation of HslV peptidase. J Biol Chem 277: 25976–25982 [DOI] [PubMed] [Google Scholar]
- Smith DM, Kafri G, Cheng Y, Ng D, Walz T, Goldberg AL (2005) ATP binding to PAN or the 26S ATPases causes association with the 20S proteasome, gate opening, and translocation of unfolded proteins. Mol Cell 20: 687–698 [DOI] [PubMed] [Google Scholar]
- Stover CK, De La Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, Snapper SB, Barletta RG, Jacobs WR Jr, Bloom BR (1991) New use of BCG for recombinant vaccines. Nature 351: 456–460 [DOI] [PubMed] [Google Scholar]
- Sugantino M, Zheng R, Yu M, Blanchard JS (2003) Mycobacterium tuberculosis ketopantoate hydroxymethyltransferase: tetrahydrofolate-independent hydroxymethyltransferase and enolization reactions with alpha-keto acids. Biochemistry 42: 191–199 [DOI] [PubMed] [Google Scholar]
- Tian J, Bryk R, Shi S, Erdjument-Bromage H, Tempst P, Nathan C (2005) Mycobacterium tuberculosis appears to lack alpha-ketoglutarate dehydrogenase and encodes pyruvate dehydrogenase in widely separated genes. Mol Microbiol 57: 859–868 [DOI] [PubMed] [Google Scholar]
- Unno M, Mizushima T, Morimoto Y, Tomisugi Y, Tanaka K, Yasuoka N, Tsukihara T (2002) The structure of the mammalian 20S proteasome at 2.75 A resolution. Structure 10: 609–618 [DOI] [PubMed] [Google Scholar]
- Wang J, Song JJ, Franklin MC, Kamtekar S, Im YJ, Rho SH, Seong IS, Lee CS, Chung CH, Eom SH (2001) Crystal structures of the HslVU peptidase–ATPase complex reveal an ATP-dependent proteolysis mechanism. Structure 9: 177–184 [DOI] [PubMed] [Google Scholar]
- Wolf S, Nagy I, Lupas A, Pfeifer G, Cejka Z, Müller SA, Engel A, De Mot R, Baumeister W (1998) Characterization of ARC, a divergent member of the AAA ATPase family from Rhodococcus erythropolis. J Mol Biol 277: 13–25 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S1 Legend