

Abstract
Loss of the epithelial polarity gene scribble in clones of Drosophila imaginal disc cells can cooperate with Ras signaling to induce malignant tumors. Such mutant tissue overproliferates, resists apoptosis, leaves its place of origin and invades other organs, ultimately causing lethality. We show that increased Jun N-terminal kinase (JNK) signaling resulting from the loss of scribble promotes the movement of transformed cells to secondary sites. This effect requires Fos-dependent transcriptional activation of a matrix metalloprotease gene mmp1 downstream of JNK. Expression of the Mmp inhibitor Timp or Mmp RNAi knockdown suppresses cell invasiveness. The proinvasive function of the JNK pathway is revealed in a tumor context when active Ras signaling prevents the apoptotic response to JNK activity as it occurs in nontransformed cells. Based on these results, we present a model that explains the oncogenic cooperation between JNK and Ras, and describes how aberrant regulation of cell survival, proliferation and mobilization cooperate to incite malignant tumor formation.
Keywords: Fos, JNK, Mmp, oncogene cooperation, Ras
Introduction
A key determinant for the malignancy of tumors is their propensity to shed metastases. Metastatic tumor cells can move actively and have the ability to penetrate surrounding extracellular matrix (ECM) stroma and epithelial basement membranes. The ability of tumor-derived cells to leave an organ, survive, colonize and proliferate at ectopic locations relies on reprogramming of multiple signaling pathways that control not only viability and growth but also cell shape, adhesion and motility (Hanahan and Weinberg, 2000). The complexity of such oncogenic programming explains why in order for a malignant tumor to arise, multiple cooperating oncogenic mutations have to occur (Land et al, 1983).
Cooperation of multiple oncogenic events has been successfully reconstructed in the fruit fly Drosophila melanogaster, where imaginal discs can develop tumors displaying characteristics of metastatic cancer (Brumby and Richardson, 2005; Ferres-Marco et al, 2006). Disruption of tumor suppressor gene, such as scribble (scrib), lethal(2) giant larvae or discs large, together with the activation of the Ras signaling pathway (achieved by expression of active RasV12 or Rafact variants) transforms clones of normally proliferating imaginal disc cells into malignant, lethal tumors (Brumby and Richardson, 2003; Pagliarini and Xu, 2003; Uhlirova et al, 2005). This malignancy results from a significant growth advantage of the tumor cells over surrounding tissues combined with their ability to move actively from their place of origin and to penetrate basement membrane barriers. As a result, rasV12, scrib−/− cells can leave the eye/antennal imaginal disc and colonize distal parts of the brain, for example, the ventral nerve cord (VNC) or, rarely, other parts of the larval body (Pagliarini and Xu, 2003). The genetic and molecular principles of these migratory and invasive cellular behaviors have not yet been identified.
The Jun N-terminal kinase (JNK) signaling pathway is an important regulator of cellular phenotypes that are relevant in the context of tumorigenesis. In addition to its well-known role in apoptosis (Adachi-Yamada et al, 1999; Behrens et al, 1999; Kuan et al, 1999; Tournier et al, 2000; Igaki et al, 2002; Moreno et al, 2002; Ryoo et al, 2004; McEwen and Peifer, 2005), JNK has been implicated in the control of cell motility and adhesion during developmental as well as wounding-induced cell movements. For example, JNK-deficient mice exhibit defects in neural tube and eyelid closure; growth factor-stimulated embryonic stem cell migration is also affected (Xia et al, 2000; Yujiri et al, 2000; Weston et al, 2003, 2004; Zhang et al, 2003). Components of the Drosophila JNK pathway are required for extension and sealing of embryonic epidermal sheets during dorsal closure (Riesgo-Escovar et al, 1996; Sluss et al, 1996; Glise and Noselli, 1997; Hou et al, 1997; Kockel et al, 1997; Martin-Blanco et al, 1998; Homsy et al, 2006). During metamorphosis, JNK is required for shape remodeling of imaginal epithelia, involving their eversion, spreading and fusion at the thorax dorsal midline (Agnes et al, 1999; Zeitlinger and Bohmann, 1999; Martin-Blanco et al, 2000; Pastor-Pareja et al, 2004). JNK activity also promotes cell shape changes and re-epithelialization of wounds in larval and adult epidermis (Rämet et al, 2002; Galko and Krasnow 2004) or in regenerating imaginal discs (Rämet et al, 2002; Bosch et al, 2005; Mattila et al, 2005).
JNK signaling is undoubtedly implicated in mammalian cancer (Huang et al, 2003; Ventura et al, 2004; Hagemann et al, 2005; Kwei et al, 2006), and notable JNK activity has also been found in Drosophila rasV12, scrib−/− clonal tumors (Brumby and Richardson, 2005). However, the precise contribution of JNK activity to tumor cell invasiveness remains only partially understood. It has been shown that JNK exerts its effects on cell migration and epithelial movements at least in part by modulating gene expression (Jasper et al, 2001; Homsy et al, 2006). One possibility is that JNK might promote expression of matrix metalloproteases (MMP), enzymes with clear links to tumor cell motility. MMPs degrade basement membrane and ECM components, and normally function during developmental tissue remodeling and wound healing (McCawley and Matrisian, 2000; Vu and Werb, 2000). Various human tumors display increased MMP expression in correlation with metastasis formation (Stetler-Stevenson et al, 1993; Nelson et al, 2000). Studies in a range of mammalian cell lines have identified JNK-activated transcription factors of the Fos and Jun family (AP-1) as regulators of MMP gene expression (Westermarck and Kahari, 1999; Chakraborti et al, 2003). However, the genetic evidence for the causal relationship between JNK, AP-1, MMP expression and tumor invasiveness in vivo is scarce.
Here, we employ the Drosophila model to show that activation of the JNK signaling pathway is essential for the invasive behavior of tumors in vivo. In cooperation with two oncogenic events, activated Ras and disrupted epithelial integrity, JNK enhances tumor malignancy via Fos-dependent activation of the gene encoding Mmp1.
Results
JNK is required for the invasive phenotype of rasV12, scrib−/− tumor cells
Consistent with previous reports (Brumby and Richardson, 2003; Pagliarini and Xu, 2003; Uhlirova et al, 2005; Igaki et al, 2006), we find that clones of eye/antennal imaginal disc cells deficient for the epithelial polarity gene scribble or overexpressing the oncogenic ras allele (RasV12) give rise to a mild tumor phenotype with no detectable invasiveness. Only when both oncogenic lesions combine, prominent cell invasion becomes evident. Malignant cells can be tracked by the GFP marker that is expressed along with RasV12 in the scrib-deficient clones induced by eyeless-driven FLP. Starting on day 6 after egg laying (AEL), a significant fraction of the GFP-labeled imaginal disc cells appear in the VNC. As the eyeless driver is inactive in the VNC, GFP-marked cells residing in it must have migrated there from the eye/antennal discs or from the optic lobes (Pagliarini and Xu, 2003) and, thus, their presence indicates abnormal cell migration or invasiveness. On day 9 AEL, the invading cells obscure the eye/antennal discs, brain lobes and the VNC, such that they are no longer distinguishable. (Figure 1 and Supplementary Figure 1; Pagliarini and Xu, 2003; Uhlirova et al, 2005; Igaki et al, 2006). The invading tumor cells are morphologically distinct from the epithelial cells and neurons that normally populate the VNC. They display a prominent actin cytoskeleton network as shown by phalloidin staining (Figure 1A, right panel). Such an abundant actin polymerization is typical of cells that are actively moving or undergoing shape changes. JNK signaling is a known regulator of actin dynamics in Drosophila and other species (Xia and Karin, 2004). To explore a possible contribution of the JNK pathway to cell invasiveness, we expressed a dominant-negative form of the Drosophila JNK, Basket (BskDN) in clonal tumors of the rasV12, scrib−/− genotype.
