

Abstract
The entry and enzymatic activity of the anthrax edema factor (EF) in different cell types was studied by monitoring EF-induced changes in intracellular cAMP with biochemical and microscopic methods. cAMP was imaged in live cells, transfected with a fluorescence resonance energy transfer biosensor based on the protein kinase A regulatory and catalytic subunits fused to CFP and YFP, respectively. The cAMP biosensor was located either in the cytosol or was membrane-bound owing to the addition of a tag determining its myristoylation/palmitoylation. Real-time imaging of cells expressing the cAMP biosensors provided the time course of EF catalytic activity and an indication of its subcellular localization. Bafilomycin A1, an inhibitor of the vacuolar ATPase proton pump, completely prevented EF activity, even when added long after the toxin. The time course of appearance of the adenylate cyclase activity and of bafilomycin A1 action suggests that EF enters the cytosol from late endosomes. EF remains associated to these compartments and its activity shows a perinuclear localization generating intracellular cAMP concentration gradients from the cell centre to the periphery.
Keywords: anthrax, cAMP, cell entry, edema toxin, FRET imaging
Introduction
Anthrax is an infection caused by pathogenic strains of the sporulating bacterium Bacillus anthracis (Turnbull, 2002). The progress of the disease depends on the route of entry of B. anthracis spores into the body: skin abrasions, alimentary tract and lungs (Dixon et al, 1999). Skin anthrax and gastro-intestinal anthrax are less dangerous than inhalational anthrax, which very frequently leads to death. In this case, the spores are phagocytosed by alveolar macrophages or dendritic cells which migrate to regional lymph nodes. The spores germinate within the phagocytes and bacteria are released. They multiply in the lymphatic system and enter the bloodstream, causing septicemia and toxemia (Dixon et al, 1999; Ascenzi et al, 2002; Mock and Mignot, 2003; Moayeri and Leppla, 2004).
B. anthracis secretes a three-components toxic complex consisting of the protective antigen (PA, 87 kDa), the lethal factor (LF, 90 kDa) and the edema factor (EF, 89kDa) (Collier and Young, 2003). PA binds to two different cell surface receptors: Endothelial Marker 8 (TEM8) and Capillary Morphogenesis Protein 2 (CMG2), with a rather wide distribution among tissues and cells (Bradley et al, 2001; Collier and Young, 2003; Scobie et al, 2003; Rainey et al, 2005). PA binds CMG2 stronger than TEM8 (Bradley et al, 2001; Wigelsworth et al, 2004, Rainey et al, 2005). Both TEM8 and CMG2 interact with LDL receptor-related protein 6 (LRP6), which is essential for the following step of endocytosis (Wei et al, 2006). PA is proteolytically activated by removal of a 20 kDa N-terminal domain and self-associates into heptamers capable of binding LF and EF (Moayeri and Leppla, 2004). PA+LF complexes enter surface rafts (Abrami et al, 2003), are endocytosed and reach late endosomal compartments, whose acid luminal pH causes a conformational change, which results in LF release into the cell cytosol (Abrami et al, 2004, 2005; Gruenberg and van der Goot, 2006). LF is a zinc metalloprotease that cleaves mitogen-activated protein kinase kinases (MAPKK) (Duesbery et al, 1998; Vitale et al, 1998, 2000), thereby interfering with the MAPK cascade, a major signalling pathway triggered by surface receptors, controlling cell proliferation and survival (Dong et al, 2002; Gaestel, 2006). LF inhibits the immune response by acting on a variety of immune cells via MAPKK cleavage (Pellizzari et al, 1999; Park et al, 2002; Agrawal et al, 2003; Paccani et al, 2005; Tournier et al, 2005; Baldari et al, 2006).
EF is a calcium/calmodulin-activated adenylate cyclase, which catalyses the formation of cAMP (Drum et al, 2000; Shen et al, 2005), thus altering cell signalling and tissue ion fluxes; the ensuing edema causes failure of different organs leading to rapid death (Firoved et al, 2005). EF was recently shown to strongly inhibit the release of cytokines from dendritic cells and the activation and proliferation of T lymphocytes with a strong synergism of action with LF (Paccani et al, 2005; Tournier et al, 2005). Much less is known about the process of entry of EF into cells and of rise in cAMP cellular level. In the fibroblast CHO cells, EF causes a detectable rise of cAMP after 30 min of incubation, which increased many fold after 60 min (Gordon et al, 1989). An assay of the cAMP-induced Cl− current in polarized epithelial T84 cells treated with PA+EF showed a similar time course (Beauregard et al, 1999).
Stimulated by the findings that EF is a potent effector in immune cells, we have decided to investigate the time course of the EF adenylate activity inside different cell lines. cAMP was determined both by the conventional biochemical assay and by using fluorescence-based cAMP biosensors and optical imaging (Zaccolo et al, 2000; Evellin et al, 2004). The fluorescent cAMP probes allowed us to measure in real time and at the single-cell level the EF-induced changes in cAMP concentration and to monitor the intracellular distribution of changes in cAMP in living cells. The results obtained are consistent with a translocation of EF from late endosomal compartments and show that the rise in cAMP is prolonged and unevenly distributed in the cytosol with a decreasing concentration from the perinuclear area to the cell periphery.
Results
The time course of EF-induced cytosolic cAMP changes suggests that EF enters the cytosol from late endosomal compartments
In order to display their enzymatic activities, both EF and LF have to reach the cell cytosol where their substrates and activators reside. PA is essential in such a process because it mediates cell surface binding and endocytosis of the toxins. Two PA receptors are known and we have tested by RT–PCR that at least one of them is expressed by the three cells lines used here, which were chosen because of their relation to major in vivo cell targets of EF+PA (Friedlander 1986; Beauregard et al, 1999; Baldari et al, 2006). Supplementary Figure S1 shows that HeLa and Jurkat cells express predominantly TEM8, whereas the RAW264.7 macrophages express only CMG2. After endocytosis, PA undergoes a low-pH triggered structural change with formation of a trans-membrane channel, which assists the membrane translocation of EF and LF (Collier and Young, 2003; Krantz et al, 2005). Results obtained with different experimental approaches indicate that LF translocates into the cytosol from late endosomal compartments (Abrami et al, 2005), which undergo an intracellular trafficking involving sequential entry into early endosomes (EE), multivesicular carrier vesicles and late endosomes (Abrami et al, 2004; Gruenberg and van der Goot, 2006). Similar information is lacking in the case of EF.
