

Abstract
Angiotensin (Ang) II participates in the pathogenesis of heart failure through induction of cardiac hypertrophy. Ang II-induced hypertrophic growth of cardiomyocytes is mediated by nuclear factor of activated T cells (NFAT), a Ca2+-responsive transcriptional factor. It is believed that phospholipase C (PLC)-mediated production of inositol-1,4,5-trisphosphate (IP3) is responsible for Ca2+ increase that is necessary for NFAT activation. However, we demonstrate that PLC-mediated production of diacylglycerol (DAG) but not IP3 is essential for Ang II-induced NFAT activation in rat cardiac myocytes. NFAT activation and hypertrophic responses by Ang II stimulation required the enhanced frequency of Ca2+ oscillation triggered by membrane depolarization through activation of DAG-sensitive TRPC channels, which leads to activation of L-type Ca2+ channel. Patch clamp recordings from single myocytes revealed that Ang II activated DAG-sensitive TRPC-like currents. Among DAG-activating TRPC channels (TRPC3, TRPC6, and TRPC7), the activities of TRPC3 and TRPC6 channels correlated with Ang II-induced NFAT activation and hypertrophic responses. These data suggest that DAG-induced Ca2+ signaling pathway through TRPC3 and TRPC6 is essential for Ang II-induced NFAT activation and cardiac hypertrophy.
Keywords: angiotensin, cardiac hypertrophy, diacylglycerol, L-type Ca2+ channel, TRPC
Introduction
Regulators of cardiac function such as vasoactive neurotransmitters and hormones activate phospholipase C (PLC) and thereby generate inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG). These agonists elevate the concentration of cytoplasmic free Ca2+ ([Ca2+]i) in cardiomyocytes, which induces positive inotropic effects on the heart and activates several transcriptional pathways that lead to cardiac hypertrophy (Wilkins and Molkentin, 2004; Woodcock and Matkovich, 2005). NFAT is one of the transcriptional factors regulated by [Ca2+]i (Crabtree and Olson, 2002). The relevance of the NFAT signaling pathway to cardiac hypertrophy is underscored by the observation that cardiac-targeted transgenic animals expressing constitutively activated forms of either calcineurin or NFAT produced ventricular hypertrophy (Molkentin et al, 1998; Taigen et al, 2000). The Ca2+-sensitive serine/threonine phosphatase (calcineurin) primarily regulates NFAT activity by rapid dephosphorylation of NFAT proteins and their translocation to the nucleus. A drop in nuclear Ca2+ deactivates calcineurin and allows one of several NFAT kinases to rephosphorylate NFAT, causing it to leave the nucleus and thereby inactivating transcription (Timmerman et al, 1996; Dolmetsch et al, 1997). Therefore, a sustained elevation of [Ca2+]i is required for NFAT-dependent transcription.
The importance of agonists that activate PLC for cardiac hypertrophy is well established (Molkentin and Dorn, 2001). Many lines of evidence have shown that stimulation of PLC-linked G protein-coupled receptors, such as α1-adrenergic receptor (Maruyama et al, 2002), Ang II receptor (Nishida et al, 2005) and endothelin receptor (Arai et al, 2003), induce hypertrophic growth of rat cardiac myocytes. More clinically relevant, hypertrophied hearts induced by volume overload are commonly characterized by high levels of IP3-generating agonists such as Ang II (Dostal et al, 1992; Sadoshima et al, 1993). Numerous studies have demonstrated the need for sustained or periodic increases in [Ca2+]i to cause the nuclear localization of NFAT (Dolmetsch et al, 1997; Tomida et al, 2003). In nonexcitable cells, IP3 is generally accepted to function as a mediator of sustained Ca2+ responses (Timmerman et al, 1996; Dolmetsch et al, 1997). The sustained Ca2+ signaling requires the store-operated Ca2+ channel (SOC), which opens in response to depletion of intracellular stores through IP3 receptor (IP3R). Therefore, it is currently believed that Ca2+ entry through SOC regulates NFAT translocation. In the heart, however, the expression level of IP3R is much lower than that of ryanodine receptor (Moschella and Marks, 1993). Voltage-dependent L-type Ca2+ channel and ryanodine receptor function as the major source of Ca2+ for normal Ca2+-induced Ca2+ release of excitation–contraction (E–C) coupling, but many reports do not support the idea that the increase in [Ca2+]i through E–C coupling between L-type Ca2+ channel and ryanodine receptor is coupled to NFAT activation (Wilkins and Molkentin, 2004).
A possible source of Ca2+ for activation of calcineurin is Ca2+ influx through transient receptor potential (TRP) proteins that are involved in store-operated Ca2+ entry (Clapham, 2003). Upregulation of canonical transient receptor potential (TRPC) proteins is recently reported to contribute to the development of cardiac hypertrophy (Seth et al, 2004). Other groups reported that TRPM7 regulates Mg2+ homeostasis, and TRPM6 and TRPM7 are differentially regulated by Ang II in vascular smooth muscle cells (He et al, 2005; Touyz et al, 2006). However, it is still unknown whether TRP channels contribute to receptor-stimulated activation of calcineurin-NFAT pathway in the heart. In this study, we investigated the mechanism of how Ang II stimulation induces the sustained Ca2+ signaling leading to NFAT activation and hypertrophic growth of rat neonatal cardiomyocytes.
Results
Essential role of DAG in Ang II-induced NFAT activation and cardiac hypertrophy
We first examined whether IP3 or DAG is involved in Ang II-induced NFAT activation in rat neonatal cardiomyocytes. As it has been reported that pressure overload- and Ang II-induced cardiac hypertrophy are attenuated in NFAT4 (NFATc3)-null mice (Wilkins et al, 2002), the translocation of NFAT4 was determined in this study. Stimulation of cardiac myocytes with Ang II for 30 min increased the maximal nuclear predominant fluorescence of GFP-fused amino-terminal region of NFAT4 protein (GFP-NFAT4) (Figure 1A–C). The Ang II-induced NFAT translocation was completely suppressed by the expression of DAG kinase β (DGKβ), an enzyme that decreases the cellular DAG level by converting DAG to phosphatidic acid. Treatment with RHC80267, a DAG lipase inhibitor, significantly increased the Ang II-induced nuclear translocation of GFP-NFAT4. However, treatment with xestospongin C, an IP3R blocker, did not affect the Ang II-induced translocation of GFP-NFAT4 to the nucleus. To directly inhibit IP3-mediated signaling, we expressed the ligand-binding region of type 1 IP3R (IP3-sponge) (Uchiyama et al, 2002). The Ang II-induced transient increase in [Ca2+]i (or Ca2+ release) was completely suppressed by the treatment with xestospongin C and by the expression of IP3-sponge but not DGKβ (Supplementary Figure S1), suggesting the efficient inhibition of IP3-mediated Ca2+ signaling. The Ang II-induced increase in NFAT-dependent luciferase reporter activity was suppressed by DGKβ, but not by xestospongin C and IP3-sponge (Figure 1D and E). Treatment with RHC80267 promoted the Ang II-induced NFAT activation (Figure 1E). These results suggest the involvement of DAG in Ang II-induced NFAT activation. We also examined the involvement of DAG in Ang II-induced hypertrophic responses. Expression of DGKβ, but not IP3-sponge, completely suppressed Ang II-induced hypertrophic responses, such as actin reorganization (Figure 1F), protein synthesis (Figure 1G), and expression of brain natriuretic peptide (BNP) (Figure 1H). These results suggest that DAG, but not IP3, is essential for Ang II-induced NFAT activation and hypertrophic responses of neonatal cardiomyocytes.
Figure 1.