Figure 1.
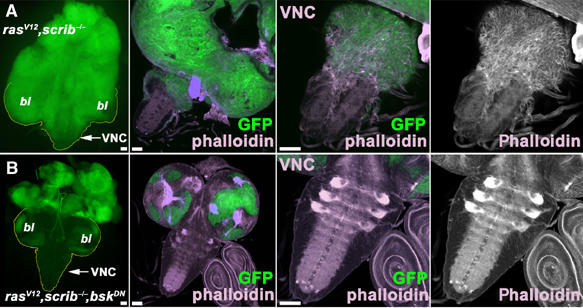
The JNK signaling pathway is required for tumor cell invasiveness. The eyeless-FLP/MARCM system was used to induce GFP-marked clones (green) of the indicated genotypes. The left panels of (A, B) show epifluorescence images of eye/antennal/brain complexes. All other panels show confocal micrographs (projections of multiple sections or single confocal sections for magnification of VNC, respectively) of third instar larval brains on day 6 AEL. Brains were stained with phalloidin to visualize actin filaments. (A) rasV12, scrib−/− double mutant cells proliferate strongly and overgrow wild-type tissue in the brain lobes (bl) and the eye/antennal discs. The tumor cells display pronounced migratory behavior. At higher magnification (A, right panels), the elongated shape and the prominent actin cytoskeleton (phalloidin stain) in the clonal cells invading the VNC are visible. The tumor cell morphology differs strikingly from that of normal brain cells. (B) Blocking JNK signaling by BskDN expression in rasV12, scrib−/− cells completely suppresses tumor cell invasiveness and the VNC is free of GFP-positive clonal cells. Scale bars=50 μm.
Strikingly, inhibition of JNK signaling by expression of BskDN blocked invasiveness completely (Figure 1B, see also Figure 5 for statistical evaluation). Clones of the rasV12, scrib−/−, bskDN genotype still overgrew the eye/antennal discs (Figure 1A and 2B). However, clonal cells never left the structure (Figure 1B). Similarly, overexpression of the JNK-specific phosphatase Puckered (Puc), which specifically dephosphorylates and inactivates JNK/Basket (Martin-Blanco et al, 1998), suppressed the invasiveness of tumor cells into the VNC (Figure 5 and Supplementary Figure 2). Evidently, a functional JNK pathway is essential for the full manifestation of the invasive phenotype. This observation is in agreement with a recent report by Igaki et al (2006), which describes a function for JNK in a similar model.
Figure 5.
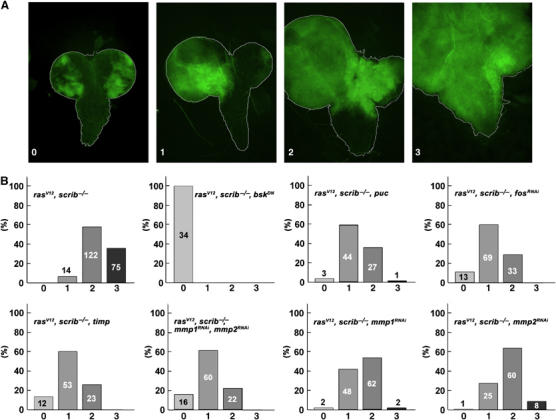
Quantification of tumor cell invasiveness among various genotypes. (A) Eye/antennal/brain complexes dissected from third instar larvae (8 days AEL, except rasV12, scrib−/−, bskDN, 6 days AEL) carrying GFP-marked clones of the indicated genotypes were analyzed by fluorescent microscopy and divided into four categories, depicted by the examples, based on their degree of invasiveness: (0) noninvasive, (1) clonal cells overgrow one of the optic lobe and invade the VNC, (2) both optic lobes are overgrown by mutant tissue and cells enter the VNC from both sides and (3) optic lobes and most of the VNC are invaded by clonal tissue. (B) The histograms show percentages of brains classified into each of the four categories; the number of brains analyzed is indicated in each column.
Figure 2.
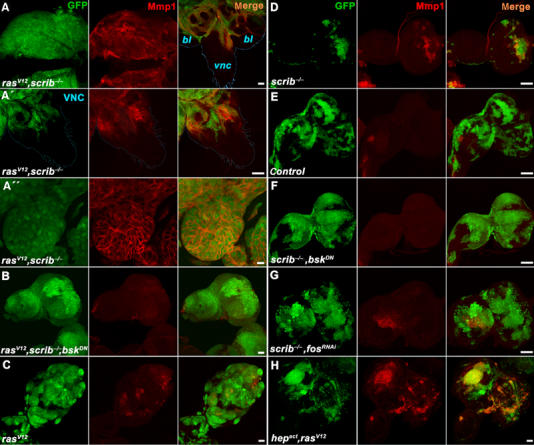
JNK-dependent expression of Mmp1 in rasV12, scrib−/− tumors. Third instar eye/antennal imaginal discs carrying GFP-marked clones (green) of the indicated genotypes were immunostained with an anti-Mmp1 antibody (red) and analyzed by confocal microscopy. (A) Strong upregulation of Mmp1 protein expression in rasV12, scrib−/− clones is seen not only in overgrown eye/antennal discs but also in cells invading distal parts of the brain such as the VNC (A, right). Magnification of the VNC with invading Mmp1-positive cells (A′). Mmp1 protein is enriched on surface of malignant rasV12, scrib−/− cells (A″). (B) Mmp1 expression is lost when JNK activity is suppressed by the expression of BskDN in rasV12, scrib−/− clones. Note that the clonal tissue still overgrows the entire eye/antennal imaginal disc. (C–E) While significantly smaller, scrib−/− clones show prominent Mmp1 staining (D) compared to the bigger wild-type control (E) or rasV12 (C) clones. (F, G) Mmp1 expression in scrib−/− clones is lost when JNK signaling is suppressed by BskDN (F) or when Fos function is inhibited by RNAi (G) in these clones. Note that the scrib−/− clones grow bigger in size when their JNK activity is suppressed. (H) Activation of the JNK pathway by Hepact is sufficient to induce Mmp1 expression in rasV12 clones that have normal scrib function. Except for the images in (A–A″), which are single confocal sections, all other panels show projections of multiple sections. Scale bars=50 μm in (A, A′, B–H) and 10 μm in (A″).
The consequences of invasive tumor growth for the organism were profound. Animals bearing malignant rasV12, scrib−/− clones progressed normally throughout larval development, but never pupated. Instead of initiating metamorphosis by pupariating on day 6 AEL, they persisted as third instar larvae, increasing their body size and finally dying around day 13 AEL. In contrast, the expression of BskDN in the potentially malignant rasV12, scrib−/− clones allowed all larvae to pupariate on day 6 AEL. They progressed through most of metamorphosis and died as pharate adults. Thus, interfering with JNK activity in rasV12, scrib−/− clones rescued much of the normal development and its timing, presumably by curbing of the invasive tumor mass. Pupariation was also restored, albeit to a lesser extent, by expression of the JNK phosphatase Puc in the rasV12, scrib−/− clones (data not shown), confirming that the suppression of the larval arrest resulted from the JNK inhibition and not from some other effect of BskDN expression.
Mmp1 is upregulated in invasive rasV12, scrib−/− tumors
MMPs are potential effectors of tumor cell motility, and previous transcriptome analyses have suggested their regulation by JNK in Drosophila (Jasper et al, 2001; Boutros et al, 2002). To explore the link between JNK signaling and the invasiveness of rasV12, scrib−/− tumors, we therefore examined the expression of both Drosophila Mmp encoding genes, mmp1 and mmp2 (Llano et al, 2000, 2002; Page-McCaw et al, 2003), in the transformed imaginal disc tissue.