Figure 1 shows the time course of the changes in intracellular cAMP concentration in different cell lines. In all cases, cAMP rises significantly after 1 h from the addition of PA+EF and remains high up to 4 h after toxin addition. The absolute cAMP level induced by EF is much higher in T cells with respect to macrophages and epithelial cell lines. This time course of the EF induced rise in cAMP includes toxin binding, endocytosis and membrane translocation and it is compatible with an entry of EF from late endosomes, as previously found for LF (Menard et al, 1996; Abrami et al, 2004). If this is the case, the EF-induced rise of cAMP must be abolished by the specific v-ATPase inhibitor bafilomycin A1, which prevents the cellular activity of LF (Menard et al, 1996). Moreover, by adding this inhibitor at different times after intoxication (bafilomycin shift assay), information about the endocytic step wherefrom the toxin enters the cytosol can be obtained (Papini et al, 1993).
Figure 1.
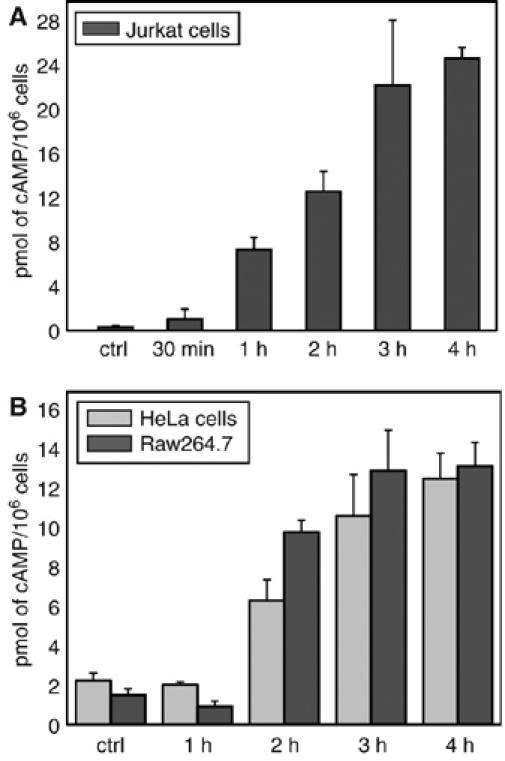
EF generates a prolonged cAMP increase in different cell lines. Cells were treated with EF 10 nM and PA 20 nM and incubated for the indicated time periods at 37°C. After centrifugation, the supernatant was removed and cells were lysed to measure intracellular cAMP with the Amersham immuno assay kit. (A) Jurkat T cells, (B) HeLa cells (grey bars) and Raw264.7 macrophages (dark grey bars). Data are the average of two or more different experiments run in triplicates and bars represent±s.d.
Figure 2 shows the results of such an assay performed on the three cell lines used here with bafilomycin A1 being capable of about 50% inhibition when introduced after 1 h from PA+EF. These results are closely similar to those obtained on macrophage death induced by PA+LF (Menard et al (1996) and by monitoring the MAPKK3 cleavage by LF, reported in Supplementary Figure S2). LF has been well documented to travel along the endocytic pathway until late endosomes (Abrami et al, 2004, 2005). That EF follows a similar intracellular pathway is further supported by the inhibition of the EF-induced rise of cAMP by nocodazole (Figure 2C). This agent inhibits the membrane trafficking from early to late endosome by inducing microtubule depolymerization (Gruenberg et al, 1989). Taken together, these data indicate that EF follows the same pathway of entry into cells as LF, with a time course closely similar in different cell lines. Therefore, it appears that the main determinant of the final organelle destination of EF or LF is PA, their common cell binding and membrane translocating partner. It also appears that the different expression of the PA receptors does not determine a different kinetics of rise of cellular cAMP in the three different cell lines.
Figure 2.
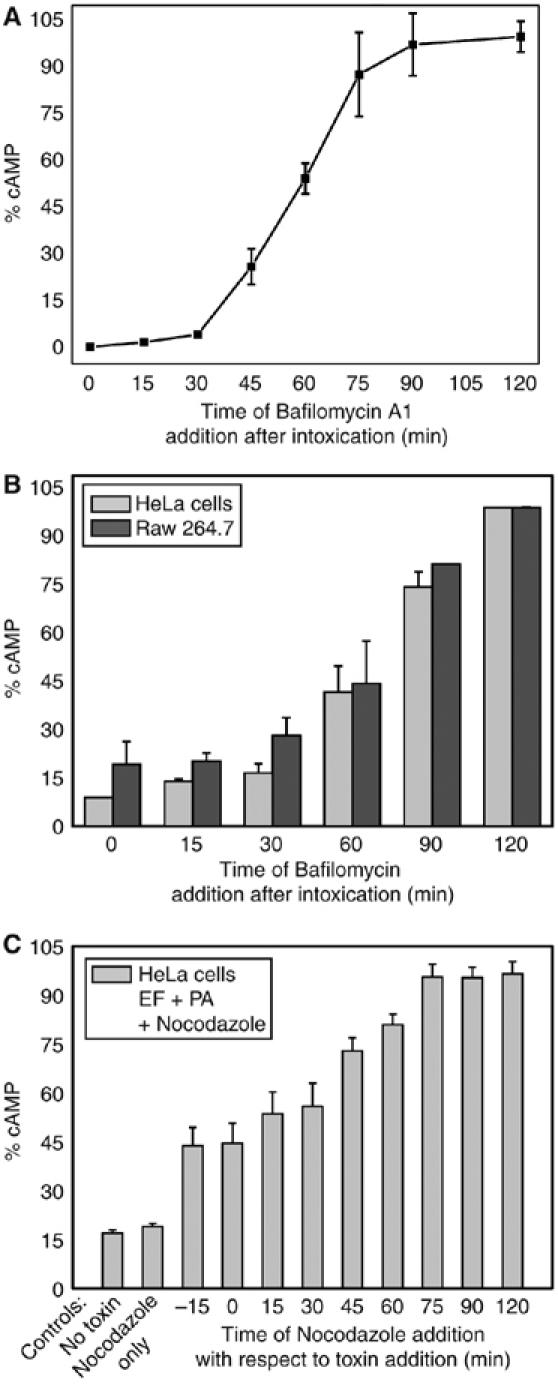
The v-ATPase proton pump bafilomycin A1 prevents EF activity long after toxin addition to cells. (A) Jurkat cells were treated with EF 10 nM plus PA 20 nM at time 0 and incubated at 37°C for 2 h; bafilomycin A1 (0.5 μM) was added at the indicated time intervals after the addition of the toxin. Cells were centrifuged, and lysed, after removal of the supernatant, to measure intracellular cAMP with the Amersham immunoassay kit. (B) The same experiment was performed with HeLa cells (grey bars) and with the Raw264.7 macrophages (dark grey bars). cAMP determined at any given time is reported as percentage of the maximum value. (C) cAMP content of HeLa cells treated with 10 μM nocodazole (Sigma) added at the indicated time intervals before or after toxin addition. The partial initial inhibitory effect of nocodazole is likely to be due to the fact that some time is required for microtubule depolymerization to take place. Data are the average of two or more different experiments run in triplicates and bars represent±s.d.