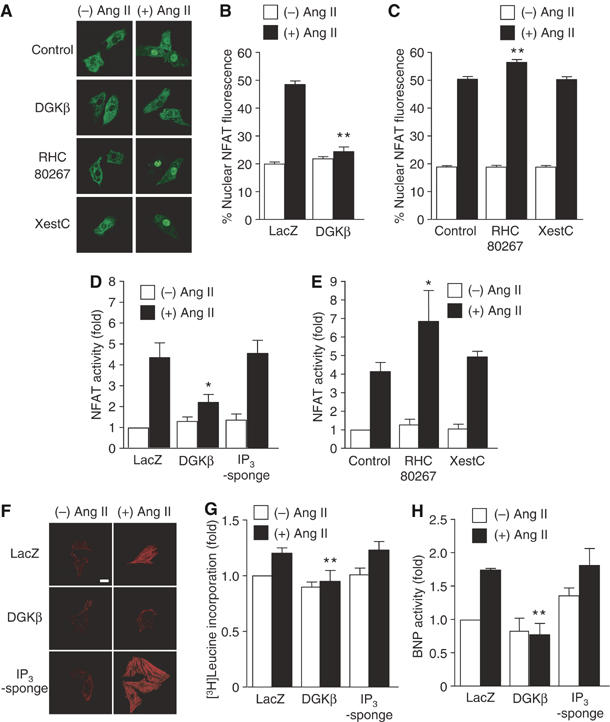
Essential role of DAG in Ang II-induced cardiomyocyte hypertrophy. (A) Nuclear translocation of GFP-NFAT4 by Ang II stimulation. A portion of cells was treated with RHC80267 (30 μM) or xestospongin C (XestC, 20 μM) for 30 min before the addition of Ang II (100 nM), and a portion of cells was infected with DGKβ for 48 h before Ang II stimulation. (B, C) Quantification of nuclear predominant fluorescence of GFP-NFAT4 after Ang II stimulation. (D, E) Effects of DGKβ, RHC80267, and XestC on the increase in NFAT-dependent luciferase activity by Ang II stimulation for 6 h. The fold activation was calculated by the values of untreated cells set as 1. (F–H) Effects of DGKβ and GFP-IP3-sponge on Ang II-induced actin reorganization (F), protein synthesis (G), and BNP expression (H). Scale bar=20 μm. *P<0.05, **P<0.01 versus control or LacZ-expressing cells.
Involvement of Ang II type 1 receptor, Gαq, and PLC in Ang II-induced NFAT activation
In contrast to the absence of extracellular Ca2+ (Supplementary Figure S1A), myocytes showed spontaneous increases in [Ca2+]i in the presence of extracellular Ca2+. Treatment with Ang II induced the transient increase in [Ca2+]i followed by sustained oscillatory increase in [Ca2+]i (Figure 2A; the former can more clearly be seen in Supplementary Figure S1A). The Ca2+ oscillation represents a spontaneous activity of myocytes, and Ang II stimulation increased its frequency (Supplementary Figure S1C). The Ang II-induced Ca2+ response and NFAT activation were greatly suppressed by U73122, a PLC inhibitor, but not by U73343, an inactive analog of U73122 (Figure 2A–C). Thus, PLC primarily regulates Ang II-induced Ca2+ signal generation. The Ang II-induced translocation of GFP-NFAT4 was suppressed by CV11974, an Ang II type 1 receptor (AT1R) blocker, but not by PD123319, an AT2R blocker (Figure 2D). These results indicate that AT1R-mediated PLC activation is involved in Ang II-induced NFAT4 activation. We next examined which G proteins are involved in Ang II-induced NFAT activation. It has been generally believed that Gαq plays an important role in agonist-induced cardiac hypertrophy (Molkentin and Dorn, 2001). To examine the involvement of Gαq, we expressed regulator of G protein signaling (RGS) domain that is ∼200 amino acids, specifically binds GTP-bound form of Gα and accelerates GTPase activity. When RGS domain is expressed in cells, it competes with activated form of Gα for endogenous effectors and accelerates turn-off reaction of Gα. Therefore, RGS domain can work as a specific inhibitor of Gα. As expected, the expression of a Gαq-specific RGS domain of G protein-coupled receptor kinase 2 (GRK2-RGS) completely suppressed the Ang II-induced translocation of GFP-NFAT4 (Figure 2E). However, the expression of a Gα12/13-specific RGS domain of p115RhoGEF (p115-RGS) did not affect the Ang II-induced translocation of GFP-NFAT4. Pertussis toxin (PTX) and carboxyl terminal region of GRK2 (GRK2-ct), a βγ subunit of G protein (Gβγ)-sequestering polypeptide, did not inhibit the Ang II-induced translocation of GFP-NFAT4 (Figure 2E). Thus, these results support the evidence that agonist-induced Ca2+-dependent NFAT activation is predominantly regulated by Gαq, but not by Gα12/13, Gi or Gβγ in cardiomyocytes.
Figure 2.
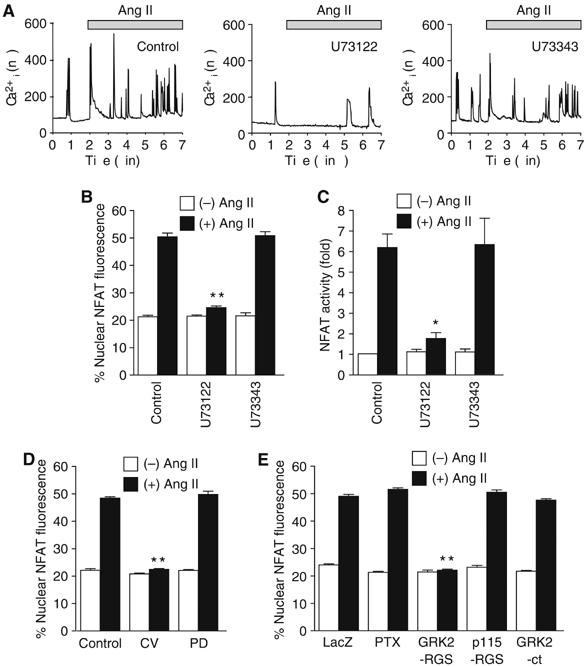
Involvement of AT1R, Gαq, and PLC in Ang II-induced NFAT activation. (A–C) Effects of U73122 and U73343 on Ang II-induced Ca2+ responses (A), translocation of GFP-NFAT4 (B), and NFAT activation (C). (A) Effects of U73122 and U73343 on the increases in the frequency of Ca2+ oscillation during 5 min Ang II stimulation. The digital images were obtained every 1 s. (D) Effects of CV11974 and PD123319 on Ang II-induced NFAT translocation. Cells were treated with U73122 (5 μM), U73343 (5 μM), CV11974 (CV, 5 μM), or PD123319 (PD, 5 μM) for 30 min before the addition of Ang II (100 nM). (E) Effects of PTX, GRK2-RGS, p115-RGS, and GRK2-ct on Ang II-induced NFAT translocation. Cells were infected with adenovirus encoding LacZ (100 MOI), p115-RGS or GRK2-ct (100 MOI), or GRK2-RGS (300 MOI) for 48 h. A portion of cells was treated with PTX (100 ng/ml) for 24 h before Ang II stimulation. *P<0.05, **P<0.01 versus Ang II stimulation of control or LacZ-expressing cells.