Immunostaining of wild-type eye/antennal imaginal discs finds Mmp1 protein expression restricted to a small area of the disc stalk (Figure 2E). In contrast, Mmp1 expression was markedly increased in invasive rasV12, scrib−/− clones regardless of their location within the disc (Figure 2A). Mmp1 was detected at varying levels in almost all tumor cells, notably in those that invaded the distal parts of the brain (Figure 2A (right) and A′). The expression pattern of Mmp1 in the invading tissue appeared dynamic, and a significant fraction of presumably secreted Mmp1 protein remained associated with cell surface (Figure 2A″). Interestingly, in areas where tumorigenic clones with high levels of Mmp1 expression came to close proximity with the basement membrane, its integrity was disrupted as shown by staining for laminin A (Supplementary Figure 3). Destruction of basement membrane barriers is one of the hallmarks of invasive tumors as it permits the penetration of secondary organs.
Mmp1 protein levels in tumorigenic clones decreased significantly when JNK was inhibited by coexpression of either BskDN or Puc (Figure 2B and Supplementary Figure 2). The above results show that Mmp1 expression in rasV12, scrib−/− clones requires JNK pathway activity and is associated with invasiveness and disruption of the basement membrane barrier. Unlike mmp1, mmp2 mRNA expression stayed at its basal level in all tested genotypes (Supplementary Figure 4).
Mmp1 upregulation is due to the loss of scribble
To discern which of the oncogenic lesions, Ras activation or the loss of scrib, triggers JNK-mediated Mmp1 expression, we examined eye discs bearing clones that either expressed RasV12 or that were deficient for scrib. Compared to discs with combined rasV12, scrib−/− clones (Figure 2A), discs bearing overproliferating but noninvasive rasV12 clones displayed only sporadic patches of increased Mmp1 expression relative to the sizeable GFP-marked clonal areas (Figure 2C and Supplementary Figure 6D). In addition, Mmp1 was not detectable in clones expressing activated Raf (Supplementary Figure 5A). In contrast, Mmp1 was prominently expressed in clones lacking scrib, although these clones did not overgrow to the extent of the RasV12 (or Rafact) tumors (Figure 2D) and showed increased level of apoptosis (Brumby and Richardson, 2003; Uhlirova et al, 2005; Supplementary Figure 6A). We conclude that the major cause of Mmp1 activation in tumors is the loss of scrib. This assumption is supported by RT–PCR data, showing similarly high mmp1 mRNA levels in discs bearing scrib−/− and rasV12, scrib−/− clones (Supplementary Figure 4A).
Mmp1 expression in tumors requires JNK and Fos
To confirm that the loss of scribble causes activation of Mmp1 expression in a JNK-dependent manner, we measured Mmp1 levels in scrib−/− clones overexpressing BskDN or Puc. As before, in the case of rasV12, scrib−/− cells, suppression of JNK activity abolished the elevated Mmp1 expression (Figure 2F and Supplementary Figures 2B and 6B). We also investigated whether JNK-mediated induction of Mmp1 in scrib-deficient cells required AP-1 transcription factors. Inhibition of Drosophila Fos activity by transgenic expression of an RNAi construct decreased Mmp1 levels in scrib−/− clones to a similar extent as did suppression of Basket activity (Figure 2G and Supplementary Figure 6C). Interestingly, and similar to scrib−/− clones in which JNK activity was blocked with BskDN (Brumby and Richardson, 2003; Uhlirova et al, 2005) or Puc (Supplementary Figures 2A and 6B), scrib−/−, fosRNAi clones grew much bigger in size (Figure 2G) and apoptosis was suppressed in them (Supplementary Figure 6C). These results suggest that besides regulation of mmp1, Fos is also involved in the execution of cell death in response to JNK signals. We conclude that the loss of the epithelial polarity gene scribble induces a pathway encompassing JNK and Fos to activate mmp1 gene expression and apoptosis.
Transcriptional activation of mmp1 by JNK
To establish that Mmp1 is induced downstream of JNK as its transcriptional target in vivo, we undertook two complementary approaches. First, we utilized a heat-inducible engrailed-Gal4 system to activate JNK by expressing its constitutively active kinase Hepact in the wing imaginal discs. The conditional system was used to avoid lethality due to massive apoptosis that would result from constitutive Hepact expression. Increased levels of mmp1 mRNA were clearly detected by in situ hybridization in the domain of engrailed activity in the posterior half of the wing discs (Figure 3A) upon Hepact induction, while no such increase was observed in control samples. This result shows that JNK activation is sufficient to trigger mmp1 expression at the mRNA level. Second, we used an mmp1-lacZ transgenic reporter to assess transcriptional regulation of mmp1 gene directly in the tumor tissue. The reporter construct was derived from the first mmp1 intron, in which we identified three putative AP-1 binding sites (Figure 3B). In two independent transgenic lines, this mmp1 reporter perfectly recapitulated the expression pattern of endogenous mmp1 as determined by in situ experiments (Llano et al, 2000; Page-McCaw et al, 2003) (data not shown). Importantly, the activity of mmp1-lacZ in tumorigenic clones mirrored expression of the endogenous Mmp1 protein and was likewise restricted to the area of oncogenic rafact, scrib−/− clones (Figure 3C). We conclude that mmp1 is transcriptionally induced in rafact, scrib−/− (and rasV12, scrib−/−, not shown) cells and that the pattern of mmp1 gene activation is largely congruent with the clonal area. Only rarely do we observe cells within tumorigenic clones that do not show detectable levels of Mmp1 activity.
Figure 3.
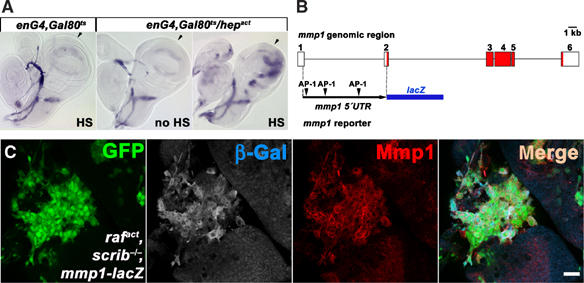
Transcriptional activation of mmp1 by JNK. (A) mmp1 mRNA is specifically upregulated in the posterior compartment of the wing disc upon activation of JNK signaling by conditional expression of Hepact in the Engrailed domain. The approximate anterior/posterior boundary is indicated by arrows, and posterior is to the right. Note that endogenous mmp1 mRNA is expressed in a crescent shape in the wing pouch and trachea. (B) Schematic representation of the mmp1-lacZ reporter construct. Open and red boxes indicate noncoding and coding exons, respectively, in the mmp1 genomic region. A 4.78-kb fragment spanning the first intron with three putative AP-1 binding sites was inserted upstream of the lacZ coding sequence. (C) The mmp1-lacZ reporter activity is upregulated in rafact, scrib−/− clones (green) as shown by anti-β-Gal staining. Note the close correlation between the reporter activity and the endogenous Mmp1 expression as monitored with antibody staining (red). The scale bar corresponds to 20 μm.
Mmp1 is required for tumor invasiveness
Next, we sought to confirm the suspected contribution of Mmp1 expression to tumor invasiveness. To this end, we suppressed Mmp function in tumorigenic clones by overexpression of Timp, a Drosophila homolog of the tissue inhibitor of metalloproteases. Timp can inhibit the activity of both Drosophila metalloproteases Mmp1 and Mmp2 as shown by in vitro studies and by functional genetic interactions in vivo (Pohar et al, 1999; Llano et al, 2000; Page-McCaw et al, 2003).
Control clones in eye/antennal discs overexpressing Timp alone had a normal appearance and were completely viable (data not shown). The effect of Timp expression on the invasiveness of rasV12, scrib−/− tumors was striking. Late third instar larvae bearing rasV12, scrib−/−, timp clones showed extensive GFP-marked clonal mass to an extent comparable or even exceeding that observed in larvae with rasV12, scrib−/− clones (compare Figure 4D and F). However, dissected brains (8 days AEL) revealed that the invasiveness of tumor cells into the VNC was greatly diminished by Timp expression (compare Figure 4E, E′ and G, G′). Eye/antennal discs of the rasV12, scrib−/−, timp genotype overgrew significantly and became even bigger than the entire tumor mass of rasV12, scrib−/− larvae (compare Figure 4A and B). Nevertheless, the structures of the optic lobes were clearly distinguishable and could be easily separated from the overgrown eye/antennal discs (compare Figure 4E′ and G′). When quantitatively analyzed, tumor cell invasiveness was significantly inhibited in the presence of ectopic Timp expression (Figure 5). These data indicate that Mmp activity is critical for cell invasiveness, but is not required for cell survival and proliferation. Importantly, the massive size of the rasV12, scrib−/−, timp tumors further supports the notion that invasiveness is not just an indirect consequence of tumor overgrowth.
Figure 4.
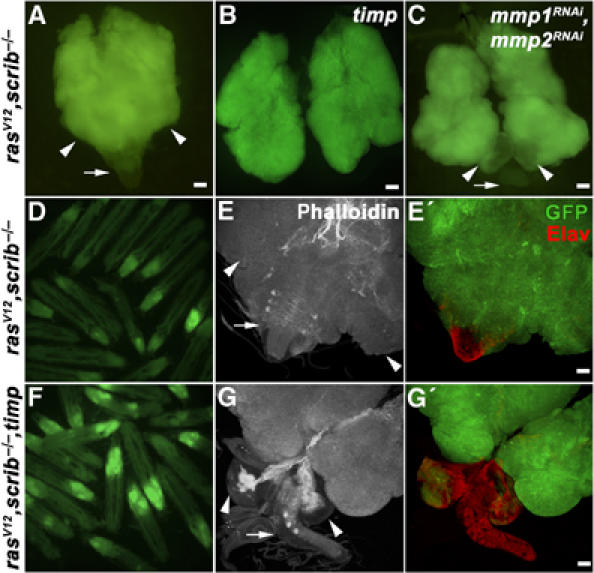
Overexpression of the Mmp inhibitor Timp or Mmp RNAi suppresses invasiveness of rasV12, scrib−/− tumors but does not interfere with tumor growth. Fluorescent microscopy of eye/antennal/brain complexes (8 days AEL) (A–C) and live 9-day-old larvae (D, F), carrying GFP-marked clones of the indicated genotypes shows that suppression of Mmp function by Timp expression or RNAi does not interfere with the growth of malignant eye disc tissue. The extent of clonal overgrowth in rasV12, scrib−/−, timp eye/antennal discs (B) and of rasV12, scrib−/− discs expressing Mmp1 and Mmp2 RNAi (C) exceeds that of the entire rasV12, scrib−/− eye/antennal/brain complex (A). (D–G′) Examination of dissected eye/antennal/brain complexes (8 days AEL) by confocal microscopy reveals no invasive clonal tissue in the VNC (arrows) in the rasV12, scrib−/−, timp genotype (G′) as opposed to rasV12, scrib−/− (E′). The clones are marked with GFP (green), the actin cytoskeleton is visualized using phalloidin staining (white) and Elav marks differentiated neurons (red). The arrows and arrowheads indicate VNC and brain optic lobes, respectively. Images C, C′ and G, G′ represent projections of multiple confocal sections. Scale bars=50 μm.
As pointed out above, Drosophila Timp inhibits both Mmp1 and Mmp2. To assess the contribution of each protease to tumor invasiveness individually, we employed an RNA interference strategy. Expressing the Mmp1RNAi and Mmp2RNAi constructs in the tracheal system or the developing dorsal body wall (under the control of breathless-Gal4 driver or pannier-Gal4, respectively) faithfully recapitulates published phenotypes of mmp1 and mmp2 null alleles such as breaks and shortening of dorsal tracheal trunks and cleft thorax phenotype, respectively (Page-McCaw et al, 2003; Supplementary Figure 7). Upon expression of either mmp1 or mmp2 RNAi in rasV12, scrib−/− clones, invasiveness was significantly suppressed, while tumors still overgrew the entire eye/antennal discs in both cases (Figure 5). When both Mmp1 and Mmp2 were knocked down simultaneously, the invasiveness of the rasV12, scrib−/− cells was reduced as efficiently as after Timp overexpression (Figures 4C and 5). We conclude that, even though only Mmp1 expression is significantly upregulated by JNK, both Mmp1 and Mmp2 contribute to the invasiveness of rasV12, scrib−/− clones. Consistent with the observation that Fos is required for Mmp1 expression in tumorigenic clones (Figure 2G), we found that FosRNAi in rasV12, scrib−/− clones phenocopied the negative effect of Mmp inhibition on invasiveness (Figure 5). These data support the central role of a JNK–AP-1–Mmp1 pathway in tumor invasiveness.
JNK activation is sufficient for Mmp1 expression, cell motility and invasiveness
The results presented so far suggest a mechanism through which scribble deficiency cooperates with oncogenic Ras activity to induce invasive tumors: Loss of scribble leads to JNK activation and ensuing Fos-dependent Mmp1 expression. Mmp1 then supports cell invasiveness, while Ras signaling is necessary to suppress the apoptotic effect of JNK activity and to stimulate cell growth. To test this model further, we asked whether artificial activation of JNK signaling in the presence of normal scrib function might be sufficient to induce Mmp1 and to confer an invasive phenotype to clones of imaginal disc cells that express RasV12.
Expression of the constitutively active JNKK (Hepact) in eye/antennal imaginal disc clones causes massive apoptosis, which can be partially suppressed by simultaneous activation of the Ras/Raf/ERK pathway (Uhlirova et al, 2005; Supplementary Figure 6E and F). Thus, compared to rasV12 or rafact alone, rasV12, hepact or rafact, hepact clones stay relatively small due to the residual apoptosis induced by the strong gain of function mutant Hepact. However, such clones express significantly more Mmp1 than rasV12 or rafact clones (compare Figure 2C and H and Supplementary Figures 5 and 6D–F). Thus, JNK is not only necessary for Mmp1 expression under conditions of scrib deficiency, but is also sufficient to induce it in the presence of wild-type scrib function. Next, we examined if JNK was also able to mediate the tumorigenic effect of scrib deficiency in a Ras gain-of-function background.
When we examined brains of larvae bearing clones of the rasV12 and rasV12, hepact genotypes, we found that some of the surviving cells of the latter genotype elongated and migrated to ectopic regions of the brain (Figure 6C). Invasiveness was not very efficient in this genotype, however, presumably because the proapoptotic function of Hepact cannot be suppressed completely by RasV12 and thus leaves few surviving cells to invade. Consistently, much more marked invasion was observed when JNK signaling was induced moderately by expression of the wild-type form of Drosophila JNKK, Hepwt (Figure 6B). As visualized by phalloidin staining (blue in Figure 6B and B′), the activation of JNK coincided with a marked build-up of F-actin structures in the invading cells. Thus, the clones have the appearance of cells that are engaged in active movement and cytoskeletal remodeling. We conclude that in conjunction with oncogenic Ras activation, JNK activity is sufficient to induce Mmp1 expression and promote cell motility and invasiveness.
Figure 6.
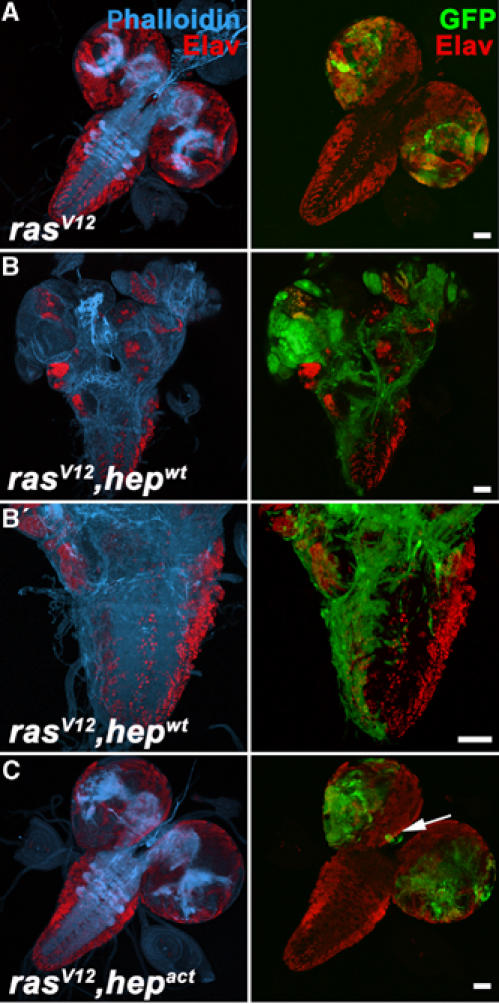
JNK activation cooperates with oncogenic Ras to cause cell invasiveness. (A–C) Brains dissected from third instar larvae, in which mutant clones of the indicated genotypes were induced using the eyeless-FLP/MARCM system were analyzed by confocal microscopy for the presence of clonal tissue (green) in the VNC. Phalloidin staining serves to visualize the actin cytoskeleton (blue). Staining for the neuronal marker Elav (red) marks terminally differentiated cells. While rasV12-expressing clones overgrow, clonal cells remain restricted to the eye/antennal discs and optic lobes (A). The brain morphology and neuronal differentiation are not affected. In contrast, rasV12, hepwt-expressing cells effectively invade the VNC (B) (26 positive cases out of 89 analyzed brains, 10 days AEL). Neuronal differentiation is disrupted and the invasive cells show an extensive F-actin network (B′). Invasiveness of rasV12, hepact cells, in contrast, is rather inefficient due to excessive apoptosis (C). However, occasionally clonal cells are found at ectopic locations (arrow) of the brain. Scale bars=50 μm.
Ras cooperates with JNK by suppressing apoptosis
Next, we wanted to dissect the contribution of oncogenic Ras to cooperative tumor induction in rasV12, scrib−/− or rasV12, hepact clones. Our model predicts that the antiapoptotic function of Ras will permit scrib-deficient cells with high levels of JNK activity to survive long enough to engage into significant cell migration and ultimately tissue invasion. To test this prediction, we asked whether selective substitution of the cell survival function of Ras by expressing the apoptosis inhibitor p35 in a JNK gain of function context might at least partially reproduce a malignant and invasive tumor phenotype. To this end, we employed wing imaginal discs and coexpressed Hepact together with p35 in a defined subset of cells using the dpp-Gal4 driver. Overexpression of p35 alone had no notable effect on cell morphology (Figure 7B). However, the combined expression of Hepact and p35 recreated aspects of malignant cell phenotypes. The hepact, p35 cells did not remain in the compact stripe of the Dpp expression domain along the anterior/posterior boundary as the control cells did. Instead, they scattered into the adjacent area of the wing disc. Furthermore, hepact, p35 cells displayed extensive actin reorganization and extended actin rich filopodia (Figure 7A and data not shown) characteristic of invasive cells. They also expressed elevated levels of Mmp1. All these phenotypes are associated with malignancy, indicating that our model captures integral aspects of cooperative cell transformation.
Figure 7.
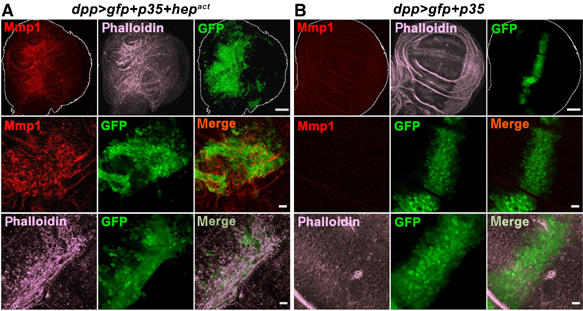
JNK-mediated cell migration becomes evident when apoptosis is blocked. p35 and GFP were either expressed together with Hepact (A) or alone (B) under the control of the dpp-Gal4 driver in the wing imaginal disc (green). Third instar wing discs were stained with Mmp1 antibody (red) and phalloidin to visualize F-actin filaments (magenta) and analyzed by confocal microscopy. Except for the images in the top row, which are projections of multiple sections, all other panels show single confocal sections. Scale bars=50 μm (top row) and 10 μm (middle and bottom rows), respectively.
Discussion
Genetic experiments using Drosophila imaginal discs have yielded valuable information about the interplay between mechanisms that regulate cell proliferation, apoptosis and motility in the genesis of malignant tumors. In the present study, we begun to dissect the downstream signaling events that give rise to invasive malignant tumors when oncogenic alleles of Ras pathway components cooperate with loss of the epithelial polarity gene scrib in clones of eye imaginal disc tissue. By exploring and then attempting to reproduce cellular effects elicited by either of these two oncogenic genotypes, we pinpointed essential mechanisms contributing to tumorigenesis. The data emerging from these experiments suggest that the activation of JNK in cells lacking scribble and the suppression of apoptosis by oncogenic Ras signaling are central events in the establishment of the transformed invasive phenotype (Figure 8).
Figure 8.
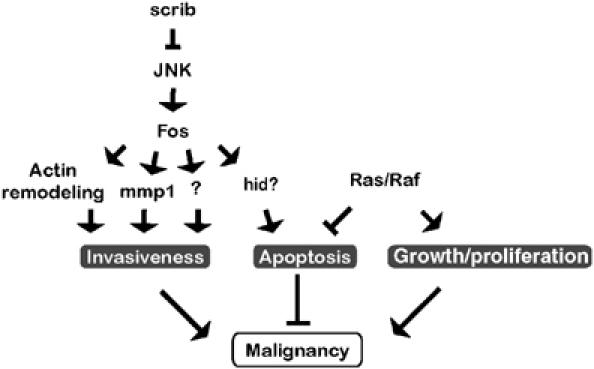
Model of oncogene cooperation in malignant rasV12, scrib−/− tumors. The functional readout of JNK activation is determined by the status of the cooperating Ras signaling pathway. Loss of the apical–basal polarity in clones of imaginal epithelium lacking scribble function causes elimination of mutant cells through JNK-dependent apoptosis, possibly involving the transcriptional activation of hid (X Luo, D Bohmann and H Jasper, unpublished observation). Simultaneously, JNK stimulates actin cytoskeleton rearrangements. A third effect of JNK is the destabilization of the tissue environment and the basement membrane by mmp1 activation. Other, yet to be identified downstream targets of JNK may contribute to the development of malignancy. Survival signals delivered by activation of Ras/Raf/ERK signaling in scribble clones counteract apoptosis, enabling other JNK effectors to promote cell mobilization and invasion to ectopic sites. Multiple JNK functions require the Fos transcription factor.
At this point, it is not clear what causes the activation of JNK in scribble-deficient cells, and further experiments are required to elucidate this question. One credible speculation would be the involvement of cell competition in which faster growing wild-type cells eliminate slower growing scrib−/− neighbors from the eye imaginal disc epithelium via inducing JNK-dependent apoptosis. The loss of epithelial polarity would further sensitize the scrib−/− cells to cell competition (Thompson et al, 2005). As predicted by this hypothesis, clonal growth is restored when apoptosis of scrib−/− clones is blocked by expression of the caspase inhibitor p35 (Brumby and Richardson, 2003) or DIAP (this study). Similarly, it is conceivable that cell competition caused by the difference in proliferation rate coupled with abnormal cell–cell interactions would underlie the observation of occasional areas of JNK activity (evidenced by mmp1 gene induction, this study and puc-lacZ expression (data not shown; Igaki et al, 2006)) in imaginal discs harboring clones of over proliferating RasV12 cells. The interaction between clones of transformed cells and their normal cell environment is probably of key relevance in malignant tumor progression and the model system used in this study offers a great opportunity to explore such relationships.
In scribble-deficient cells, active JNK signaling stimulates both invasiveness and cell death. Interestingly, either response requires the AP-1 transcription factor Fos. RNAi-mediated knockdown of Fos function inhibits both the invasiveness of rasV12, scrib−/− tumors as well as apoptosis of scrib-deficient cells. Fos-mediated control of apoptosis might be achieved by transcriptional activation of the hid gene (X Luo, H Jasper and D Bohmann, unpublished data).
Because of the apoptotic function of JNK signaling, its stimulatory effect on cell mobilization only becomes evident, and can be functionally dissected, when the proapoptotic effect of the pathway is repressed. This occurs under conditions of increased Ras signaling, or experimentally when apoptosis is inhibited by the expression of p35. Figure 8 depicts a model of the signaling interactions between Ras gain-of-function and scribble loss-of-function mutations that would explain the cooperative induction of tumor invasiveness and malignancy.
A plausible transcriptional target of JNK-Fos signaling with relevance to cell invasiveness in the tumor model under study here is the mmp1 gene. We show that JNK and Fos are required for the induction of Mmp1 in malignant tumor tissue. It has to be stressed that this JNK-induced Mmp1 expression is essential for the establishment of the invasive phenotype, as shown by using Timp expression or Mmp1 RNAi, but that Mmp1 activation alone is most likely not sufficient for this effect. We find that overexpression of Mmp1 in a raf gain-of-function, but scrib wild-type background does not give rise to an invasive phenotype (data not shown). It appears that JNK activation causes multiple cellular changes that are important for cell mobilization. In addition to the loss of basement membrane integrity that correlates with elevated Mmp1 expression, changes in the actin cytoskeleton are observed as a consequence of JNK activation in rasV12, scrib−/−-transformed cells. This cytoskeletal rearrangement phenotype is not due to Mmp1 induction, since blocking of Mmp activity by Timp or RNAi does not influence it, a conclusion that is also supported by the observation that in the embryo JNK induces actin reorganization without turning on the mmp1 gene. Interestingly, the only other mmp gene in Drosophila, mmp2, has recently been implicated in Src-induced and JNK-mediated mobilization of wing imaginal disc cells (Vidal et al, 2006). Our analysis shows clearly that mmp2 is not transcriptionally induced by JNK. However, the RNAi experiments indicate that both Mmp1 and Mmp2 act in conjunction to mediate cell invasiveness. This might occur by a threshold effect where the combined activity of Mmp1 and Mmp2 has to exceed a certain level required for cell invasion. Alternatively, the two Drosophila Mmps might have nonoverlapping substrate specificities, which are both essential for the tumor cell to escape from its tissue of origin. Strikingly, under conditions of Timp expression and Mmp downregulation by RNAi growth of the tumor tissue was not reduced, and even seemed to be more pronounced compared to rasV12, scrib−/−. This accumulation of clonal tissue could be explained simply by the fact that proliferation and growth of rasV12, scrib−/−, timp and rasV12, scrib−/−, mmp1RNAi, mmp2RNAi clones is similar to rasV12, scrib−/−; however, their reduced ability to degrade ECM, spread and penetrate new organs forces them to expand in volume.
The study presented here illustrates how the derailing of several signaling systems is required to create a malignant cancer phenotype in which apoptosis is suppressed, epithelial integrity breaks down, tissue barriers are breached and cells proliferate and mobilize. Using the eye imaginal disc model, it is possible to deconstruct these complex signaling mechanisms and their interplay from the molecular to the tissue level. Future studies that can take full advantage of the experimental power of Drosophila genetics may improve understanding and ultimately the management of pathological cell movements.
Materials and methods
Fly stocks
For generation of eye mosaics, the following mutant and transgenic fly strains were used:
(1) ey-FLP1; Act>y+>Gal4, UAS-GFP; FRT82B, Tub-Gal80, (2) w; UAS-rasV12; FRT82B scrib1/TM6B, (3) w; UAS-rasV12; FRT82B (4) w; FRT82B UAS-rafact, (5) w; FRT82B scrib1/TM6B, (6) w; FRT82B scrib1UAS-rafact/TM6B, (7) w; FRT82B, (8) w; UAS-hepactUAS-rasV12; FRT82B, (9) w; UAS-rasV12; FRT82B scrib1UAS-bskDN/TM6B, (10) w; FRT82B scrib1UAS-bskDN/TM6B, (11) w; UAS-rasV12; FRT82B scrib1UAS-timp/TM6B, (12) w; UAS-fosRNAi47; FRT82B scrib1UAS-fosRNAi35/19, (13) w; FRT82B scrib1UAS-diap, (14) w; UAS-rasV12UAS-mmp1RNAi; FRT82B scrib1UAS-mmp1RNAi, (15) w; UAS-rasV12UAS-mmp1RNAi; FRT82B scrib1UAS-mmp2RNAi, (16) w; UAS-rasV12; FRT82B scrib1UAS-fosRNAi35/19, (17) w; UAS-rasV12; FRT82B scrib1UAS-mmp2RNAi, (18) w; UAS-rasV12; FRT82B scrib1UAS-puc, (19) w; FRT82B scrib1UAS-puc, (20) w; UAS-hepwtUAS-rasV12; FRT82B. To conduct experiments in the wing imaginal discs we used the following fly strains: (1) w; dpp-GAL4, UAS-GFP/TM6, (2) w; UAS-hepact; UAS-p35 and (3) w; UAS-p35 as a control.
UAS-timp transgenic flies were a gift from A Page-McCaw (Page-McCaw et al, 2003). UAS-fosRNAi47 and UAS-fosRNAi35/19 transgenic flies were a gift from C Yanicostas (Hyun et al, 2006).
Conditional activation of Hepact was achieved by using the Gal4/Gal80ts system (McGuire et al, 2003). enGal4, UAS-gfp; tub-Gal80ts females were crossed to UAS-hepact or w1118 males and kept at 20°C. Gal80ts was inactivated by incubation at 37°C for 60 min. Larvae were kept at 25°C for 3 h before dissection.
To generate UAS-mmp1RNAi and UAS-mmp2RNAi flies, cDNA fragments from the mmp1 and mmp2 genes were amplified using the following primer sets: mmp1, 5′-TGAGTGCCATCGAGGAGTTCCA-3′ and 5′-ATGCTCGCTCTCCACGAACTTG-3′; mmp2, 5′-TGCAGACCGCCCTGGACGT-3′ and 5′-CATACATGAGTCCGGCTTGGG-3′. The fragments were cloned as inverted repeats into the pWIZ vector (Lee and Carthew, 2003) and transgenic lines were established by standard P-element germline transformation.
Mosaic analyses
The eyeless-FLP/MARCM system (Lee and Luo, 2001) was utilized to induce positively marked GFP clones in eye/antennal discs. In all experiments, ey-FLP1; Act>y+>Gal4, UAS-GFP; FRT82B, Tub-Gal80 females were crossed with males of the desired genotype.
mmp1-lacZ reporter construct
To generate the mmp1-lacZ reporter construct, a 4.78-kb mmp1 genomic DNA fragment was amplified with the following PCR primers: 5′-ACCACCAAGATCTAATCGCCATCG-3′ and 5′-ATCTCGAGTGGTTCCACTTGCCGCTGGCA-3′ containing BglII and XhoI restriction sites underlined, respectively, and inserted into the pPelican transformation vector that encompasses the lacZ coding sequence without a minimal promoter (Barolo et al, 2000). Three putative AP-1 binding sites map to positions −1730, −3534 and −4581 upstream of the predicted Mmp1 translation start. Transgenic flies were generated with standard P-element-mediated transformation.
Immunohistochemistry
Imaginal discs and brains were fixed and stained according to standard protocols. The following antibodies and their dilutions were used: rabbit anti-active Caspase-3 (1:200; Cell Signaling Technology); mouse anti-Mmp1 (1:300; Page-McCaw et al, 2003); rabbit anti-β-Gal (1:500; Cappel); rabbit anti-Laminin A (1:1000; a gift from S Baumgartner); rat anti-Elav (1:300; Developmental Studies Hybridoma Bank); rhodamine (TRITC) or CY5-conjugated secondary antibodies (Jackson Immunoresearch) were used. Alexa 633-labeled phalloidin was added to the penultimate wash and incubated with the tissue for 40 min. Samples were analyzed by laser confocal microscopy (Leica SP2). Images represent either single confocal sections or projections of several sections as indicated in the figure legends.
In situ hybridization
mmp1 mRNA was detected in third instar wing imaginal discs with a digoxigenin-labeled antisense RNA probe as described previously (Tautz and Pfeifle, 1989).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Material
Acknowledgments
We are grateful to A Page-McCaw, H Richardson, R Pagliarini, T Xu, C Yanicostas, S Baumgartner, the Bloomington Stock Center and the Developmental Studies Hybridoma Bank for fly stocks and antibodies. We also thank C Sommers for technical support, H Jasper for comments and M Jindra for critical reading and suggestions to this manuscript. This work was supported by NIH Grant No. 1RO1EY014624 to DB.
References
- Adachi-Yamada T, Fujimura-Kamada K, Nishida Y, Matsumoto K (1999) Distortion of proximodistal information causes JNK-dependent apoptosis in Drosophila wing. Nature 400: 166–169 [DOI] [PubMed] [Google Scholar]
- Agnes F, Suzanne M, Noselli S (1999) The Drosophila JNK pathway controls the morphogenesis of imaginal discs during metamorphosis. Development 126: 5453–5462 [DOI] [PubMed] [Google Scholar]
- Barolo S, Carver LA, Posakony JW (2000) GFP and beta-galactosidase transformation vectors for promoter/enhancer analysis in Drosophila. Biotechniques 29: 726, 728, 730, 732 [DOI] [PubMed] [Google Scholar]
- Behrens A, Sibilia M, Wagner EF (1999) Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet 21: 326–329 [DOI] [PubMed] [Google Scholar]
- Bosch M, Serras F, Martin-Blanco E, Baguna J (2005) JNK signaling pathway required for wound healing in regenerating Drosophila wing imaginal discs. Dev Biol 280: 73–86 [DOI] [PubMed] [Google Scholar]
- Boutros M, Agaisse H, Perrimon N (2002) Sequential activation of signaling pathways during innate immune responses in Drosophila. Dev Cell 3: 711–722 [DOI] [PubMed] [Google Scholar]
- Brumby AM, Richardson HE (2003) scribble mutants cooperate with oncogenic Ras or Notch to cause neoplastic overgrowth in Drosophila. EMBO J 22: 5769–5779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumby AM, Richardson HE (2005) Using Drosophila melanogaster to map human cancer pathways. Nat Rev Cancer 5: 626–639 [DOI] [PubMed] [Google Scholar]
- Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T (2003) Regulation of matrix metalloproteinases: an overview. Mol Cell Biochem 253: 269–285 [DOI] [PubMed] [Google Scholar]
- Ferres-Marco D, Gutierrez-Garcia I, Vallejo DM, Bolivar J, Gutierrez-Avino FJ, Dominguez M (2006) Epigenetic silencers and Notch collaborate to promote malignant tumours by Rb silencing. Nature 439: 430–436 [DOI] [PubMed] [Google Scholar]
- Galko MJ, Krasnow MA (2004) Cellular and genetic analysis of wound healing in Drosophila larvae. PLoS Biol 2: E239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glise B, Noselli S (1997) Coupling of Jun amino-terminal kinase and Decapentaplegic signaling pathways in Drosophila morphogenesis. Genes Dev 11: 1738–1747 [DOI] [PubMed] [Google Scholar]
- Hagemann T, Wilson J, Kulbe H, Li NF, Leinster DA, Charles K, Klemm F, Pukrop T, Binder C, Balkwill FR (2005) Macrophages induce invasiveness of epithelial cancer cells via NF-kappa B and JNK. J Immunol 175: 1197–1205 [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA (2000) The hallmarks of cancer. Cell 100: 57–70 [DOI] [PubMed] [Google Scholar]
- Homsy JG, Jasper H, Peralta XG, Wu H, Kiehart DP, Bohmann D (2006) JNK signaling coordinates integrin and actin functions during Drosophila embryogenesis. Dev Dyn 235: 427–434 [DOI] [PubMed] [Google Scholar]
- Hou XS, Goldstein ES, Perrimon N (1997) Drosophila Jun relays the Jun amino-terminal kinase signal transduction pathway to the Decapentaplegic signal transduction pathway in regulating epithelial cell sheet movement. Genes Dev 11: 1728–1737 [DOI] [PubMed] [Google Scholar]
- Huang C, Rajfur Z, Borchers C, Schaller MD, Jacobson K (2003) JNK phosphorylates paxillin and regulates cell migration. Nature 424: 219–223 [DOI] [PubMed] [Google Scholar]
- Hyun J, Becam I, Yanicostas C, Bohmann D (2006) Control of G2/M transition by Drosophila Fos. Mol Cell Biol (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T, Kanda H, Yamamoto-Goto Y, Kanuka H, Kuranaga E, Aigaki T, Miura M (2002) Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J 21: 3009–3018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igaki T, Pagliarini RA, Xu T (2006) Loss of cell polarity drives tumor growth and invasion through JNK activation in Drosophila. Curr Biol 16: 1139–1146 [DOI] [PubMed] [Google Scholar]
- Jasper H, Benes V, Schwager C, Sauer S, Clauder-Munster S, Ansorge W, Bohmann D (2001) The genomic response of the Drosophila embryo to JNK signaling. Dev Cell 1: 579–586 [DOI] [PubMed] [Google Scholar]
- Kockel L, Zeitlinger J, Staszewski LM, Mlodzik M, Bohmann D (1997) Jun in Drosophila development: redundant and nonredundant functions and regulation by two MAPK signal transduction pathways. Genes Dev 11: 1748–1758 [DOI] [PubMed] [Google Scholar]
- Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA (1999) The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron 22: 667–676 [DOI] [PubMed] [Google Scholar]
- Kwei KA, Finch JS, Ranger-Moore J, Bowden GT (2006) The role of Rac1 in maintaining malignant phenotype of mouse skin tumor cells. Cancer Lett 231: 326–338 [DOI] [PubMed] [Google Scholar]
- Land H, Parada LF, Weinberg RA (1983) Tumorigenic conversion of primary embryo fibroblasts requires at least two cooperating oncogenes. Nature 304: 596–602 [DOI] [PubMed] [Google Scholar]
- Lee T, Luo L (2001) Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci 24: 251–254 [DOI] [PubMed] [Google Scholar]
- Lee YS, Carthew RW (2003) Making a better RNAi vector for Drosophila: use of intron spacers. Methods 30: 322–329 [DOI] [PubMed] [Google Scholar]
- Llano E, Adam G, Pendas AM, Quesada V, Sanchez LM, Santamaria I, Noselli S, Lopez-Otin C (2002) Structural and enzymatic characterization of Drosophila Dm2-MMP, a membrane-bound matrix metalloproteinase with tissue-specific expression. J Biol Chem 277: 23321–23329 [DOI] [PubMed] [Google Scholar]
- Llano E, Pendas AM, Aza-Blanc P, Kornberg TB, Lopez-Otin C (2000) Dm1-MMP, a matrix metalloproteinase from Drosophila with a potential role in extracellular matrix remodeling during neural development. J Biol Chem 275: 35978–35985 [DOI] [PubMed] [Google Scholar]
- Martin-Blanco E, Gampel A, Ring J, Virdee K, Kirov N, Tolkovsky AM, Martinez-Arias A (1998) puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev 12: 557–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Blanco E, Pastor-Pareja JC, Garcia-Bellido A (2000) JNK and decapentaplegic signaling control adhesiveness and cytoskeleton dynamics during thorax closure in Drosophila. Proc Natl Acad Sci USA 97: 7888–7893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattila J, Omelyanchuk L, Kyttala S, Turunen H, Nokkala S (2005) Role of Jun N-terminal Kinase (JNK) signaling in the wound healing and regeneration of a Drosophila melanogaster wing imaginal disc. Int J Dev Biol 49: 391–399 [DOI] [PubMed] [Google Scholar]
- McCawley LJ, Matrisian LM (2000) Matrix metalloproteinases: multifunctional contributors to tumor progression. Mol Med Today 6: 149–156 [DOI] [PubMed] [Google Scholar]
- McEwen DG, Peifer M (2005) Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development 132: 3935–3946 [DOI] [PubMed] [Google Scholar]
- McGuire SE, Le PT, Osborn AJ, Matsumoto K, Davis RL (2003) Spatiotemporal rescue of memory dysfunction in Drosophila. Science 302: 1765–1768 [DOI] [PubMed] [Google Scholar]
- Moreno E, Yan M, Basler K (2002) Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr Biol 12: 1263–1268 [DOI] [PubMed] [Google Scholar]
- Nelson AR, Fingleton B, Rothenberg ML, Matrisian LM (2000) Matrix metalloproteinases: biologic activity and clinical implications. J Clin Oncol 18: 1135–1149 [DOI] [PubMed] [Google Scholar]
- Page-McCaw A, Serano J, Sante JM, Rubin GM (2003) Drosophila matrix metalloproteinases are required for tissue remodeling, but not embryonic development. Dev Cell 4: 95–106 [DOI] [PubMed] [Google Scholar]
- Pagliarini RA, Xu T (2003) A genetic screen in Drosophila for metastatic behavior. Science 302: 1227–1231 [DOI] [PubMed] [Google Scholar]
- Pastor-Pareja JC, Grawe F, Martin-Blanco E, Garcia-Bellido A (2004) Invasive cell behavior during Drosophila imaginal disc eversion is mediated by the JNK signaling cascade. Dev Cell 7: 387–399 [DOI] [PubMed] [Google Scholar]
- Pohar N, Godenschwege TA, Buchner E (1999) Invertebrate tissue inhibitor of metalloproteinase: structure and nested gene organization within the synapsin locus is conserved from Drosophila to human. Genomics 57: 293–296 [DOI] [PubMed] [Google Scholar]
- Rämet M, Lanot R, Zachary D, Manfruelli P (2002) JNK signaling pathway is required for efficient wound healing in Drosophila. Dev Biol 241: 145–156 [DOI] [PubMed] [Google Scholar]
- Riesgo-Escovar JR, Jenni M, Fritz A, Hafen E (1996) The Drosophila Jun-N-terminal kinase is required for cell morphogenesis but not for DJun-dependent cell fate specification in the eye. Genes Dev 10: 2759–2768 [DOI] [PubMed] [Google Scholar]
- Ryoo HD, Gorenc T, Steller H (2004) Apoptotic cells can induce compensatory cell proliferation through the JNK and the Wingless signaling pathways. Dev Cell 7: 491–501 [DOI] [PubMed] [Google Scholar]
- Sluss HK, Han Z, Barrett T, Davis RJ, Ip YT (1996) A JNK signal transduction pathway that mediates morphogenesis and an immune response in Drosophila. Genes Dev 10: 2745–2758 [DOI] [PubMed] [Google Scholar]
- Stetler-Stevenson WG, Aznavoorian S, Liotta LA (1993) Tumor cell interactions with the extracellular matrix during invasion and metastasis. Annu Rev Cell Biol 9: 541–573 [DOI] [PubMed] [Google Scholar]
- Tautz D, Pfeifle C (1989) A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals translational control of the segmentation gene hunchback. Chromosoma 98: 81–85 [DOI] [PubMed] [Google Scholar]
- Thompson BJ, Mathieu J, Sung HH, Loeser E, Rorth P, Cohen SM (2005) Tumor suppressor properties of the ESCRT-II complex component Vps25 in Drosophila. Dev Cell 9: 711–720 [DOI] [PubMed] [Google Scholar]
- Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288: 870–874 [DOI] [PubMed] [Google Scholar]
- Uhlirova M, Jasper H, Bohmann D (2005) Non-cell-autonomous induction of tissue overgrowth by JNK/Ras cooperation in a Drosophila tumor model. Proc Natl Acad Sci USA 102: 13123–13128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventura JJ, Kennedy NJ, Flavell RA, Davis RJ (2004) JNK regulates autocrine expression of TGF-beta1. Mol Cell 15: 269–278 [DOI] [PubMed] [Google Scholar]
- Vidal M, Larson DE, Cagan RL (2006) Csk-deficient boundary cells are eliminated from normal Drosophila epithelia by exclusion, migration, and apoptosis. Dev Cell 10: 33–44 [DOI] [PubMed] [Google Scholar]
- Vu TH, Werb Z (2000) Matrix metalloproteinases: effectors of development and normal physiology. Genes Dev 14: 2123–2133 [DOI] [PubMed] [Google Scholar]
- Westermarck J, Kahari VM (1999) Regulation of matrix metalloproteinase expression in tumor invasion. FASEB J 13: 781–792 [PubMed] [Google Scholar]
- Weston CR, Wong A, Hall JP, Goad ME, Flavell RA, Davis RJ (2003) JNK initiates a cytokine cascade that causes Pax2 expression and closure of the optic fissure. Genes Dev 17: 1271–1280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston CR, Wong A, Hall JP, Goad ME, Flavell RA, Davis RJ (2004) The c-Jun NH2-terminal kinase is essential for epidermal growth factor expression during epidermal morphogenesis. Proc Natl Acad Sci USA 101: 14114–14119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Y, Karin M (2004) The control of cell motility and epithelial morphogenesis by Jun kinases. Trends Cell Biol 14: 94–101 [DOI] [PubMed] [Google Scholar]
- Xia Y, Makris C, Su B, Li E, Yang J, Nemerow GR, Karin M (2000) MEK kinase 1 is critically required for c-Jun N-terminal kinase activation by proinflammatory stimuli and growth factor-induced cell migration. Proc Natl Acad Sci USA 97: 5243–5248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yujiri T, Ware M, Widmann C, Oyer R, Russell D, Chan E, Zaitsu Y, Clarke P, Tyler K, Oka Y, Fanger GR, Henson P, Johnson GL (2000) MEK kinase 1 gene disruption alters cell migration and c-Jun NH2-terminal kinase regulation but does not cause a measurable defect in NF-kappa B activation. Proc Natl Acad Sci USA 97: 7272–7277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlinger J, Bohmann D (1999) Thorax closure in Drosophila: involvement of Fos and the JNK pathway. Development 126: 3947–3956 [DOI] [PubMed] [Google Scholar]
- Zhang L, Wang W, Hayashi Y, Jester JV, Birk DE, Gao M, Liu CY, Kao WW, Karin M, Xia Y (2003) A role for MEK kinase 1 in TGF-beta/activin-induced epithelium movement and embryonic eyelid closure. EMBO J 22: 4443–4454 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Figure 6
Supplementary Figure 7
Supplementary Material