Fractionation of cells treated with anthrax edema toxin or LF
Previous studies of the membrane insertion and translocation of EF and LF as a function of pH (Kochi et al, 1994; Guidi-Rontani et al, 2000) raised the possibility that EF remains associated to the cytosolic face of the late endosomal membranes after translocation. If this is the case, on the basis of the present findings, EF should be detected on isolated late endosomal compartments. Ultracentrifugation of lysed HeLa cells gave a organelle-free postnuclear supernatant (PNS) and a organelle-containing suspension, which was then further fractionated on a sucrose density gradient into a heavy membrane fraction (HM), EE and late endosomes (Gruenberg et al, 1989; Gruenberg, 2001; Kobayashi et al, 2002). Figure 3 shows that in HeLa cells treated with EF and LF, the major proportion of EF is associated to the LE fraction and a slightly proportion is associated to the EE fraction, with no EF found in the PNS. At the same time, the major part of LF is in the PNS with some of it present in the EE fraction. Apart from the fact that the EE fraction may include some LE, as judged by the presence of the rab7 LE marker, the EF and LF presence in EE is due to the fact that these experiments were performed under the same conditions used for imaging (see next paragraph), which cannot include a low-temperature binding and synchronization step for technical reasons. However, the differential presence of EF and LF in the LE and PNS fractions is clear enough to support the view that, after sorting out of late endosomes, LF mainly disperses in the cytosol and EF mainly associates to the cytosolic surface of LE. The appearance and localization of the EF adenylate cyclase activity can be monitored in time by novel techniques of cAMP imaging, which have been recently introduced. In addition, this approach, which uses living cells, allows one to test the possibility that some redistribution of EF among cellular fractions may have occurred during cell lysis.
Figure 3.
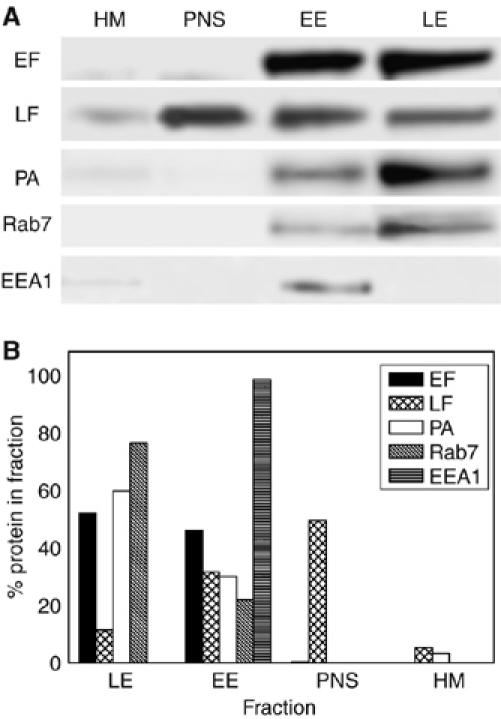
Different subcellular localizations of anthrax LF and EF. HeLa cells were treated either with LF+PA (10 and 20 nM, respectively) or with EF+PA (10 and 20 nM, respectively) for 1 h at 37°C and were then lysed and separated into a supernatant (PNS), which includes the cell cytosol, and a membrane fraction that was fractionated on a sucrose density gradient. Three fractions were obtained: HM, heavy membranes, EE, early endosomes and LE, late endosomes. Ten microgram proteins of these fractions were subjected to SDS–PAGE, immunoblotted and developed as described in the Materials and methods. (A) Toxin and markers distribution among the different fractions in a representative experiment, while (B) show the corresponding quantification of the bands, which includes a normalization to the total amount of proteins of each of the four fractions examined. Rab7 is a marker of late endosomes and EEA1 is a marker of early endosomes. Note the largely different distribution of LF and EF, with LF being present mainly in the cell cytosol fraction and EF on endosomes. The significant amount of LF and EF still present in the early endosomal fraction is due to the fact that the experiment was performed without low-temperature synchronization of the binding step, under the same conditions required for imaging (see text).
Imaging of anthrax edema toxin-induced cAMP changes
Cellular cAMP can be imaged in terms of both concentration and localization with a novel fluorescent biosensor (Zaccolo et al, 2000; Lissandron et al, 2005) based on the cAMP binding protein kinase A (PKA) and exploiting the phenomenon of fluorescence resonance energy transfer (FRET). Such sensor was generated by fusing the regulatory RII-β subunit and the catalytic C-α subunit of PKA to the cyan (CFP) and the yellow (YFP) variants of the green fluorescent protein, respectively. Under resting conditions, the R-CFP and C-YFP subunits are associated in a holotetrameric complex and FRET occurs among them. In the presence of cAMP, the C-YFP subunits are released and diffuse apart from R-CFP thus lowering FRET. Monitoring of fluorescence emission changes provides a real-time image of the changes and distribution of cAMP generated by the adenylate cyclase activity of EF within a cell. When expressed in transfected cells, the cAMP sensor described above evenly distributes in the cytosol (see Figure 4A and B1, insets and Figure 5A and B). A variant of such sensor was generated by fusing to the N-terminus of the RII-CFP subunit, a short peptide containing consensus sequences for fatty acid acylation (Zacharias et al, 2002). Post-transcriptional myristoylation and palmitoylation of the RII-CFP chimera effectively targets the cAMP biosensor mainly to the plasma membrane (Figure 4C, D2 and D3, insets and Figure 6A and B), as found by Zacharias et al (2002) in MDCK cells.
Figure 4.