Requirement of Ca2+ influx through L-type Ca2+ channels and nonselective cation channels in Ang II-induced NFAT activation
It has been reported that DAG induces Ca2+ influx through activation of cation channels (Hofmann et al, 1999; Clapham, 2003). As the Ang II-induced periodic increase in [Ca2+]i likely results from enhanced spontaneous activity of myocytes (which are dependent on extracellular Ca2+; see above), and these were suppressed by DGKβ (Supplementary Figure S1), we next examined whether Ca2+ influx is involved in DAG-mediated responses. Treatment of cardiac myocytes with Ang II or with a DAG derivative, 1-oleoyl-2-acyl-sn-glycerol (OAG), increased the nuclear translocation of GFP-NFAT4 and NFAT activity, both of which were almost completely suppressed by the voltage-dependent Ca2+ channel blocker nitrendipine and a receptor-activated cation channel (RACC) inhibitor SK&F96365 (Figure 3A–C). As OAG-induced NFAT activation was also completely suppressed by cyclosporine A, a calcineurin inhibitor (Figure 3C), DAG increases NFAT activity through calcineurin activation. These results suggest that RACC and Ca2+ influx through L-type Ca2+ channel mediate Ang II- or DAG-induced NFAT activation.
Figure 3.
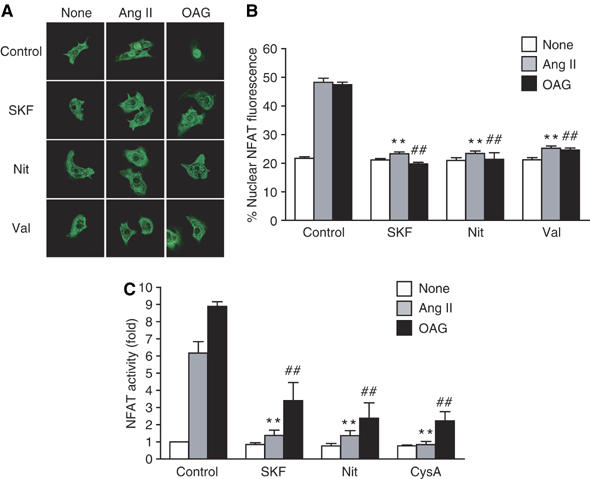
Requirement of RACC and L-type Ca2+ channel in DAG-mediated NFAT translocation. (A) Effects of SK&F96365 (SKF), nitrendipine (Nit) and valinomycin (Val) on Ang II- or OAG-induced NFAT translocation. (B) Quantification of the nuclear predominant fluorescence of GFP-NFAT4 without (None) or with Ang II or OAG stimulation. (C) Effects of SKF, Nit, and cyclosporine A (CysA) on Ang II- or OAG-induced increase in NFAT-luciferase activity. Cells were treated with SKF (10 μM), Nit (1 μM), Val (1 μM), or CysA (1 μM) for 30 min before the addition of Ang II (100 nM) or OAG (25 μM). **P<0.01 versus Ang II stimulation of control cells. ##; P<0.01 versus OAG stimulation of control cells.
Ang II activates DAG-sensitive cation channels in cardiac myocytes
To directly demonstrate that Ang II activates DAG-sensitive RACC, whole-cell patch-clamp experiments were performed. In quasi-physiological ionic conditions, administration of Ang II into the bath activated inward currents at −80 mV, which were further enhanced by RHC80267 (Figure 4A and B). These currents were completely abolished by N-methyl-D-glucamine substitution for all external cations (data not shown), and showed an outward-rectifying property with the reversal potential of ca. 0 mV (1.0±1.0 mV, n=6), when Cs+ was intracellularly dialyzed via patch pipette and TTX (3 μM) and nitrendipine (1 μM) were added into K+-free external solution to block voltage-dependent K+, Na+, and L-type Ca2+ channels, respectively (see inset in Figure 4C). Administration of OAG (25 μM) also activated inward currents showing indistinguishable properties from those activated by Ang II, whereas application of myo-IP3 (10 μM) in the internal solution was unable to activate any discernible currents by itself (data not shown). These results collectively suggest that Ang II activates DAG-sensitive nonselective cation currents in cardiomyocytes via an IP3-independent pathway, which bears considerable resemblance to heterologously expressed TRPC channels.
Figure 4.
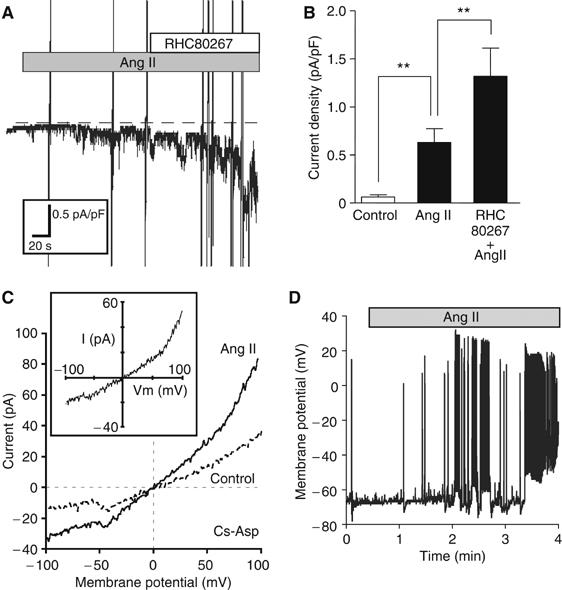
Activation of DAG-sensitive currents from single cardiomyocytes by Ang II stimulation. (A) Representative traces of ionic currents recorded from Ang II-treated cardiomyocytes at a holding potential of −80 mV under conventional whole-cell patch-clamp with K+-internal solution. The nonselective cation currents are activated by Ang II (1 μM), and potentiated by RHC80267 (30 μM). The dotted line represents the zero current level. (B) Current density of inward current (at −80 mV) averaged for the period of 60–65 s after Ang II stimulation or RHC80267 treatment (n>8). **P<0.01. (C) I–V relationship of ionic currents from unstimulated (Control) and Ang II-stimulated myocytes with Cs+-internal solution containing myo-IP3 (10 μM). TTX (3 μM) and nitrendipine (1 μM) are included in the K+-free external solution. (Inset) I–V relationship of TRPC-like currents induced by Ang II (differences between Ang II and Control). (D) Representative traces of time-dependent changes in the membrane potential and the frequency of action potential by Ang II stimulation in the current-clamp mode.
In the next step, we examined Ang II-induced changes in membrane potential by using the current-clamp technique, since the treatment with valinomycin, a K+ ionophore, which causes inactivation of voltage-dependent channels via stabilization of membrane potential (Linares-Hernandez et al, 1998), completely suppressed the Ang II-induced translocation of GFP-NFAT4 (Figure 3A and B), and in general, the activation of RACC causes membrane depolarization (Large, 2002). As expected, membrane potential recording from single myocytes with current-clamp mode clearly demonstrated that Ang II increased the frequency of action potentials, which eventually led to continuous burstic firing superimposed on concomitant sustained depolarization (22.2±5.6 mV, n=5) (Figure 4D). It is noteworthy that the time course of these effects is very similar to that observed for the enhanced frequency of Ca2+ oscillations induced by Ang II (see above).