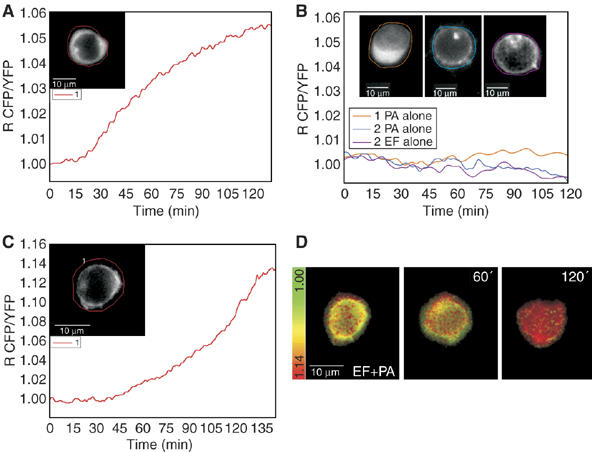
Imaging of the EF-induced rise of cAMP with PKA fluorescent probes in Jurkat cells. Jurkat cells expressing the catalytic PKA subunit coupled to YFP and the regulatory PKA subunit coupled to CFP in the cytosol or in the plasma membrane depending on the presence of a membrane localization sequence were imaged after treatment with EF 10 nM+PA 20 nM (time zero). During microscopic observations, cells were maintained in 2 ml of a balanced salt solution inside a microscope-adapted micro-incubator at 37°C and constant 5% CO2 pressure. Images were acquired every 10 s and the ratio between CFP and YFP emissions was calculated. An increasing ratio corresponds to increasing cAMP concentrations. Similar traces were recorded in other cells and they do not depend on cell size. (A) Change of cAMP with time in a cell expressing the cytosolic probe; the inset shows the fluorescence of CFP at time O indicating a cytosolic distribution of the probe. (B) cAMP remains low in cells treated with PA only or EF only. This is revealed by both the cytosolic PKA fluorescent probe (orange trace corresponding to the cell of inset 1 which shows the CFP fluorescence at time 0) and by the membrane localized PKA probe (inset 2, blue trace, and inset 3, magenta trace, show the CFP fluorescence taken at time 0 of cells treated with PA or Ef, respectively). (C) The change of cAMP with time in a Jurkat cell expressing the membrane localized PKA probe; the inset shows the fluorescence of membrane-bound CFP at time O. (D) The Jurkat cell of (C) as pseudo-colours, which reflect the increasing cAMP concentration from green (low cAMP) to red (high cAMP) at the indicated time points of incubation with PA+EF.
Figure 5.
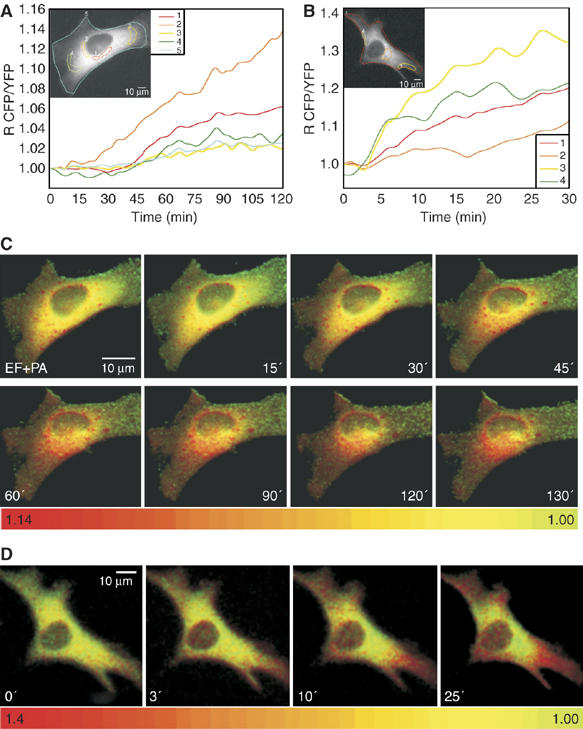
Anthrax edema toxin creates c-AMP microdomains in HeLa cells. (A) HeLa cells expressing the cytosolic PKA-based probe cAMP fluorescence biosensor were treated with EF 10+PA 20 nM (time zero) and maintained in 2 ml of balanced salt solution at 37°C during microscopic observations. CFP/YFP ratios were measured in the indicated areas, identified with different colour contours: perinuclear regions (1, red trace; 2, orange trace) and cell periphery (3, yellow trace; 4, green trace). Notice the lower cAMP rising in the peripheral areas. (B) HeLa cell expressing the cAMP cytosolic probe treated with the B. pertussis CyaA adenylate cyclase toxin, which enters from the plasma membrane. Notice the faster rise of the ratiometic signal in the sub-plasma membrane areas identified by different colours, which are the same of those of the corresponding traces. (C, D) Pseudo-colour images, generated by CFP/YFP ratio imaging, of the intracellular cAMP at the given time points of the cell of (A) treated with PA+EF and of the cell of (B) treated with CyaA.
Figure 6.
Different routes of entry of EF and of the CyaA adenylate cyclase of B. pertussis. HeLa cells expressing the membrane-bound PKA-based biosensor for cAMP were treated with 1 nM CyaA (A) or with EF 10 nM+PA 20 nM (B) and the CFP/YFP ratio was measured in plasma membrane areas of the cells (see contours in the cells of the insets) as a function of time.
Jurkat T cells can be reproducibly transfected with the chimeric fluorescent PKA subunits and Figure 4 shows representative time courses of the fluorescence emission ratio changes, corresponding to increasing levels of cytosolic cAMP concentration, recorded in transfected Jurkat cells exposed to PA+EF. The time course of such cAMP imaging increase is comparable to that measured with the conventional biochemical assay (Figure 1). Small differences indicating cell-to-cell variability in the responses to the EF toxin become detectable with the imaging approach, but the general trend is well represented by the cell in Figure 4A. In addition, the EF activity assay by fluorescence imaging provides a much better time-resolved record of the phenomenon. A comparison of the time courses of the rise of cAMP induced in Jurkat cells expressing the cytosolic (Figure 4A) and membrane (Figure 4C) probes shows that they are delayed. This difference is highly reproducible and suggests that the EF adenylate cyclase activity is not evenly distributed across the cytosol of T cells. This result is in keeping with EF being associated to LE, which has a perinuclear localization and a consequent higher local concentration of cAMP around the nucleus.
cAMP microdomains in HeLa cells treated with the anthrax edema toxin or with the Bordetella pertussis Cya adenylate cyclase toxin
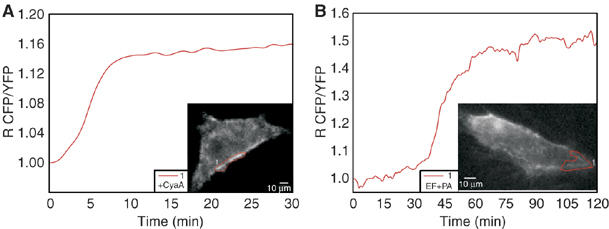
Jurkat cells are not the most appropriate test system to evaluate intracellular cAMP concentration gradients, owing to their limited cytoplasm. Therefore, we have extended our studies to the HeLa cells. Figure 5A shows a typical EF response of a cell transfected with the cytosolic PKA probe. The pseudo-colour image frames in Figure 5C, where the transition from yellow to red indicates an increasing cAMP concentration, document a higher signal in the perinuclear area, where late endosomal compartments concentrate. This result is very representative of many cells and is not affected by an uneven distribution of the PKA probe as cAMP changes are given by the ratio between the emission intensities generated by the two subunits of the sensor, thereby eliminating artefacts due to different probe concentration. The evidence of a gradient of cAMP concentration from the perinuclear region to the plasma membrane is reinforced by an analysis of the time course of the FRET changes monitored in different intracellular domains of HeLa cells, showing a steeper rise of signal close to the nucleus (orange trace in Figure 5A), with lower and delayed rises in peripheral areas (green and yellow traces in Figure 5A). This conclusion is further supported by the use of the B. pertussis adenylate cyclase toxin (CyaA), which is well documented to enter cells directly from the plasma membrane (Ladant and Ullmann, 1999; Hewlett et al, 2000). The cAMP increase induced by CyaA in HeLa cells, documented in Figure 5B and D, shows an inverted gradient with respect to that induced by EF. CyaA induces an earlier signal increase underneath the plasma membrane (yellow and green traces), while a delayed and lower cAMP signal in a perinuclear area (orange trace). The pseudo-colour images well describe the opposite intracellular cAMP gradients created by B. anthracis EF (Figure 5C) and by B. pertussis CyaA (Figure 5D).
HeLa cells were also transfected with the membrane probe described previously for MDCK cells by Zacharias et al (2002). Also in the case of the HeLa cells, this probe localizes mainly on the plasma membrane with little presence in intracellular membranes. The different behaviour of the two adenylate cyclase toxins is also highlighted by the membrane-bound PKA probe, with CyaA causing a rapid rise of cAMP signal below the plasma membrane (Figure 6A) and EF causing a delayed response (Figure 6B). The ensemble of these results also indicate that the cell fractionation study, described in the previous paragraph, reflected the distribution of EF present in the living cells after 1 h of incubation, and proves that at least a fraction of late endosomal EF is present on the cytosolic side of the organelle.
Discussion
The present work was aimed at characterizing the process of cell entry of the anthrax EF and of the modification of the concentration and distribution of cAMP that EF induces in living cells. These goals were reached by using fluorescent PKA-based probes for optical imaging of cAMP with subcellular spatial resolution and appropriate biochemical assays. An important finding presented here is that EF reaches its cytosolic substrate ATP with a time course, determined with both biochemical and imaging assays, similar to that of the anthrax LF. LF was previously shown to enter the cytosol via the endocytic route at the late endosomal stage (Abrami et al, 2005). Similarly to LF, EF entry depends strictly on the activity of the vacuolar ATPase proton pump; the effect of its specific inhibitor bafilomycin A1, added at different time periods after the toxin, also indicates a sorting out of EF from late endosomes. Taken together, these results provide strong evidence that EF is endocytosed together with PA and that it reaches the cytosol from a late endocytic compartment. EF binds to PA, the toxin component responsible for cell binding and translocation of both LF and EF similarly, but not identically, to LF (Shen et al, 2005) and therefore the fact that the two anthrax toxins follow the same entry route was not completely expected. This is the first experimental evidence for such a conclusion and it is based on completely different experimental approaches. Moreover, very similar time courses were found in three different type of cells: T cells, macrophages and epithelial cell lines, notwithstanding their differences in terms of PA receptors, cell shape, amount of cytosol, etc.
Previous experiments suggested that EF, after translocation across the endosomal membrane, refolds in the cytosolic and neutral pH side of the membrane and remains associated to it (Kochi et al, 1994; Guidi-Rontani et al, 2000). Consequently, EF would become a novel enzyme of the cytosolic surface of late endosomes with the possibility of generating cAMP gradients across the cytosol. This possibility was supported by cell fractionation, which showed EF associated to endosomal compartments, while LF was mainly found in the cytosol. This result is fully consistent with previous findings on LF (Menard et al, 1996; Abrami et al, 2004). However, cell fractionation may allow for some relocation of a ligand adsorbed on an organelle surface. Therefore, to test the hypothesis in cells in vivo, we performed imaging of cAMP with biosensors targeted to different intracellular compartments. With both a cytosolic and a membrane-bound PKA-based fluorescent probe, we obtained evidence that in the flat HeLa cells, which are very appropriate for detecting cytosolic gradients, there is a cAMP gradient of decreasing concentration from the perinuclear late endosomal area to the sub-plasmalemma cytosol. In other words, the time course of the EF-induced rise of cAMP is more rapid in the perinuclear cytosol than in the sub-plasma membrane area. The converse is true in cells treated with the CyaA adenylate cyclase toxin of B. pertussis, which is known to enter the cytosol from the plasma membrane (Ladant and Ullmann, 1999; Hewlett et al, 2000). These data indicate that the methods introduced here to image cAMP in cells exposed to EF and CyaA can be extended with profit to all bacterial toxins acting via increasing intracellular cAMP concentration. This is an important experimental achievement and its use will provide relevant information on the mode of action of the large group of microbial toxins that increase the cytosolic cAMP concentration, which include very important virulence factors such as cholera toxin, Eschesichio coli heat labile toxin and pertussis toxin.
Materials and methods
Protein cloning, expression and purification
The EF gene was PCR-amplified using the following primers: 5′AAAGGATCCATGAATGAACATTACACTGAG3′ (forward) and 5′AAAGAGCTCTTATTTTTCATCAATAATTTTTTGG3′ (reverse), digested with BamHI and SacI and inserted in pRSETa (Invitrogen) downstream from an N-terminal His-tag coding region. The sequence was confirmed by DNA sequencing. The protein was expressed in Escherichia coli BL21(DE3)-Codon Plus-RIL (Stratagene) grown at 37°C in LB broth containing 100 μg/ml of ampicillin (Sigma). After 4 h of induction with 1 mM isopropyl-1-thio-a-D-galactopyranoside at 30°C, the pellet was resuspended in buffer A (50 mM Na2HPO4, 500 mM NaCl, pH 8) and lysozyme (0.1 mg/ml). Bacterial cells were disrupted by ultrasonic dispersion, centrifuged and the supernatant was loaded onto a Hi-trap column charged with Cu2+ and equilibrated with buffer A. The column was washed with buffer A and the protein was eluted with a 0–100 mM imidazole gradient, and the fractions containing EF were pooled and dialysed against binding buffer (50 mM Tris, 20 mM NaCl and 1 mM EDTA, pH 7.5) to remove imidazole and NaCl. The dialysed sample was loaded onto a monoQ anion-exchange column (Amersham Biosciences) previously equilibrated with binding buffer. The protein was eluted with a linear gradient of 0–1 M NaCl in the same buffer. EF eluted at approximately 400 mM NaCl. Throughout these processes, identity and purity of EF were assessed by immunoblotting with anti-His tag and anti-EF monoclonal antibodies (kind gift of M Mock, Institut Pasteur, Paris). Purified EF was dialysed against 10 mM HEPES, 50 mM NaCl, pH 7.5 and stored in aliquots at −80°C after quick freezing in liquid nitrogen.