Properties of DAG induced membrane depolarization in rat cardiac myocytes
Current-clamp recordings were technically little feasible to monitor the membrane potential for a long period of time, because of rhythmical contractions of myocytes evoked by Ang II. To circumvent this problem, we adopted a voltage-sensitive fluorescent probe DiBAC4(3). After DiBAC4(3) enters the cells, it binds to cellular proteins and membrane lipids. Then, DiBAC4(3) enhances fluorescence. Because of its slow dissociating nature, DiBAC4(3) can only detect slow cumulative changes in resting potential rather than rapid changes in membrane potential generated by action potential. Ang II stimulation gradually increased the fluorescence intensity of DiBAC4(3) (Figure 5A and B), indicating the shift of membrane potential to positive (BACzkó et al, 2004). The averaged changes in membrane potential induced by Ang II were estimated to be ∼15 mV. Treatment with RHC80267 enhanced the Ang II-induced increases in the fluorescence intensity of DiBAC4(3) (Figure 5C). These results indicate that DAG generated by Ang II stimulation shifts the membrane potential of cardiac myocytes more positively. DAG also activates other signaling molecules including protein kinase C (PKC). PKC is known to potentiate the extent of L-type Ca2+ channel activation, and both OAG and phorbor 12-myristrate 13-acetate (PMA) have been reported to increase the channel open probability in rat cardiomyocytes (Guinamard et al, 2004). However, treatment with PMA did not increase the fluorescence intensity of DiBAC4(3) (Figure 5A and B) and OAG-induced translocation and activation of NFAT were not affected by bisindolylmaleimide, a selective PKC inhibitor (Supplementary Figure S2). It is possible that the metabolites of DAG work as mediators for NFAT translocation. However, treatment with arachidonic acid (AA) or phospholipase A2 (PLA2) inhibitors did not affect Ang II-induced NFAT translocation (Supplementary Figure S2). These results suggest that PKCs and DAG metabolites do not participate in Ang II-induced depolarization and NFAT translocation. The Ang II-induced increases in the fluorescence intensity of DiBAC4(3) were completely suppressed by SK&F96365, but not by nitrendipine and xestospongin C (Figure 5D).
Figure 5.
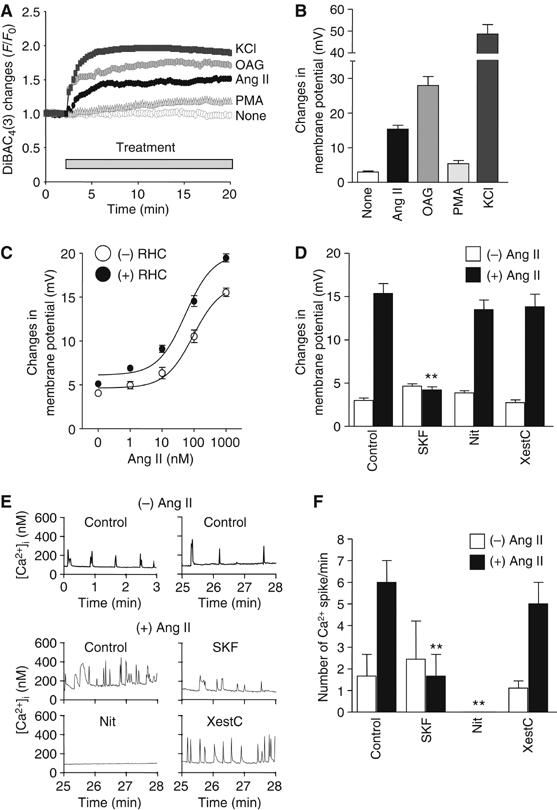
Changes in membrane potential through RACC activation by DAG. (A) Representative time courses of changes in Ang II-, OAG-, or PMA-induced F/F0 of DiBAC4(3) fluorescence from time course experiments. Cells were stimulated with Ang II (1 μM), OAG (25 μM), PMA (1 μM), or KCl (10 mM). F0 means the initial value of fluorescence. (B) Maximal changes in resting membrane potential calculated from the changes in DiBAC4(3) fluorescence intensity during 15 min drug treatment. For the in vivo calibration of the membrane potentials, the KCl-induced maximal changes in fluorescence were fitted to the theoretical potentials obtained from Nernst equation, and then the changes in membrane potential by Ang II stimulation was calculated based on the fitting fomula. (C) Effects of RHC80267 on the concentration-dependent changes in resting membrane potentials induced by Ang II stimulation. (D) Involvement of RACC in Ang II-induced increases in the resting membrane potential. Cells were treated with SK&F96365 (SKF, 10 μM), nitredipine (Nit, 1 μM), or xestospongin C (XestC, 20 μM) for 30 min before the addition of Ang II. **P<0.01 versus Ang II stimulation of control cells. (E) Effects of SK&F96365 (SKF), nitrendipine (Nit), and xestospongin C (XestC) on Ang II-induced Ca2+ responses. The digital images were obtained every 1 s during 0–3 min under basal conditions and during 25–28 min after Ang II stimulation. (F) Number of Ca2+ spikes was normalized to per minute. **P<0.01 versus Ang II stimulation of control cells.
We next examined whether periodic increase in [Ca2+]i is regulated by RACC. The myocytes showed spontaneous Ca2+ oscillations in the presence of extracellular Ca2+ (top panel in Figure 5E). The frequency of Ca2+ oscillations was increased by Ang II stimulation and this was suppressed by SK&F96365 and nitrendipine, but not by xestspongin C (middle and bottom panels in Figure 5E and F). These results support the idea that DAG generated by Ang II-induced PLC activation causes membrane depolarization through RACC activation and thereby secondarily activates L-type Ca2+ channel, leading to increased frequency of Ca2+ oscillations.
Requirement of TRPC3 and TRPC6 in Ang II-induced membrane depolarization
TRPC proteins are thought to be molecular candidates for RACC (Clapham, 2003). We found the expression of at least five TRP canonical (TRPC) mRNAs (TRPC1, TRPC3, TRPC4, TRPC5, TRPC6, and TRPC7) in rat neonatal cardiomyocytes by RT–PCR analysis (data not shown). Recent reports have demonstrated that three TRPC channels (TRPC3, TRPC6, and TRPC7) are activated directly by DAG (Hofmann et al, 1999; Clapham, 2003). Thus, we next examined which DAG-sensitive TRPC protein is involved in Ang II-induced NFAT activation. We overexpressed TRPC3, TRPC6, or TRPC7, and examined the Ang II-induced changes in membrane potential with DiBAC4(3) (Figure 6A and B). Among three TRPC proteins, Ang II-induced increases in the fluorescence intensity of DiBAC4(3) were significantly enhanced by the expression of TRPC3 and TRPC6 but not by TRPC7 (Figure 6B), although the latter enhanced OAG-induced [Ca2+]i increases to the same extent as the former two did (Supplementary Figure S3). These results indicate that TRPC3 and TRPC6, but not TRPC7, likely regulate the Ang II-induced membrane depolarization. This conclusion was further corroborated by siRNA-mediated knockdown of TRPC3 (siRNA 1397, 1992, and 2043) and TRPC6 (siRNA 1609 and 1786) in the cardiomyocytes; this procedure decreased the expression level of endogenous TRPC3 and TRPC6 proteins without affecting other TRPC proteins (Figure 6C–F), and simultaneously caused significant suppression of Ang II-induced increases in the fluorescence intensity of DiBAC4(3) (Figure 6G). Taken together, the above results strongly suggest that DAG-mediated activation of TRPC3 and TRPC6 channels contributes to the enhanced Ca2+ oscillation by Ang II via their membrane depolarizing actions.
Figure 6.