The adenylate cyclase toxin (CyaA) from B. pertussis was expressed in E. coli and purified to homogeneity as previously described (Karimova et al, 1998).
Cell cultures
The macrophage cell line RAW264.7 was maintained in RPMI-1640 containing 10% heat-inactivated foetal calf serum (FCS, Euroclone), penicillin (100 U/ml) and streptomycin (100 μg/ml). HeLa cells were cultivated in DMEM (Gibco) supplemented with 10% FCS, penicillin (100 U/ml) and streptomycin (100 μg/ml). The human lymphoma T cell line Jurkat was grown in RPMI-1640 supplemented with 10% FCS, penicillin, streptomycin and glutamine (Gibco stock solution diluted 100-fold).
Anthrax edema toxin adenylate cyclase activity
5 × 104 Jurkat cells or 1.5 × 104 HeLa or 2 × 104 RAW264.7 cells were incubated with EF 10 nM and PA 20 nM in appropriate complete medium at 37°C for different time periods in a 96-well plate. After removal of the culture medium, the cells were lysed and processed for intracellular cAMP measurement by enzyme-linked immunoassay following the manufacturer's instruction (Biotrak EIA, Amersham Biosciences).
Effect of bafilomycin A1 and of nocodazole on EF translocation
5 × 104 Jurkat cells or 1.5 × 104 HeLa or 2 × 104 Raw264.7 cells were plated the day before the assay in a 96-well plate. Cells were preincubated with phosphodiesterase inhibitor 3-Isobutyl-1-methylxanthine 100 μM (Sigma) at 37°C for 15 min before toxin addition. Bafilomycin A1 0.5 μM (Sigma) or nocodazole 10 μM was added at different times before and after the addition of PA+EF (20 and 10 nM, respectively). The cellular cAMP content was measured after 2 h of incubation with the toxin.
Cell transfections and FRET imaging of cAMP intracellular dynamics
Jurkat cells (1 × 106 cells) were transfected by electroporation with 20 μg each of two pCDNA3 plasmid, one carrying the catalytic (C) subunit of PKA fused to YFP (C-YFP) and one carrying either the regulatory (R) subunit of PKA fused to CFP (RII-CFP) (Lissandron et al, 2005) or a MyrPalm tagged version of RII-CFP. In MyrPalmRII-CFP, a myristoylated and palmitoylated peptide (MGCIKSKRKDNLNDD) from the Lyn kinase (Zacharias et al, 2002) was fused at the N-terminus of the RII-CFP chimera. After transfection, T cells were plated at 5 × 105 cells/ml density on fibronectin-coated glass coverslips. HeLa cells (1.2 × 105 cells/ml) were co-transfected with 1 μg each of the above plasmids by using the transfection vehicle FuGENE6 (Roche Diagnostic Corporation) as described (Mongillo et al, 2004). At 24–48 h after transfection, cells were incubated in a balanced salt solution (NaCl 135 mM, KCl 5 mM, KH2PO4 0.4 mM, MgSO4 1 mM, HEPES 20 mM, CaCl2 1.8 mM, glucose 5.4 mM, pH 7.4) in a microscope-adapted micro-incubator equipped with a temperature controller (HTC, Italy) at 37°C and constant 5% CO2 pressure. Toxins were added (EF 10 nM+PA 20 nM or CyaA 1 nM) after 15 min of imaging, and then images were taken every 10 s for the required time periods. Integration time was 300 ms or lower. At each time point, the intracellular cAMP level was estimated by measuring the ratio between the background subtracted cyan emission image (480 nm) and the yellow emission image (545 nm) upon excitation at 430 nm (R CFP/YFP) (Mongillo et al, 2005). Images were acquired using an oil immersion × 100 PlanApo 1.30 NA objective for Jurkat cells and × 40 PlanApo 1.00 NA for HeLa cells on an Olympus IX70 microscope, equipped with a Polychrome IV monochromator (TILL Photonics, Germany) and a CCD camera (PCO SensiCam QE). The acquisition software was TILLvisION v.3.3 (TILL Photonics). Recorded images were processed with Vimmaging developed under MATLAB™ (MathWorks, Natick, MA), and WCIFImageJ v1.35 (http://rsb.info.nih.gov/ij).
Cell fractionation
Confluent HeLa cells grown on Petri dishes were intoxicated with 10 nM EF or 10 nM LF and 20 nM PA. After 1 h of incubation, cells were treated with trypsin (0.05% w/v) (Sigma) at 37°C for 1 min to remove surface-bound toxins. Trypsin activity was blocked in PBS 10% FCS. Cells were washed with ice-cold PBS and scraped in PBS with the addition of protease inhibitors cocktail 1 × (Roche). Cells were lysed in 600 μl of homogenization buffer (HB; 8.5% sucrose, 3 mM imidazole, pH 7.4) (Kobayashi et al, 2002) with the addition of protease inhibitors cocktail 1 × (Roche). The lysed cell suspension was centrifuged, the pellet was discarded, and 50 μl of the supernatant were diluted with HB to 200 μl in a TLA-120.1 Beckman rotor and ultracentrifuged at 80 000 r.p.m. for 30 min at 4°C in a Beckman XL-100 ultracentrifuge to obtain a clear postnuclear supernatant (PNS) fraction. The remaining 550 μl of supernatant were adjusted to 40.6% sucrose, loaded at the bottom of an SW55 tube, and overlaid sequentially with 35 and 25% sucrose solutions in 3 mM imidazole, pH 7.4, and then HB as previously described (Gruenberg et al, 1989; Kobayashi et al, 2002). The gradient was centrifuged for 60 min at 35 000 r.p.m. with an SW55 rotor. Late endosomal fractions (LE) were collected at the 25%/HB interface, EE at the 35/25% and the HM at the 40.6/35% interface. PNS fractions were collected from the clear, ultracentrifuged PNS, obtained as described above. The proteins content in each fraction was quantified with Bradford assay (Biorad) as described elsewhere (Simpson and Sonne, 1982). Ten micrograms of proteins from each fraction were subjected to SDS–PAGE and analysed by Western blot. The same blotting membrane was probed sequentially with the different primary antibodies against the proteins of interest. None of the bands was saturated, according to the Quantity One® software (Biorad) that was used to analyse the results.
Western blotting
Ten microgram protein samples were boiled and subjected to SDS–PAGE on 4–12% polyacrylamide gradient gels (Invitrogen) and blotted onto nitrocellulose membranes, which were then incubated with the following antibodies. Anti-EF and anti LF monoclonal antibodies were a kind gift of P Goossens (Institute Pasteur, Paris). Anti-PA human monoclonal antibody was donated by A Lanzavecchia (IRB, Bellinzona). Anti-Rab7 polyclonal antibody was a kind gift of S Meresse (CIML, Marseille). The anti EEA1 polyclonal antibody was from Abcam (Cambridge, UK). Samples were developed with ECL plus detection system (Amersham Biosciences) and chemiluminescence emission was detected with ChemiDoc™ XRS (Biorad). Images were elaborated with WCIFImageJ v1.35 (http://rsb.info.nih.gov/ij) and band intensities were quantified with the Quantity One® software (Biorad).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary information
Acknowledgments
This work was supported by grants from the EC Anthrax Euronet Program, from the Istituto Superiore di Sanità Italy-USA protocol on ‘Programmi di Ricerca sulla Malattie di Grande Rilievo Sociale—Meccanismi tossinogenici e preparazione di antigeni vaccinali' and from the University of Padova to CM, by Telethon Italy (TCP00089, GGPO5113), the Italian Cystic Fibrosis Research Foundation, the Fondazione Compagnia di San Paolo and the HFSPO (RGP1/2005) to MZ, and grants form Institut Pasteur and from the EU 6th Framework Programme Contract LSHB-CT-2004-503582 (Theravac)' to DL.
References
- Abrami L, Liu S, Cosson P, Leppla SH, Van der Goot FG (2003) Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J Cell Biol 160: 321–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L, Lindsay M, Parton RG, Leppla SH, Van der Goot FG (2004) Membrane insertion of anthrax protective antigen and cytoplasmic delivery of lethal factor occur at different stages of the endocytic pathway. J Cell Biol 166: 645–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrami L, Reig N, Van der Goot FG (2005) Anthrax toxin: the long and winding road that leads to the kill. Trends Microbiol 13: 72–78 [DOI] [PubMed] [Google Scholar]
- Agrawal A, Lingappa J, Leppla SH, Agrawal S, Jabbar A, Quinn C, Pulendran B (2003) Impairment of dendritic cells and adaptive immunity by anthrax lethal toxin. Nature 424: 329–334 [DOI] [PubMed] [Google Scholar]
- Ascenzi P, Visca P, Ippolito G, Spallarossa A, Bolognesi M, Montecucco C (2002) Anthrax toxin: a tripartite lethal combination. FEBS Lett 531: 384–388 [DOI] [PubMed] [Google Scholar]
- Baldari T, Tonello F, Rossi-Paccani S, Montecucco C (2006) Anthrax toxins: a paradigm of bacterial immune suppression. Trends Immunol 27: 434–440 [DOI] [PubMed] [Google Scholar]
- Beauregard KE, Wimer-Mackin S, Collier RJ, Lencer WI (1999) Anthrax toxin entry into polarized epithelial cells. Infect Immun 67: 3026–3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA (2001) Identification of the cellular receptor for anthrax toxin. Nature 414: 225–229 [DOI] [PubMed] [Google Scholar]
- Collier RJ, Young JA (2003) Anthrax toxin. Annu Rev Cell Dev Biol 19: 45–70 [DOI] [PubMed] [Google Scholar]
- Dixon TC, Meselson M, Guillemin J, Hanna PC (1999) Anthrax. N Engl J Med 341: 815–826 [DOI] [PubMed] [Google Scholar]
- Dong C, Davis RJ, Flavell RA (2002) MAP kinases in the immune response. Annu Rev Immunol 20: 55–72 [DOI] [PubMed] [Google Scholar]
- Drum CL, Yan SZ, Sarac R, Mabuchi Y, Beckingham K, Bohm A, Grabarek Z, Tang WJ (2000) An extended conformation of calmodulin induces interactions between the structural domains of adenylyl cyclase from Bacillus anthracis to promote catalysis. J Biol Chem 275: 36334–36340 [DOI] [PubMed] [Google Scholar]
- Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, Vande Woude GF (1998) Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 280: 734–737 [DOI] [PubMed] [Google Scholar]
- Evellin S, Mongillo M, Terrin A, Lissandron V, Zaccolo M (2004) Measuring dynamic changes in cAMP using fluorescence resonance energy transfer. Methods Mol Biol 284: 259–270 [DOI] [PubMed] [Google Scholar]
- Firoved AM, Miller GF, Moayeri M, Kakkar R, Shen Y, Wiggins JF, McNally EM, Tang WJ, Leppla SH (2005) Bacillus anthracis edema toxin causes extensive tissue lesions and rapid lethality in mice. Am J Pathol 167: 1309–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander AM (1986) Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J Biol Chem 261: 7123–7126 [PubMed] [Google Scholar]
- Gaestel M (2006) MAPKAP kinases—MKs—two's company, three's a crowd. Nat Rev Mol Cell Biol 7: 120–130 [DOI] [PubMed] [Google Scholar]
- Gordon VM, Young WW Jr, Lechler SM, Gray MC, Leppla SH, Hewlett EL (1989) Adenylate cyclase toxins from Bacillus anthracis and Bordetella pertussis. Different processes for interaction with and entry into target cells. J Biol Chem 264: 14792–14796 [PubMed] [Google Scholar]
- Gruenberg J (2001) The endocytic pathway: a mosaic of domains. Nat Rev Mol Cell 2: 721–730 [DOI] [PubMed] [Google Scholar]
- Gruenberg J, Griffiths G, Howell KE (1989) Characterization of the early endosome and putative endocytic carrier vesicles in vivo and with an assay of vesicle fusion in vitro. J Cell Biol 108: 1301–1316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenberg J, van der Goot G (2006) Mechanism of pathogen entry thorugh the endosomal compartments. Nat Rev Mol Cell Biol 7: 495–504 [DOI] [PubMed] [Google Scholar]
- Guidi-Rontani C, Weber-Levy M, Mock M, Cabiaux V (2000) Translocation of Bacillus anthracis lethal and oedema factors across endosome membranes. Cell Microbiol 2: 259–264 [DOI] [PubMed] [Google Scholar]
- Hewlett EL, Kim KJ, Lee SJ, Gray MC (2000) Adenylate cyclase toxin from Bordetella pertussis: current concepts and problems in the study of toxin functions. Int J Med Microbiol 290: 333–335 [DOI] [PubMed] [Google Scholar]