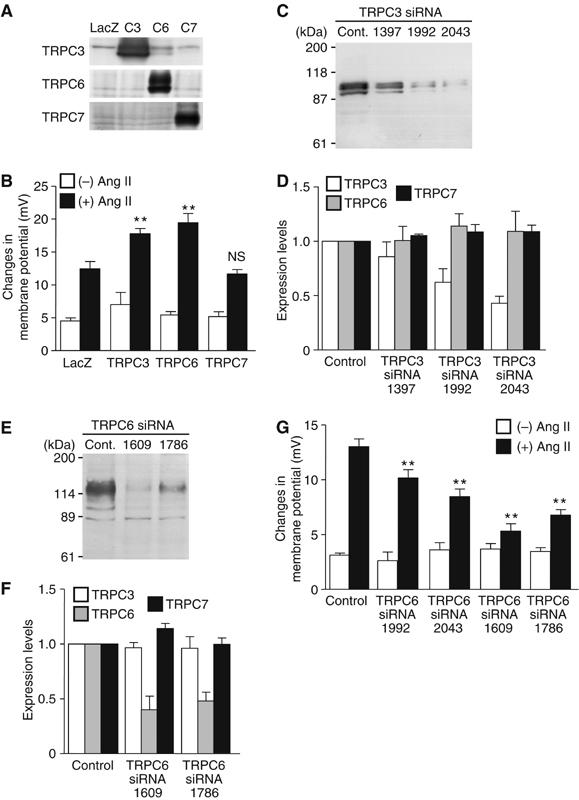
Requirement of TRPC3 and TRPC6 in Ang II-induced increases in membrane potential. (A) Western blots of the respective TRPC proteins. To identify the sizes of TRPC3 (C3), TRPC6 (C6), and TRPC7 (C7), each TRPC was overexpressed with recombinant adenoviruses. (B) Potentiating effects of TRPC3 and TRPC6 on changes in membrane potential by Ang II stimulation in LacZ-, TRPC3-, TRPC6-, and TRPC7-expressing cells. **P<0.01 versus Ang II stimulation of LacZ-expressing cells. NS means no significance from LacZ-expressing cells. (C–F) Effects of TRPC3 siRNAs (C, D) and TRPC6 siRNAs (E, F) on the expression of the respective TRPC proteins. (C, E) Representative Western blots with anti-TRPC3 (C) and anti-TRPC6 (E). (D, F) Effects of siRNAs of TRPC3 and TRPC6 on the average expression of native TRPC3, TRPC6, and TRPC7 proteins. (G) Effects of siRNAs of TRPC3 and TRPC6 on the maximal changes in DiBAC4(3) fluorescence intensity by Ang II (100 nM). Data are shown as the changes in membrane potentials (mV) calculated by in vivo calibration. **P<0.01 versus Ang II stimulation of control siRNA-treated cells (Control).
In addition, siRNA silencing of TRPC3 and TRPC6 also significantly suppressed Ca2+ entry-mediated [Ca2+]i elevation induced by the addition of Ca2+ into the bath after Ang II stimulation (Supplementary Figure S3). Thus, some role of direct Ca2+ entry via TRPC3/TRPC6-associated pathway cannot completely be excluded in the Ang II-enhanced Ca2+ oscillation.
Requirement of TRPC3 and TRPC6 in Ang II-induced NFAT translocation and hypertrophic responses
We next examined whether TRPC3 and TRPC6 are involved in Ang II-induced hypertrophic responses. Treatment with siRNAs of TRPC3 and TRPC6 significantly suppressed Ang II-induced NFAT translocation (Figure 7A and B). Furthermore, both TRPC3 and TRPC6 siRNAs suppressed Ang II-induced actin reorganization and protein synthesis (Figure 7C and D). We further examined the involvement of TRPC6 in Ang II-induced cardiomyocyte hypertrophy by using two dominant negative TRPC6 mutants (Hofmann et al, 2002; Hisatsune et al, 2004). Expression of TRPC6-Δ(N) and TRPC6-3A significantly suppressed Ang II-induced NFAT activation, actin reorganization, and protein synthesis (Supplementary Figure S4). These results suggest that TRPC3 and TRPC6 play a critical role in Ang II-induced hypertrophic responses in rat neonatal cardiomyocytes.
Figure 7.
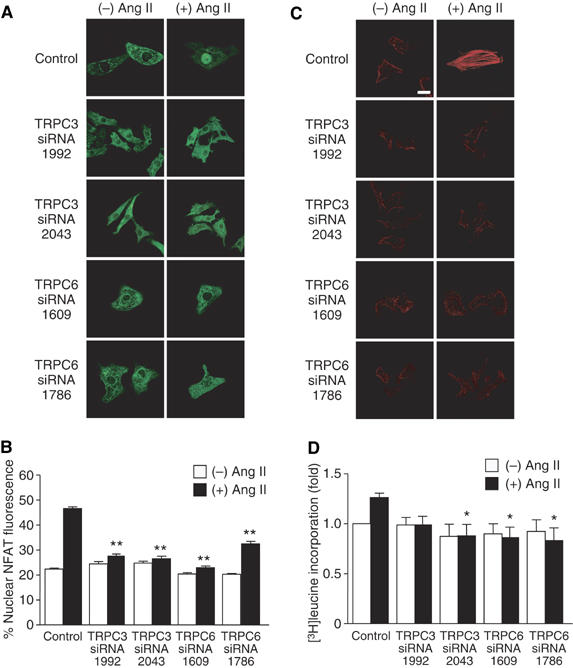
Requirement of TRPC3 and TRPC6 in Ang II-induced hypertrophic responses. Effects of siRNAs of TRPC3 and TRPC6 on Ang II-induced NFAT translocation (A, B), actin reorganization (C), and protein synthesis (D). Scale bar=20 μm. *P<0.05, **P<0.01 versus Ang II stimulation of control siRNA-treated cells (Control).
Discussion
This study reveals the role of DAG in Ang II-induced NFAT activation and hypertrophic responses. DAG produced by Ang II-induced PLC activation directly activates TRPC3 and TRPC6, and the resulting cation (Na+, Ca2+) influx changes membrane potential to positive, leading to activation of voltage-dependent L-type Ca2+ channel possibly through the generation of action potential. The increase in Ca2+ influx through L-type Ca2+ channel can activate calcineurin/NFAT pathway and hypertrophic responses in rat neonatal cardiomyocytes (Figure 8).
Figure 8.
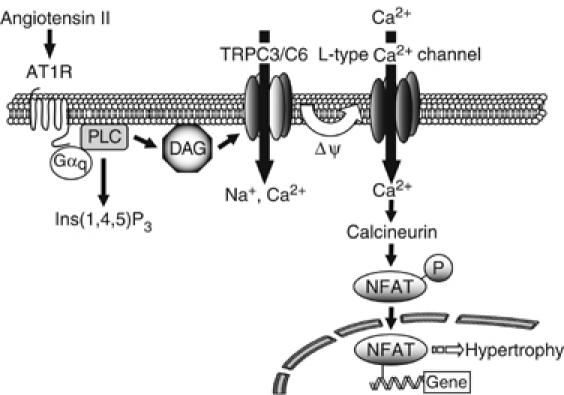
Schema of Ang II-induced NFAT activation in cardiac myocytes. In cardiac myocytes, stimulation of AT1R induces NFAT activation through Gαq-PLC signaling pathway. DAG, generated by PLC activation, directly activates TRPC3 and TRPC6 (TRPC3/C6). Activation of TRPC3/C6 causes slow increases in the membrane potential to a positive direction (ΔΨ) and concomitantly increases the frequency of spontaneous firing due to activation of L-type Ca2+ channel. The Ca2+ influx though L-type Ca2+ channel activates calcineurin-NFAT pathway, which leads to hypertrophic responses in cardiomyocytes.
The physiological role of TRPC was first identified in the vascular smooth muscle cells (Inoue et al, 2001). In the vascular system, activation of TRPC6 contributes to membrane depolarization and regulates myogenic tone of resistance arteries (Large, 2002; Welsh et al, 2002). In the present study, we demonstrated that TRPC3 and TRPC6 activated by DAG contributes to the shift of membrane potential and subsequent Ca2+ signal generation through voltage-dependent Ca2+ channel in cardiac myocytes. The role of DAG-induced TRPC3 and TRPC6 activation in membrane depolarization has been reported in vascular smooth muscle cells (Reading et al, 2005; Soboloff et al, 2005). The novel finding of the present study is to characterize the pathophysiological significance of TRPC3 and TRPC6 in Ang II-induced hypertrophic responses of the heart.
We cannot determine the subtype(s) of TRPC proteins activated by Ang II from the I–V relationship of native RACC, as inward current activated by Ang II was too small (Figure 4). Previous report has shown that fulfenamate inhibits TRPC3 but enhances TRPC6 channel activity (Inoue et al, 2001). The Ang II-induced inward current was slightly inhibited by flufenamate (data not shown). However, the similar behavior of currents to the present study was reported in TRPC3/C6-co-expressing HEK293 cells (Maruyama et al, 2006). TRPC3-like currents were observed by coexpression of TRPC3 and TRPC6. As Ang II-induced responses were inhibited both by siRNAs of TRPC3 and TRPC6 (Figures 6 and 7), we speculate that TRPC3 and TRPC6 form heterotetramers to regulate DAG-sensitive native cationic currents in cardiac myocytes.
In our hands, the expression of TRPC7 did not enhance Ang II-induced membrane depolarization (Figure 6B). This may be explained by differential spatial organization and dynamics in the receptor-transduction systems (Delmas et al, 2002). As OAG-induced increases in [Ca2+]i, but Ang II-induced shift of membrane potential, was not enhanced in TRPC7-expressing cells (Supplementary Figure S3, Figure 6B), Ang II signaling microdomain may contain TRPC6 and TRPC3, but not TRPC7. This idea is supported by the reports that stimulation of AT1R activates TRPC6 (Large, 2002; Winn et al, 2005).
Previous report suggested that capacitative Ca2+ entry contributes to the nuclear translocation of NFAT and hypertrophy in cardiomyocytes (Hunton et al, 2002). In contrast with the present study, they showed that IP3-mediated store depletion triggers the activation of SOC and activates hypertrophic responses. Although we cannot explain the discrepancy between their report and the present study, we clearly demonstrated that xestospongin C or IP3-sponge did not affect Ang II-induced changes in fluorescence intensity of DiBAC4(3), NFAT activation, and hypertrophic responses (Figures 1 and 5D). We also confirmed that the application of high concentration of IP3 did not activate whole-cell currents (data not shown). In addition, treatment with caged-IP3 did not affect the localization of GFP-NFAT4 upon UV irradiation, although caged-IP3 induced a marked increases in [Ca2+]i (Supplementary Figure 5). We observed that the treatments with thapsigargin and ionomycin induce NFAT activation through store-operated Ca2+ influx. However, Ang II-stimulated increase in [Ca2+]i through IP3-mediated Ca2+ release is 25–30% of those induced by thapsigargin and ionomycin treatment. This IP3-mediated increase may not be enough for the activation of SOC. These results suggest that IP3-mediated Ca2+ signaling, including SOC, is not responsible for Ang II-induced NFAT activation.
Whole-cell current experiments revealed that the membrane currents were activated more than 1 min after Ang II stimulation (Figure 4A). However, the maximal shift of membrane potential was achieved about 2 min after Ang II stimulation (Figure 5A). The distinct delay may be explained by DAG metabolism, as RHC80267 enhanced Ang II-induced inward current (Figure 4B). The steady-state DAG lipase activity may regulate the time to elevate DAG concentration required for the activation of whole-cell currents and subsequent changes in membrane potential.
Growing evidence has indicated the involvement of L-type Ca2+ channels in the induction of cardiac hypertrophy (Lubic et al, 1994, 1995; Whitehurst et al, 1999; Liao et al, 2005). The role of L-type Ca2+ channels in excitation-transcription coupling is well established in the nervous system (Dolmetsch et al, 2001, 2003). Calmodulin is reported to be critical for conveying the Ca2+ signal to the nucleus (Dolmetsch et al, 2001). As calmodulin also regulates calcineurin activity, calmodulin may convey the signal to the nucleus in the cardiovascular system in a similar manner to the nervous system.
While this study is in review process, Bush et al (2006) reported that TRPC channels are involved in hypertrophy through pathological calcineurin/NFAT signaling. They showed that TRPC3 expression is upregulated in mice with pathological hypertrophy. We demonstrated that TRPC3 and TRPC6 mediate hypertrophic responses of neonatal myocytes by Ang II stimulation. Thus, the upregulated TRPC channels in vivo may enhance receptor-stimulated hypertrophy through the mechanism that we have demonstrated in this study.
In summary, we demonstrated for the first time that PLC-generated DAG has a pathophysiological role in activation of TRPC3 and TRPC6, and TRPC3/6 mediates NFAT-mediated hypertrophic responses through L-type Ca2+ channel.
Materials and methods
Materials, plasmid construction, and cell cultures
AT1R blocker CV11974 was provided from Takeda Chemical Industries Ltd (Osaka, Japan). PTX, SK&F96365, caged-IP3, and cPLA2 inhibitor were purchased from Calbiochem. Valinomycin, cyclosporine A, U73122, U73343, AA, PACOCF3, myo-IP3, TTX, and PD123319 were from Sigma. Fura2/AM was from Dojindo. Collagenase, Liberase (enzyme 3), and Fugene 6 were from Roche. Alexa Fluor 594-phalloidin and DiBAC4(3) were from Molecular Probe. Nitrendipine was from Wako. The cDNA coding DGKβ (KIAA0718) was obtained from Kazusa DNA Research Institute. The cDNAs coding mouse TRPC3, TRPC6, and TRPC7, and anti-TRPC7 antibody were prepared as described (Inoue et al, 2001; Nishida et al, 2003). Anti-TRPC6 and anti-TRPC3 antibodies were from Alomone. Mouse TRPC6-3A and TRPC6-Δ(N) were constructed according to the previous reports (Hofmann et al, 2002; Hisatsune et al, 2004). The cDNA coding IP3-sponge was cloned from mouse brain (Uchiyama et al, 2002), and GFP-IP3-sponge was constructed in pEGFP-C1 vector (Clontech). Isolation of rat neonatal cardiomyocytes was described (Nishida et al, 2000).
Production of adenoviruses, infection, and transfection
Recombinant adenoviruses of GFP-NFAT4, HA-tagged DGKβ, GFP-DGKβ, wild-type TRPC6, TRPC6-3A, TRPC6-Δ(N), and GFP-fused IP3-sponge (GFP-IP3-sponge) were produced by the method of He et al (1998) with a slight modification. Other adenoviruses were prepared as described previously (Nishida et al, 2000, 2005; Arai et al, 2003). Cells were infected with adenovirus(es) at 100 MOI for 48 h. Small interference RNAs (250 nM) were transfected with lipofectamine 2000 for 72 h.
Measurement of NFAT activity
Measurement of NFAT activity was performed as described previously (Fujii et al, 2005). At 2 h after adenoviral infection in serum-free medium, cardiomyocytes (1 × 106 cells) plated on 24-well dishes were transiently cotransfected with 0.45 μg pNFAT-Luc and 0.05 μg pRL-SV40 control plasmid, using Fugene 6. For measuring the translocation of GFP-NFAT4, cells (1 × 106) plated on glass-bottom 35 mm dishes were infected for 48 h with adenovirus coding GFP-NFAT4 at 100 MOI. After Ang II stimulation (100 nM) for 30 min, the localization of GFP-NFAT4 was determined with a Laser Scanning Confocal Imaging System (Carl Zeiss LSM510) as described (Fujii et al, 2005).
Measurement of [Ca2+]i and membrane potential
The intracellular Ca2+ concentration ([Ca2+]i) of cardiomyocytes was determined as described (Arai et al, 2003; Nishida et al, 2005). Briefly, cells (1 × 106) were plated on gelatin-coated glass-bottom 35 mm dishes and were loaded with 2.5 μM fura-2/AM at 37°C for 30 min. For measurement of cell membrane potential, cells were loaded with 1.5 μM DiBAC4(3) at 37°C for 30 min. The fluorescence intensity of DiBAC4(3) was measured at an excitation wavelength of 488 nm with a video image analysis system (Aquacosmos, Hamamatsu Photonics). The peak changes (ΔF/F0) of DiBAC4(3) fluorescence intensity were defined as values obtained by subtracting the basal fluorescence intensity (F0) from the maximal intensity during 19 min Ang II treatment.
Measurement of the expression of TRPC proteins
Cardiomyocytes (3 × 106 cells) plated on six-well dishes were directly harvested with 2 × SDS sample buffer. The protein samples were fractionated by 8% SDS–PAGE gel and then transferred onto PVDF membrane. The expression of endogenous TRPC proteins was assessed by Western blotting using anti-TRPC antibodies. To examine the involvement of TRPC3 and C6, knockdown experiments using siRNAs were performed (sequences of siRNAs used in this study were presented in Supplementary Table 1). We used Stealth (Invitrogen) siRNA sequence to eliminate nonspecific responses by siRNA. Transfection was performed by lipofectamine 2000.
Measurement of hypertrophic responses of cardiomyocytes
Measurement of cardiomyocyte hypertrophy was performed as described (Maruyama et al, 2002). Briefly, 24 h after infection, cardiomyocytes were stimulated with Ang II (100 nM) for 48 h. The cells were washed, fixed, and then stained with Alexa Fluor 594-phalloidin to visualize actin filaments. Protein synthesis was measured by [3H]leucine incorporation. After cells were stimulated with Ang II (100 nM) for 18 h, [3H]leucine (1 μCi/ml) was add to the culture medium and further incubated for 6 h. The incorporated [3H]leucine was measured using liquid-scintillation counter.
Electrophysiology
Single neonatal rat cardiac myocytes plated on thin coverslips for 1–2 days (3 × 10 mm; Matsunami, Japan) were used for patch-clamp experiments. The details of patch-clamp experiments are described elsewhere (Shi et al, 2004). Internal solution used for the whole-cell variant of patch clamp; K+-internal solution (mM): 140 K+, 4 Na+, 2 Mg2+, 144 Cl−, 2 EGTA, 2 ATP, 10 HEPES or Cs+-internal solution (mM): 140 Cs+, 2 Mg2+, 20 Cl−, 2 SO42−, 120 aspartate, 2 ATP, 5 EGTA (2 Ca2+ added), 10 HEPES, and 10 μM myo-IP3. For current-clamp recordings, normal external solution and K+-internal solution of the same composition as used for whole-cell voltage-clamp experiments were used. All experiments were performed at 25–28°C with the aid of a temperature control unit (Warner Instruments) to facilitate the response to Ang II.
Statistical analysis
The results are shown as means±s.e.m. All experiments were repeated at least three times. Mean values were compared with control by Student's t-test (for two groups) or one-way ANOVA followed by Dunnett's t-test (for three or more groups).
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table 1
Acknowledgments
We thank Y Ito (Department of Pharmacology, Graduate School of Medical Sciences, Kyushu University) for using Aquacosmos imaging system during early stage of this study. We also thank SM Lanier and M Sato (Louisiana State University Health Science Center) for experimental suggestion, and T Murakami for DNA construction. This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (to M Nishida and H Kurose), from Ministry of Health, Labour and Welfare of Japan, and the National Institute of Biomedical Innovation (MF-16, to YS), and by grants from The Naito Foundation, The Kanae Foundation, The Suzuken Memorial Foundation, The Uehara Memorial Foundation, Kao Foundation for Arts and Sciences, Takeda Science Foundation and Japan Heart Foundation Research Grant (to MN), and Astellas Foundation for Research on Metabolic Disorders (H Kurose).
References
- Arai K, Maruyama Y, Nishida M, Tanabe S, Takagahara S, Kozasa T, Mori Y, Nagao T, Kurose H (2003) Endothelin-1-induced MAPK activation and cardiomyocyte hypertrophy are mediated by Gα12 and Gα13 as well as Gαq and Gβγ subunits. Mol Pharmacol 63: 478–488 [DOI] [PubMed] [Google Scholar]
- BACzkó I, Giles WR, Light PE (2004) Pharmacological activation of plasma-membrane KATP channels reduces reoxygenation-induced Ca2+ overload in cardiac myocytes via modulation of the diastolic membrane potential. Br J Pharmacol 141: 1059–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush EW, Hood DB, Papst PJ, Chapo JA, Minobe W, Bristow MR, Olson EN, McKinsey TA (2006) TRPC channels promote cardiomyocyte hypertrophy through activation of calcineurin signaling. J Biol Chem, (in press) (doi: 10.1074/jbc.M605536200) [DOI] [PubMed] [Google Scholar]
- Clapham DE (2003) TRP channels as cellular sensors. Nature 426: 517–524 [DOI] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN (2002) NFAT signaling: choreographing the social lives of cells. Cell 109: S67–S79 [DOI] [PubMed] [Google Scholar]
- Delmas P, Wanaverbecq N, Abogadie FC, Mistry M, Brown DA (2002) Signaling microdomains define the specificity of receptor-mediated InsP3 pathways in neurons. Neuron 34: 209–220 [DOI] [PubMed] [Google Scholar]
- Dolmetsch R (2003) Excitation-transcription coupling: signaling by ion channels to the nucleus. Science's stke, www.stke.org/cgi/content/full/sigt rans;2003/166/pe4 [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI (1997) Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 386: 855–858 [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME (2001) Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 294: 333–339 [DOI] [PubMed] [Google Scholar]
- Dostal DE, Rothblum KN, Chernin MI, Cooper GR, Baker KM (1992) Intracardiac detection of angiotensinogen and renin: a localized rennin–angiotensin system in neonatal rat heart. Am J Physiol 263: C838–C850 [DOI] [PubMed] [Google Scholar]
- Fujii T, Onohara N, Maruyama Y, Tanabe S, Kobayashi H, Fukutomi M, Nagamatsu Y, Nishihara N, Inoue R, Sumimoto H, Shibasaki F, Nagao T, Nishida M, Kurose H (2005) Gα12/13-mediated production of reactive oxygen species is critical for angiotensin receptor-induced NFAT activation in cardiac fibroblasts. J Biol Chem 280: 23041–23047 [DOI] [PubMed] [Google Scholar]
- Guinamard R, Chatelier A, Lenfant J, Bois P (2004) Activation of the Ca2+-activated nonselective cation channel by diacylglycerol analogues in rat cardiomyocytes. J Cardiovas Electrophysiol 15: 342–348 [DOI] [PubMed] [Google Scholar]
- He TC, Zhou S, da Costa LT, Yu J, KInzler KW, Vogelstein B (1998) A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95: 2509–2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Yao G, Savoia C, Touyz RM (2005) Transient receptor potential melastatin 7 ion channels regulate magnesium homeostasis in vascular smooth muscle cells. Role of angiotensin II. Circ Res 96: 207–215 [DOI] [PubMed] [Google Scholar]
- Hisatsune C, Kuroda Y, Nakamura K, Inoue T, Nakamura T, Michikawa T, Mizutani A, Mikoshiba K (2004) Regulation of TRPC6 channel activity by tyrosine phosphorylation. J Biol Chem 279: 18887–18894 [DOI] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G (1999) Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397: 259–263 [DOI] [PubMed] [Google Scholar]
- Hofmann T, Schaefer M, Schultz G, Gudermann T (2002) Subunit composition of mammalian transient receptor potential channels in living cells. Proc Natl Acad Sci USA 99: 7461–7466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunton DL, Lucchesi PA, Pang Y, Cheng X, Dell'Italia LJ, Marchase RB (2002) Capacitative calcium entry contributes to nuclear factor of activated T cells nuclear translocation and hypertrophy in cardiomyocytes. J Biol Chem 277: 14266–14273 [DOI] [PubMed] [Google Scholar]
- Inoue R, Okada T, Onoue H, Hara Y, Shimizu S, Naitoh S, Ito Y, Mori Y (2001) The transient receptor potential protein homologue TRP6 is the essential component of vascular α1-adrenoceptor-activated Ca2+-permeable cation channel. Circ Res 88: 325–332 [DOI] [PubMed] [Google Scholar]
- Large WA (2002) Receptor-operated Ca2+-permeable nonselective cation channels in vascular smooth muscle: a physiologic perspective. J Cardiovas Electrophysiol 13: 493–501 [DOI] [PubMed] [Google Scholar]
- Liao Y, Asakura M, Takashima S, Kato H, Asano Y, Shintani Y, Minamino T, Tomoike H, Hori M, Kitakaze M (2005) Amlodipine ameliorates myocardial hypertrophy by inhibiting EGFR phosphorylation. Biochem Biophys Res Commun 327: 1083–1087 [DOI] [PubMed] [Google Scholar]
- Linares-Hernandez L, Guzman-Grenfell AM, Hicks-Gomez JJ, Gonzalez-Martinez MT (1998) Voltage dependent calcium influx in human sperm assessed by spontaneous detection of intracellular calcium and membrane potential. Biochem Biophys Acta 1372: 1–12 [DOI] [PubMed] [Google Scholar]
- Lubic SP, Giacomini KM, Giacomini JC (1994) Increased 1,4-dihydropyridine binding sites in serum-stimulated cardiomyocytes hypertrophy. J Pharmacol Exp Ther 270: 697–701 [PubMed] [Google Scholar]
- Lubic SP, Giacomini KM, Giacomini JC (1995) The effects of modulation of calcium influx through the voltage-sensitive calcium channel on cardiomyocyte hypertrophy. J Mol Cell Cardiol 27: 917–925 [DOI] [PubMed] [Google Scholar]
- Maruyama Y, Nakanishi Y, Walsh EJ, Wilson DP, Welsh DG, Cole WC (2006) Heteromultimeric TRPC6-TRPC7 channels contribute to arginine vasopressin-induced cation current of A7r5 vascular smooth muscle cells. Circ Res 98: 1520–1527 [DOI] [PubMed] [Google Scholar]
- Maruyama Y, Nishida M, Sugimoto Y, Tanabe S, Turner JH, Kozasa T, Wada T, Nagao T, Kurose H (2002) Gα12/13 mediate α1-adrenergic receptor-induced cardiac hypertrophy. Circ Res 91: 961–969 [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Dorn GW II (2001) Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev Physiol 63: 391–426 [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, Grant SR, Olson EN (1998) A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell 93: 215–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moschella MC, Marks AR (1993) Inositol 1,4,5-trisphosphate receptor expression in cardiac myocytes. J Clin Invest 120: 1137–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M, Maruyama Y, Tanaka R, Kontani K, Nagao T, Kurose H (2000) Gαi and Gαo are target proteins of reactive oxygen species. Nature 408: 492–495 [DOI] [PubMed] [Google Scholar]
- Nishida M, Sugimoto K, Hara Y, Mori E, Morii T, Kurosaki T, Mori Y (2003) Amplification of receptor signalling by Ca2+ entry-mediated translocation and activation of PLCγ2 in B lymphocytes. EMBO J 22: 4677–4688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida M, Tanabe S, Maruyama Y, Mangmool S, Urayama K, Nagamatsu Y, Takagahara S, Turner JH, Kozasa T, Kobayashi H, Sato Y, Kawanishi T, Inoue R, Nagao T, Kurose H (2005) Gα12/13- and reactive oxygen species-dependent activation of c-Jun NH2-terminal kinase and p38 MAPK by angiotensin receptor stimulation in rat neonatal cardiomyocytes. J Biol Chem 280: 18434–18441 [DOI] [PubMed] [Google Scholar]
- Reading SA, Early S, Waldron BJ, Welsh DG, Brayden JE (2005) TRPC3 mediates pyrimidine receptor-induced depolarization of cerebral arteries. Am J Physiol 288: H2055–H2061 [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Xu Y, Slayter HS, Izumo S (1993) Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell 75: 977–984 [DOI] [PubMed] [Google Scholar]
- Seth M, Sumbilla C, Mullen SP, Lewis D, Klein G, Hussain A, Soboloff J, Gill DL, Inesi G (2004) Sarco(endo)plasmic reticulum Ca2+ ATPase (SERCA) gene silencing and remodeling of the Ca2+ signaling mechanism in cardiac myocytes. Proc Natl Acd Sci USA 101: 16683–16688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Mori E, Mori Y, Mori M, Li J, Ito Y, Inoue R (2004) Multiple regulation by calcium of murine homologues of transient receptor potential proteins TRPC6 and TRPC7 expressed in HEK293 cells. J Physiol 561: 415–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soboloff J, Spassova M, Xu W, He LP, Cuesta N, Gill DL (2005) Role of endogenous TRPC6 channels in Ca2+ signal generation in A7r5 smooth muscle cells. J Biol Chem 280: 39786–39794 [DOI] [PubMed] [Google Scholar]
- Taigen T, De Windt LJ, Lim HW, Molkentin JD (2000) Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proc Natl Acd Sci USA 97: 1196–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timmerman LA, Clipstone NA, Ho SN, Northrop JP, Crabtree GR (1996) Rapid shuttling of NF-AT in discrimination of Ca2+ signals and immunosuppression. Nature 383: 837–840 [DOI] [PubMed] [Google Scholar]
- Tomida T, Hirose K, Takizawa A, Shibasaki F, Iino M (2003) NFAT functions as a working memory of Ca2+ signals in decoding Ca2+ oscillation. EMBO J 22: 3825–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touyz RM, He Y, Montezano ACI, Yao G, Chubanov V, Gudermann T, Callera GE (2006) Differential regulation of transient receptor potential melastatin 6 and 7 cation channels by ANG II in vascular smooth muscle cells from spontaneous hypertensive rats. Am J Physiol 290: R73–R78 [DOI] [PubMed] [Google Scholar]
- Uchiyama T, Yoshikawa F, Hishida A, Furuichi T, Mikoshiba K (2002) A novel recombinant hyperaffinity inositol 1,4,5-trisphosphate (IP3) absorbent traps IP3, resulting in specific inhibition of IP3-mediated calcium signaling. J Biol Chem 277: 8106–8113 [DOI] [PubMed] [Google Scholar]
- Welsh DG, Morielli AD, Nelson MT, Brayden JE (2002) Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ Res 90: 248–250 [DOI] [PubMed] [Google Scholar]
- Whitehurst RM Jr, Zhang M, Bhattacharjee A, Li M (1999) Dexamethasone-induced hypertrophy in rat neonatal cardiac myocytes involves an elevated L-type Ca2+ current. J Mol Cell Cardiol 31: 1551–1558 [DOI] [PubMed] [Google Scholar]
- Wilkins BJ, De Windt LJ, Bueno OF, Braz JC, Glascock BJ, Kimball TF, Molkentin JD (2002) Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol Cell Biol 22: 7603–7613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins BJ, Molkentin JD (2004) Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun 322: 1178–1191 [DOI] [PubMed] [Google Scholar]
- Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA, Howell DN, Vance JM, Rosenberg PB (2005) A mutation of TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science 308: 1801–1804 [DOI] [PubMed] [Google Scholar]
- Woodcock EA, Matkovich SJ (2005) Ins(1,4,5)P3 receptors and inositol phosphates in the heart-evolutionary artifacts or active signal transducers? Pharmacol Therap 107: 240–251 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Figure S5
Supplementary Table 1