- Karimova G, Fayolle C, Gmira S, Ullmann A, Leclerc C, Ladant D (1998) Charge-dependent translocation of Bordetella pertussis adenylate cyclase toxin into eukaryotic cells: implication for the in vivo delivery of CD8+ T-cell epitopes into antigen-presenting cells. Proc Natl Acad Sci USA 95: 12532–12537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Beuchat MH, Chevallier J, Makino A, Mayran N, Escola JM, Lebrand C, Cosson P, Kobayashi T, Gruenberg J (2002) Separation and characterization of late endosomal membrane domains. J Biol Chem 277: 32157–32164 [DOI] [PubMed] [Google Scholar]
- Kochi SK, Martin I, Schiavo G, Mock M, Cabiaux V (1994) The effects of pH on the interaction of anthrax toxin lethal and edema factors with phospholipid vesicles. Biochemistry 33: 2604–2609 [DOI] [PubMed] [Google Scholar]
- Krantz BA, Melnyk RA, Zhang S, Juris SJ, Lacy DB, Wu Z, Finkelstein A, Collier RJ (2005) A phenylalanine clamp catalyzes protein translocation through the anthrax toxin pore. Science 309: 777–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladant D, Ullmann A (1999) Boreatella pertussis adenylate cyclase: a toxin with multiple talents. Trends Microbiol 7: 172–176 [DOI] [PubMed] [Google Scholar]
- Lissandron V, Terrin A, Collini M, D'Alfonso L, Chirico G, Pantano S, Zaccolo M (2005) Improvement of a FRET-based indicator for cAMP by linker design and stabilization of donor–acceptor interaction. J Mol Biol 354: 546–555 [DOI] [PubMed] [Google Scholar]
- Menard A, Altendorf K, Breves D, Mock M, Montecucco C (1996) The vacuolar ATPase proton pump is required for the cytotoxicity of Bacillus anthracis lethal toxin. FEBS Lett 386: 161–164 [DOI] [PubMed] [Google Scholar]
- Moayeri M, Leppla SH (2004) The roles of anthrax toxin in pathogenesis. Curr Opin Microbiol 7: 19–24 [DOI] [PubMed] [Google Scholar]
- Mock M, Mignot T (2003) Anthrax toxins and the host: a story of intimacy. Cell Microbiol 5: 15–23 [DOI] [PubMed] [Google Scholar]
- Mongillo M, McSorley T, Evellin S, Sood A, Lissandron V, Terrin A, Huston E, Hannawacker A, Lohse MJ, Pozzan T, Houslay MD, Zaccolo M (2004) Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res 95: 67–75 [DOI] [PubMed] [Google Scholar]
- Mongillo M, Terrin A, Evellin S, Lissandron V, Zaccolo M (2005) Study of cyclic adenosine monophosphate microdomains in cells. Methods Mol Biol 307: 1–13 [DOI] [PubMed] [Google Scholar]
- Paccani SR, Tonello F, Ghittoni R, Natale M, Muraro L, D'Elios MM, Tang WJ, Montecucco C, Baldari CT (2005) Anthrax toxins suppress T lymphocyte activation by disrupting antigen receptor signaling. J Exp Med 201: 325–331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papini E, Rappuoli R, Murgia M, Montecucco C (1993) Cell penetration of diphtheria toxin. Reduction of the interchain disulfide bridge is the rate-limiting step of translocation in the cytosol. J Biol Chem 268: 1567–1574 [PubMed] [Google Scholar]
- Park JM, Greten FR, Li ZW, Karin M (2002) Macrophage apoptosis by anthrax lethal factor through p38 MAP kinase inhibition. Science 297: 2048–2051 [DOI] [PubMed] [Google Scholar]
- Pellizzari R, Guidi-Rontani C, Vitale G, Mock M, Montecucco C (1999) Anthrax lethal factor cleaves MKK3 in macrophages and inhibits the LPS/IFNgamma-induced release of NO and TNFalpha. FEBS Lett 462: 199–204 [DOI] [PubMed] [Google Scholar]
- Rainey GJ, Wigelsworth DJ, Ryan PL, Scobie HM, Collier RJ, Young JA (2005) Receptor-specific requirements for anthrax toxin delivery into cells. Proc Natl Acad Sci USA 102: 13278–13283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scobie HM, Rainey GJ, Bradley KA, Young JA (2003) Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci USA 100: 5170–5174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Zhukovskaya NL, Guo Q, Florian J, Tang WJ (2005) Calcium-independent calmodulin binding and two-metal-ion catalytic mechanism of anthrax edema factor. EMBO J 24: 929–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson IA, Sonne O (1982) A simple, rapid and sensitive method for measuring protein concentration in subcelluler membrane fractions prepared by sucrose density ultracentrifugation. Anal Biochem 119: 424–427 [DOI] [PubMed] [Google Scholar]
- Tournier JN, Quesnel-Hellmann A, Mathieu J, Montecucco C, Tang WJ, Mock M, Vidal DR, Goossens PL (2005) Anthrax edema toxin cooperates with lethal toxin to impair cytokine secretion during infection of dendritic cells. J Immunol 174: 4934–4941 [DOI] [PubMed] [Google Scholar]
- Turnbull PC (2002) Introduction: anthrax history, disease and ecology. Curr Top Microbiol Immunol 271: 1–19 [DOI] [PubMed] [Google Scholar]
- Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C (2000) Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem J 352 (Part 3): 739–745 [PMC free article] [PubMed] [Google Scholar]
- Vitale G, Pellizzari R, Recchi C, Napolitani G, Mock M, Montecucco C (1998) Anthrax lethal factor cleaves the N-terminus of MAPKKs and induces tyrosine/threonine phosphorylation of MAPKs in cultured macrophages. Biochem Biophys Res Commun 248: 706–711 [DOI] [PubMed] [Google Scholar]
- Wei W, Lu Q, Chaudry GJ, Leppla SH, Cohen SN (2006) The LDL receptor-related protein LRP6 mediates internalization and lethality of anthrax toxin. Cell 124: 1119–1121 [DOI] [PubMed] [Google Scholar]
- Wigelsworth DJ, Krantz BA, Christensen KA, Lacy DB, Juris SJ, Collier RJ (2004) Binding stoichiometry and kinetics of the interaction of a human anthrax toxin receptor, CMG2, with protective antigen. J Biol Chem 279: 23349–23356 [DOI] [PubMed] [Google Scholar]
- Zaccolo M, De Giorgi F, Cho CY, Feng L, Knapp T, Negulescu PA, Taylor SS, Tsien RY, Pozzan T (2000) A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat Cell Biol 2: 25–29 [DOI] [PubMed] [Google Scholar]
- Zacharias DA, Violin JD, Newton AC, Tsien RY (2002) Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296: 913–916 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary information