

Abstract
Homotypic yeast vacuole fusion occurs in three stages: (i) priming reactions, which are independent of vacuole clustering, (ii) docking, in which vacuoles cluster and accumulate fusion proteins and fusion regulatory lipids at a ring-shaped microdomain surrounding the apposed membranes of each docked vacuole, where fusion will occur, and (iii) bilayer fusion/compartment mixing. These stages require vacuolar SNAREs, SNARE-chaperones, GTPases, effector complexes, and chemically minor but functionally important lipids. For each, we have developed specific ligands that block fusion and conditions that reverse each block. Using them, we test whether docking entails a linearly ordered series of catalytic events, marked by sequential acquisition of resistance to inhibitors, or whether docking subreactions are cooperative and/or reversible. We find that each fusion protein and regulatory lipid is needed throughout docking, indicative of a reversible or highly cooperative assembly of the fusion-competent vertex ring. In accord with this cooperativity, vertices enriched in one fusion catalyst are enriched in others. Docked vacuoles finally assemble SNARE complexes, yet still require physiological temperature and lipid rearrangements to complete fusion.
Keywords: membrane fusion, organelle docking, yeast vacuole
Introduction
Selective membrane docking and fusion are required for subcellular compartmentation. Docking begins with tethering, mediated by Rab-family GTPases (Pfeffer, 2005). Rab/Ypt GTPases act through ‘effector' proteins that bind to their GTP form and mediate their downstream effects. Docking culminates in the assembly of fusion-competent membrane microdomains and SNARE pairing in trans.
The homotypic fusion of yeast vacuoles is a technically accessible model for studying membrane fusion mechanisms (Wickner, 2002). Vacuoles undergo constant fusion and fission, and any diminution of fusion in the face of continued fission results in an in vivo phenotype of fragmented vacuoles (Wada et al, 1992). Purified vacuoles that are incubated with ATP will also fuse, and this in vitro fusion can be measured by a colorimetric assay (Haas et al, 1994). The first step of fusion, termed ‘priming', does not require contact among vacuoles. Priming includes the disassembly of complexes of SNAREs (Ungermann et al, 1998) in ‘cis' (on the same membrane) by Sec18p (NSF) and its cochaperone Sec17p (α-SNAP) as well as the production of diacylglycerol by phospholipase C (Jun et al, 2004). Vacuole tethering requires the Ypt7p GTPase (Mayer and Wickner, 1997) and its effector complex, termed HOPS (Stroupe et al, 2006). HOPS is a heterohexameric complex of Vps 11, 16, 18, 33, 39, and 41 subunits, which has nucleotide exchange activity on Ypt7p (Wurmser et al, 2000) and binds selectively to GTP:Ypt7p (Seals et al, 2000). HOPS has direct affinity (Stroupe et al, 2006) for phosphoinositides and for Vam7p, a vacuolar SNARE that has no hydrophobic membrane anchor. Ypt7p is required for the stable association of Vam7p with the vacuole (Ungermann et al, 2000) and for Vam7p assembly into SNARE complexes (Collins et al, 2005). HOPS association with the resulting assembled SNARE complexes (Collins et al, 2005) may be mediated through its Vps33p subunit, as this is a member of the SM family of SNARE-binding proteins. In addition to these proteins, and others, fusion requires specific minor lipids of the vacuole, termed ‘regulatory lipids' (Fratti et al, 2004). These include ergosterol, which may regulate membrane microdomain fluidity and mobility (Valdez-Taubas and Pelham, 2003), diacylglycerol, which can facilitate the formation of nonbilayer lipidic fusion intermediates (Siegel et al, 1989), and 3- and 4- phosphoinositides, which can contribute to microdomain formation, serve as precursor for the generation of diacylglycerol, and serve as a binding platform for proteins.
Vacuole docking entails remarkable spatial rearrangements (Wang et al, 2002). Although vacuoles must initially touch at a point, their membranes are rapidly drawn against each other until each pair of docked vacuoles has a contact region, termed the ‘boundary domain', which resembles two closely apposed flat discs. The ‘outer membranes' of docked vacuole pairs, which are not in contact, meet the boundary membrane at a ring-shaped microdomain, which is termed the ‘vertex ring'. Fusion occurs around this vertex ring microdomain, joining the outer membranes of the two docked vacuoles and joining the boundary membranes as well. Strikingly, the product of fusion is not only a larger, fused vacuole but also, in its lumen, a vesicle that forms from the two, formerly boundary, membranes during the fusion event. Proteins (Ypt7p, HOPS, each of the vacuolar SNAREs, and others) and the regulatory lipids (ergosterol, diacylglycerol, and 3- and 4- phosphoinositides) become enriched at the vertex ring before fusion. The enrichment of proteins and lipids into the vertex ring has been studied by quantitative ratiometric fluorescence microscopy, where vacuolar proteins were visualized by attached GFP-tags and lipids by fluorophore-tagged lipid-binding probes (Wang et al, 2002, 2003; Fratti et al, 2004). Proteins depend on each other for their vertex enrichment, and each regulatory lipid depends on the others for enrichment at the vertex as well. Proteins also depend on the regulatory lipids for vertex enrichment and, most strikingly, lipid enrichment at the vertex depends on the proteins (Fratti et al, 2004). Simple models of the vertex ring as a primarily lipidic phase, which attract certain proteins, or as a proteinaceous assembly, which attracts specific lipids, are not sufficient to explain these observations.
Vacuole docking might proceed as a series of unidirectional subreactions that must be completed in a defined order to permit fusion. Alternatively, the subreactions may be interdependent, or reversible. To distinguish between these two models, we have developed a set of reversible inhibitory ligands, each with specific affinity for a protein or regulatory lipid which is required for vacuole fusion. We have now characterized these inhibitors and systematically explored whether their inhibition can be passed in a defined order during the period of reversible block by another inhibitor. We find that all the fusion lipids and proteins are required throughout docking, leading to the reversible and interdependent enrichment of select lipids and proteins at fusion-competent vertex microdomains.
Results
To assay fusion in vitro, vacuoles are purified from two yeast strains. One strain lacks the major proteases that activate vacuolar proenzymes, and thus only bears the catalytically inactive proenzyme form of Pho8p (alkaline phosphatase). The other strain has normal vacuole proteases, but bears a deletion of the PHO8 gene. While neither population of vacuoles has Pho8p activity, fusion allows the proteases from some vacuoles to gain access to the proPho8p in others and cleave them to the catalytically active Pho8p, which can be measured by its ability to hydrolyze colorless p-nitrophenylphosphate to yellow nitrophenolate. This assay has been characterized (Wickner, 2002), exploiting inhibition by ligands to specific lipids and proteins (Mayer et al, 1996). Many of these inhibitors can be reversed, but not all. SNAREs are of central importance for docking and fusion, and antibodies to the SNAREs Vam3p, Vti1p, and Nyv1p appear to be irreversible inhibitors.
We have therefore developed a SNARE-targeted reversible reaction inhibitor, comprised of the mixed soluble domains of the three vacuolar Q-SNAREs: Vti1p, Vam3p, and Vam7p (the latter has no integral membrane domain and is inherently water-soluble). The soluble domains (termed herein sSNAREs) of these three Q-SNAREs were expressed as epitope-tagged recombinant proteins in Escherichia coli and purified by affinity isolation (Figure 1A). While none of the individual sSNARE domains inhibited vacuole fusion, and pairs of these domains gave only modest inhibition, a mixture of the three sQ-SNAREs, termed 3sQ, blocked fusion completely (Figure 1B). We propose, as have others (Brandhorst et al, 2006), that 3sQ inhibits by pairing nonproductively with available R-SNAREs. In accord with this concept, the 3sQ block is prevented by the prior addition of soluble domain from the R-SNARE Nyv1p to the initial 3sQ mixture (Figure 1C). The specificity of 3sQ inhibition is underscored by the fact that vacuoles from strains that overexpress Nyv1p become relatively resistant to fusion inhibition by either antibody to Nyv1p or by 3sQ (Figure 1D). Inhibition of fusion by 3sQ is effectively reversed by added Sec18p (Figure 1E); the sensitivity of this reversal to antibody to Sec17p (see Figure 4D3 below) indicates that it reflects the normal capacity of Sec18p for Sec17p-dependent disassembly of SNARE complexes. Unexpectedly, we also find that the inhibition of fusion by 3sQ is reversed effectively by PI(4,5)P2, although little reversal is seen with phosphatidylinositol 3-phosphate (PI(3)P) (Figure 1E). Since the Vam7p component of 3sQ has direct affinity for only PI(3)P through its PX domain (Cheever et al, 2001), this reversal may reflect the essential role of PI(4,5)P2 in enriching fusion components at the vertex ring (Fratti et al, 2004). In any case, the reversal of 3sQ inhibition by PI(4,5)P2 still requires Sec17p and Sec18p action, as shown below (Figure 4D2); we do not know how PI(4,5)P2 activates the ability of Sec18p to disassemble inhibitory complexes of 3sQ.
Figure 1.
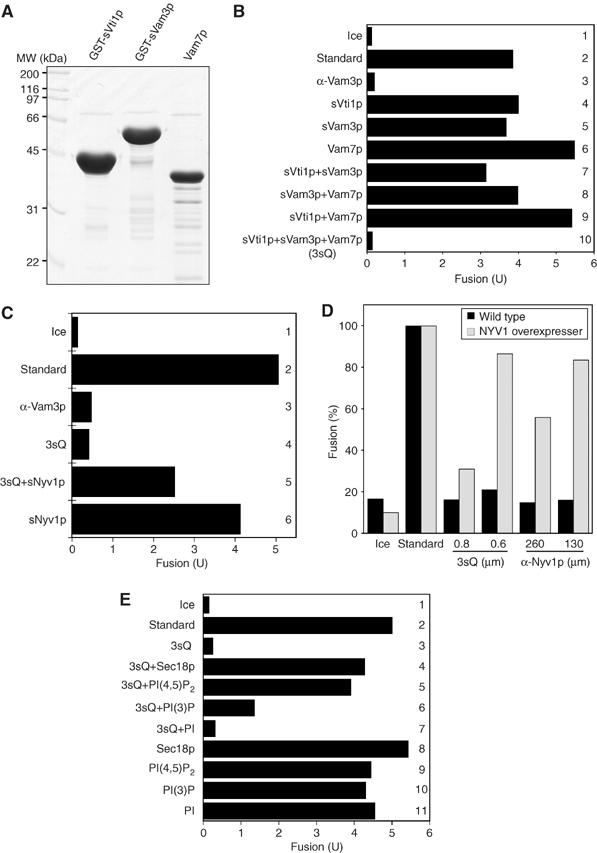
Characterization of a mixture of 3sQ-SNAREs. (A) A Coomassie-stained gel of the purified soluble domains of three Q-SNAREs. (B) Inhibition by 3sQ. Standard fusion reactions (see Materials and methods) were incubated for 90 min on ice (bar 1) or at 27°C in the absence (bar 2) or presence of 0.2 μM anti-Vam3p (bar 3), 1.1 μM of an individual sSNARE domains (bars 4–6), pairs of sSNARE domains (bars 7–9), or a mixture of the three sSNARE domains (bar 10). (C) Inhibition by 3sQ is blocked by an sR-SNARE. Fusion reactions were performed on ice (bar 1) or at 27°C without further additions (bar 2) or with anti-Vam3p (0.2 μM), 3sQ (1.1 μM), and GST-sNyv1 (2 μM) as indicated (bars 3–6). (D) Nyv1p is the target of 3sQ inhibition. Vacuoles purified from wild-type yeast or cells overexpressing Nyv1p under the ADH promoter were incubated for 90 min with the indicated amounts of 3sQ or antibody to Nyv1p. (E) Relief of 3sQ inhibition. Standard fusion reactions were incubated on ice (bar 1) or at 27°C without further addition (bar 2) or with 3sQ (1.1 μM), recombinant Sec18p (175 nM), and phosphoinositides (300 μM) as indicated.
Figure 4.
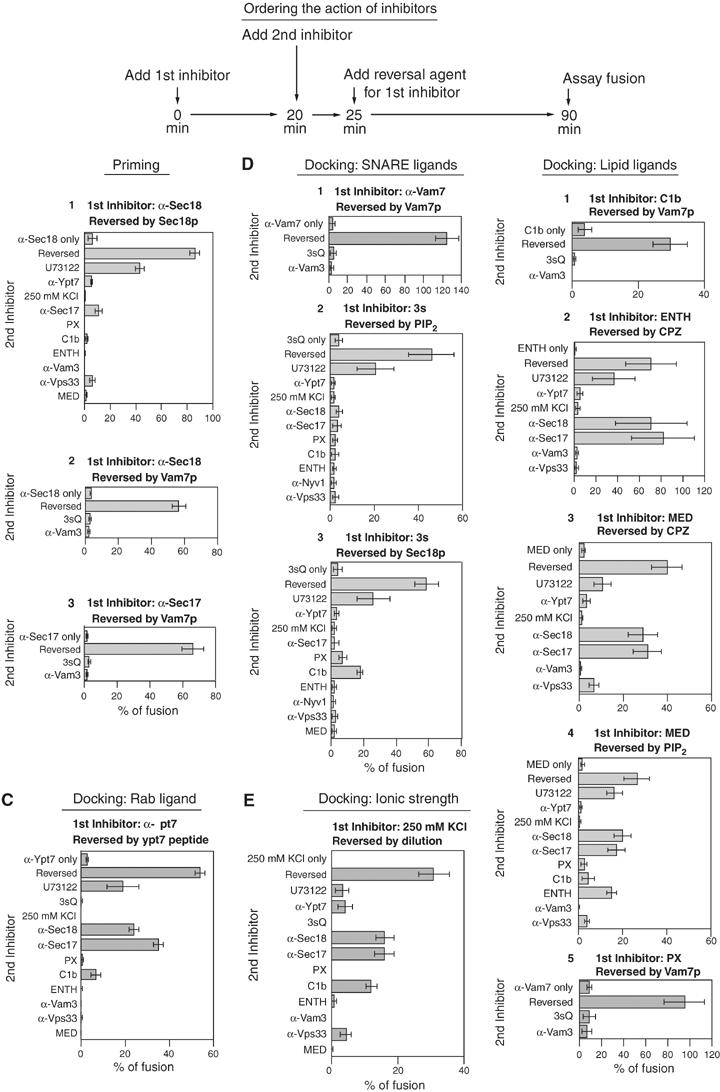
Ordering the action of fusion reaction inhibitors. (A) Scheme of the experiment. See text. (B–F) The first inhibitor was added to large-scale reactions with standard fusion components at time zero (see Figure 3 legend) and incubated at 27°C for 20 min, then aliquots (30 μl) were transferred to tubes containing second inhibitors at 27°C. Reactions were then incubated for 5 min prior to reversal agent addition. After a total time of incubation at 27°C of 90 min, alkaline phosphatase activity was assayed. See Figure 3 legend for concentrations of inhibitors and reversal agents. Anti-Nyv1p IgG (267 nM) replaced α-Vam3p as a second inhibitor in 3sQ inhibited reactions.
Like other SNARE-directed inhibitors, 3sQ blocks the fusion reaction during docking (Figure 2A). In this experiment, aliquots from a master fusion assay were removed at the indicated times and either transferred to ice to arrest fusion, mixed with 3sQ, or added to a characterized inhibitor of priming (antibody to Sec18p) or docking (antibody to Vam3p), or reaction buffer as a control. The kinetics of inhibition by 3sQ are coincident with the inhibition kinetics of the docking inhibitor αVam3p (Figure 2A). 3sQ associates with Nyv1p on the vacuole within a minute after its addition (Supplementary Figure S1), and thus its kinetics of inhibition are likely to reflect the stage of the reaction (docking) at which Nyv1p is required. Whereas normal SNARE associations require Ypt7p function (Collins et al, 2005), which is blocked by antibody to the Ypt7p effector domain (Eitzen et al, 2001), 3sQ association with R-SNAREs may not share this Ypt7p requirement. To test this, vacuoles were incubated without inhibitor, with αYpt7p, or with GDI on ice, then mixed with either buffer, 3sQ, or glutathione-S-transferase (GST)-sVti1p (the GST-tagged member of the 3sQ triad) and incubated at 27°C for 30 min. Vacuoles were then reisolated, solubilized with Triton X-100, and mixed with glutathione beads for assay of bound Nyv1p. On wild-type vacuoles, 3sQ bound to Nyv1p (Figure 2B, lane 3). Although this binding was not seen with GST-sVti1p alone (lane 6), it was unaffected by prior inhibition of Ypt7p by antibody (lane 4) or extraction of Ypt7p by Gdi1p (lane 5). The 3sQ even bound to Nyv1p on vacuoles purified from a ypt7Δ strain (lane 8). The simplest model is that 3sQ associates with R-SNAREs on the vacuole in a direct manner that does not require Ypt7p, which is normally needed for the association of the full-length, endogenous SNAREs (Collins et al, 2005). The binding of 3sQ to Nyv1p may inhibit productive Nyv1p association with the endogenous vacuole-anchored Vam3p and Vti1p.
Figure 2.
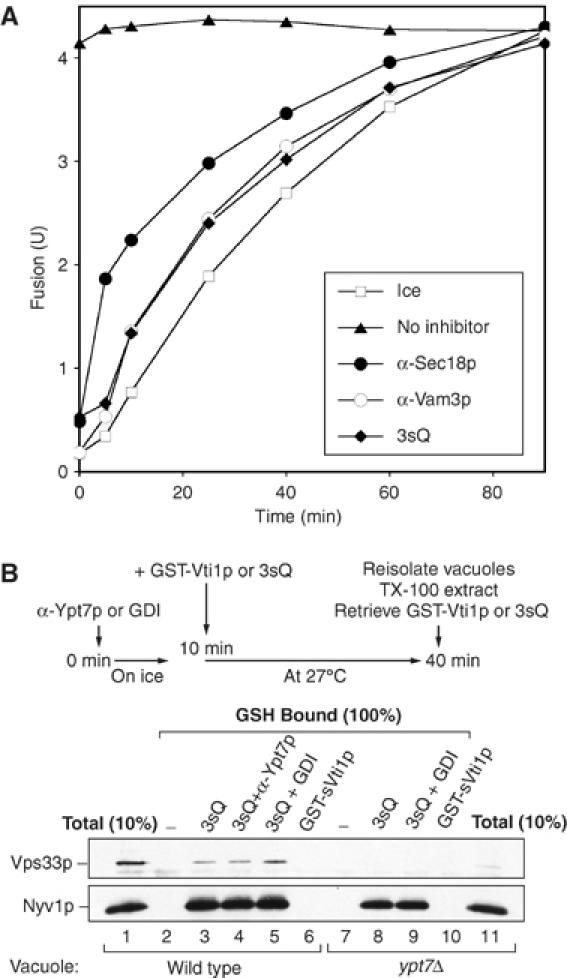
3sQ blocks docking. (A) Kinetics. A large-scale fusion reaction was incubated at 27°C. At indicated times, aliquots were transferred to tubes with indicated inhibitors and further incubated for the rest of the 90 min. Other aliquots were transferred to ice at the indicated times (open squares). (B) Ypt7p is not needed for 3sQ to bind Nyv1p. BJ3505 or BJ3505 ypt7Δ vacuoles in standard fusion buffer were incubated with anti-Ypt7p or GDI on ice for 10 min. GST-sVti1p or 3sQ (a mixture of GST-sVti1p, his6-sVam3p, and Vam7p) was added where indicated, and reactions were incubated at 27°C for 30 min. Vacuoles were reisolated and resuspended in ice-cold solubilization buffer (20 mM HEPES–KOH, pH7.4, 100 mM NaCl, 2 mM EDTA, 0.5% Triton X-100, 20% glycerol, 1 × protease inhibitor cocktail (0.46 μg/ml leupeptin, 3.5 μg/ml pepstatin, 2.4 μg/ml pefabloc-SC, 1 mM PMSF)), incubated at 4°C with rocking for 20 min. Detergent insoluble material was removed by centrifugation (TLA 120.2, 52 000 r.p.m., 11 min, 4°C). Clarified detergent extracts were incubated on a nutator for 5 h at 4°C with glutathione sepharose beads (Amersham). Beads were collected by brief centrifugation (3000 g, 2 min, 4°C) and washed five times with ice-cold solubilization buffer. Bound proteins were eluted in reducing SDS sample buffer (95°C, 3 min) for SDS–PAGE followed by immunoblotting.
Armed with the 3sQ SNARE-targeted inhibitor, we examined our collection of inhibitors of vacuole fusion for agents that can bypass or restore fusion to inhibited reactions. Vacuole fusion requires the Rab GTPase Ypt7p, SNAREs, and SNARE-chaperones such as Sec18p, Sec17p, and the SM subunit of HOPS, Vps33p. Fusion is inhibited by antibody to each of these proteins (Figure 3). Fusion has also been reported to require the Rho-family GTPases Rho1p and Cdc42p (Eitzen et al, 2001; Muller et al, 2001), and is blocked by GST-Rdi1p, which extracts Rho-family GTPases from the vacuole (Eitzen et al, 2001). Fusion is also blocked by ligands to the regulatory lipids (Figure 3; Fratti et al, 2004): PX binds to PI(3)P, Marcks effector domain (MED) and ENTH bind to PI(4,5)P2, and the C1b domain from protein kinase C binds to diacylglycerol. U73122 prevents the generation of diacylglycerol through inhibiting phospholipase C (Jun et al, 2004). Some of these inhibitors can be overcome or bypassed by the addition of PI(4,5)P2. Among the lipids that govern vacuole fusion (Fratti et al, 2004), PI(4,5)P2 is exceptionally water-soluble (Campbell et al, 2003) and thus could readily be added without detergents or organic solvents to our organelle-based assay. Although MED and ENTH are both PI(4,5)P2-binding agents, the ENTH inhibition is not reversed by exogenously added PI(4,5)P2 (Figure 3B). One possible reason is that the ENTH domain not only binds to PI(4,5)P2 but also induces membrane curvature (positive curvature, which is preferred for membrane budding/fission, but not for fusion), presumably by inserting one of its α-helices (helix 0) into outer leaflet of the lipid bilayer (Ford et al, 2002). This characteristic has not been reported for MED. Other inhibitors are overcome by Vam7p, by membrane-permeant cationic amphiphiles such as chlorpromazine, which modify membrane curvature, or by inhibitor-specific reagents. In the latter category, for example, Sec18p reverses inhibition by either 3sQ or antibody to Sec18p, and the Ypt7 effector domain peptide reverses inhibition by antibody to this peptide (Figure 3). No reversal agents have been found for antibodies to the SNAREs Vam3p or Nyv1p or to the SM subunit of HOPS, Vps33p. Untagged Rdi1p and GST-Rdi1p were equally efficient in extracting Rho-family GTPases, yet only latter inhibited fusion (Supplementary Figure S2); we therefore did not use GST-Rdi1p in the studies below. The inhibitors and reversal agents are summarized in Table I.
Figure 3.
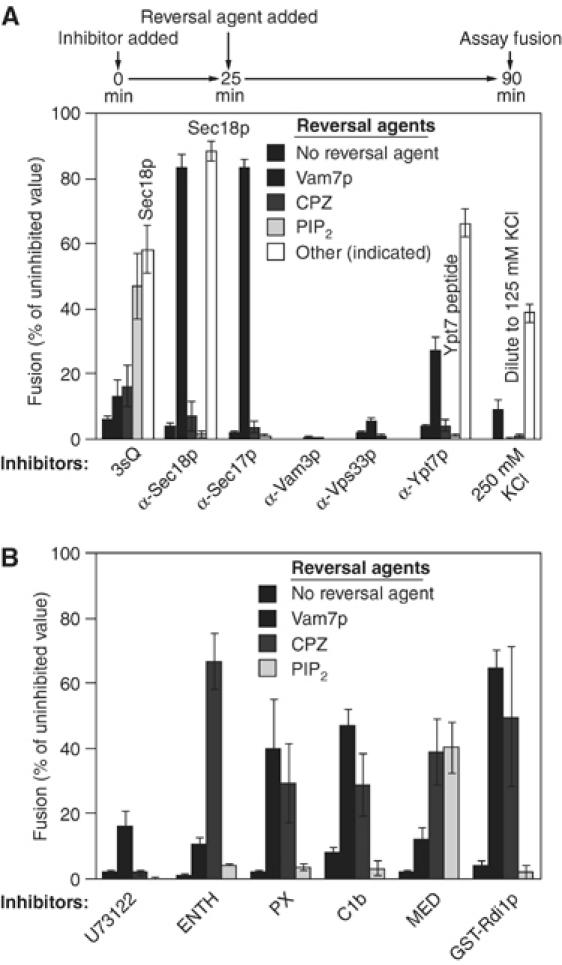
Reversal of inhibitors of vacuole fusion (A, B). Inhibitors were added at time zero with standard fusion components. After 25 min at 27°C, the reversal agent was added. Reactions were incubated at 27°C for a total of 90 min and alkaline phosphatase activity was assayed. Inhibitor concentrations: 1.23 μM 3sQ (an equimolar mixture of MBP-Vti1, GST-Vam7, and His6-Vam3), anti-Sec18p IgG (378 nM), anti-Sec17 IgG (378 nM), anti-Vam3p IgG (353 nM), anti-Vps33p IgG (32 nM), U73122 (Calbiochem) dissolved in DMSO (80 μM), ENTH domain (29 μM), GST-PX domain of Vam7 (3.9 μM), C1b domain from PKC (Johnson et al, 2000; 41 μM), MED (Marcks effector domain; Wang J et al, 2001; synthesized by Keck Center, Yale; 10 μM), and anti-Ypt7 affinity-purified IgG (133 nM). The reversal agents were added at the following concentrations: chlorpromazine (CPZ, from Sigma) dissolved in 33% DMSO (74 μM), PIP2 (di-palmitoyl C-16 D-myo-phosphatidylinositol 4,5-bisphosphate, from Echelon; 300 μM), his6-Sec18p (259 nM for reversal of 3sQ, 346 nM for reversal of anti-Sec18p), and Ypt7 peptide (66.7 μg/ml). Vam7p (prepared from a C-terminal intein construct; a generous gift from A Merz, University of Seattle, WA) was used at 833 nM for reversal of C1b, anti-Sec18p, anti-Sec17p, U73122, or anti-Ypt7p and at 370 nM for reversal of anti-Vam7p or GST-PX domain of Vam7. The percent fusion is the inhibited value minus the ice control value divided by uninhibited fusion value minus the ice value, times 100%. The average uninhibited fusion was 5.02±0.21 U (Merz and Wickner, 2004). The s.e. was calculated from three or more experiments.
Table 1.
Summary of inhibitors and reversal/bypass agents
| Inhibitor | Target | Reversal/bypass agents |
|---|---|---|
| 3sQ-SNAREs | R-SNARE | Secl8p, PI(4,5)P2 |
| αSecl8p | Secl8p | Secl8p, Vam7p |
| αVam3p | Vam3p | None known |
| αVps33p | Vps33p | None known |
| αYpt7 | Ypt7p | Ypt7p effector peptide |
| 250 mM KC1 | Unknown | Lowered salt |
| U73122 | Phospholipase C | Vam7p |
| ENTH domain | PI(4,5)P2 | Chlorpromazine (CPZ) |
| PX domain | PI(3)P | Vam7p, CPZ |
| Clb domain | Diacylglycerol | Vam7p, CPZ |
| MED peptide | PIPx | CPZ, PI(4,5)P2 |
If docking consists of a series of ordered reactions, then some early docking subreactions might be completed while late-stage subreactions are blocked. Conversely, if the subreactions of docking are interdependent or fully reversible, then the need for each protein and lipid that support fusion would continue until bilayer and content mixing are complete. To distinguish between these models, vacuoles were incubated for 25 min with each of the inhibitors under fusion reaction conditions prior to reversal of the block to fusion. A second inhibitor was added 5 min before the reversal agent (Figure 4A). Many of the earlier studies that had suggested an ordered series of docking subreactions (e.g. Eitzen et al, 2001) did not allow such an incubation period for the second inhibitor to act fully before reversing the block by the first inhibitor. The reactions that had been incubated with docking inhibitors became resistant to U73122, an inhibitor of phospholipase C, and to antibodies to Sec17p or Sec18p, priming inhibitors that prevent the ATP-dependent disassembly of cis-SNARE complexes (Figure 4B–F). For example (Figure 4C), fusion reactions were blocked by αYpt7p and mixed with other inhibitors prior to reversal of the αYpt7p block by addition of its cognate peptide. During incubation with αYpt7p, the reaction had completed priming and become resistant to αSec17, αSec18, and U73122, but remained sensitive to all tested docking inhibitors. Similar results were found with other docking inhibitors (Figure 4E2–E4 and 4F). The only second inhibitors that could be meaningfully tested in this experimental scheme are those that are not themselves bypassed by the first inhibitor's reversal agent; thus, fewer second inhibitors could be tested when the reversal agent was Vam7p, for example (Figure 4B2, 4B3, 4D1, 4E1 and 4E4). These data confirm that priming can occur when docking is blocked, as reported (Mayer et al, 1996). Although elevated ionic strength acts as a reversible inhibitor, its targets of action are not known, and limited resistance to C1b domain is acquired during the initial incubation (Figure 4E). Nevertheless, none of these reactions became resistant to any of the added second inhibitors of docking, showing that the lipids and proteins that mediate docking are interdependent or reversible in their actions.
Even reversible or interdependent subreactions may nevertheless occur in an essential order, which could be determined through kinetic studies of synchronized fusion reactions. Synchronized vacuole fusion reactions can permit a detailed evaluation of the final reaction stages (Merz and Wickner, 2004a, 2004b; Collins et al, 2005). Priming of the vacuoles by Sec17p and Sec18p, which is blocked by antibody to Sec17p (Mayer et al, 1996), is needed to provide Vam7p SNARE for trans-SNARE complex formation (Thorngren et al, 2004). Fusion reactions can be synchronized by blocking priming with antibody to Sec17p, then allowing rapid SNARE complex assembly by adding recombinant Vam7p bearing a fused GST domain on its N-terminus (Thorngren et al, 2004; Merz and Wickner, 2004a). The added GST-Vam7p binds rapidly to the vacuoles and enters into complex with Vam3p and Nyv1p (Figure 5A). The kinetics of the formation of this complex is indistinguishable from the kinetics of acquisition of the resistance of the reaction to added antibody to Vam3p (Figure 5B), a characterized measure of docking (Mayer et al, 1996; Merz and Wickner, 2004b). These SNARE complexes form in association with HOPS (Figure 5C, lane 3; also see Collins et al, 2005). Although HOPS has direct affinity for Vam7p (Stroupe et al, 2006), the sensitivity to αVam3p of the association seen here (Figure 5C) shows that HOPS is binding to the full SNARE complex rather than only to GST-Vam7p. As reported (Collins et al, 2005), the MED, a ligand to phosphoinositides, permits full SNARE:HOPS complex formation while blocking the subsequent fusion reaction (lane 8). We also find that transfer of the reaction to ice, even before the addition of GST-Vam7p, permits SNARE:HOPS complex assembly (lanes 2–4) but completely blocks fusion (Collins et al, 2005). Thus, the final mixing of aqueous contents after SNARE:HOPS complexes have formed requires physiological temperature and unliganded lipids to permit a lipid bilayer rearrangement, which depends on lipid fluidity and mobility.
Figure 5.
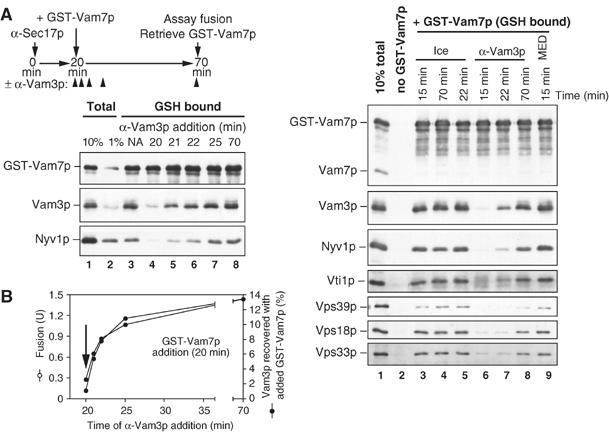
Synchronized vacuole fusion proceeds from SNARE complex assembly to temperature-dependent lipid rearrangement for lumenal compartment mixing. (A, B) Fusion reactions acquire anti-Vam3p resistance as the SNAREs pair. Vacuole fusion reactions (180 μl) containing anti-Sec17p antibodies and an ATP regeneration system were incubated at 27°C. After 20 min, GST-Vam7p (1 μg/ml) was added to bypass the anti-Sec17p antibody block (NA, no addition) (Thorngren et al, 2004). Fab fragments of antibody to Vam3p were added at the same time as GST-Vam7p (20 min) or 1, 2, 5, or 50 min after GST-Vam7p addition. After 70 min (total incubation time), the reactions were placed on ice, and fusion (B) and the SNARE associations of GST-Vam7p (A) were determined (Collins et al, 2005). An aliquot (30 μl) was withdrawn for alkaline phosphatase assay and vacuoles from the remaining 150 μl were sedimented (11 000 g, 10 min, 4°C). Pellets were resuspended in solubilization buffer (200 μl, 20 mM HEPES–KOH, pH 7.4, 100 mM NaCl, 2 mM EDTA, 0.5% Triton X-100 (Anatrace), 20% glycerol, 1 × protease inhibitor cocktail (0.46 μg/ml leupeptin, 3.5 μg/ml pepstatin, 2.4 μg/ml pefabloc-SC, 1 mM PMSF), 1 mM DTT). Detergent-insoluble material was removed by ultracentrifugation, and 10% of each clarified supernatant was removed for a loading control. GST-Vam7p was retrieved from the remaining extract using glutathione sepharose (20 μl beads, 1 h, 4°C). Unbound material was decanted, and beads were resuspended four times in solubilization buffer (800 μl each). Bound material was eluted by heating in SDS–PAGE sample buffer. Samples were separated by SDS–PAGE and immunblotted for SNARE proteins. Vam3p associations with GST-Vam7p were quantitated by densitometry. (C) Temperature-dependent lipid rearrangements and fusion occur after HOPS·SNARE complex assembly. As in (A), staged reactions containing αSec17p were bypassed with GST-Vam7p added at 20 min and the associations of GST-Vam7p were determined after 70 min. At times indicated, reactions were placed on ice or received inhibitors (anti-Vam3p Fab fragments or MED). After the incubation, vacuoles were reisolated, solubilized, and the GST-Vam7p was retrieved using glutathione agarose, as above.
Each of the lipids and proteins that are required for vacuole fusion becomes enriched at vertex rings during docking, and their enrichments have been shown to be interdependent (Wang et al, 2003; Fratti et al, 2004). However, not all vertices show equivalent enrichment (Wang et al, 2002); some are highly enriched, while others are not. The interdependent character of the docking subreactions (Figure 4) would be easier to understand if the same vertices that became enriched for one fusion protein or regulatory lipid became enriched for the others as well. We examined this question by triple-label quantitative fluorescence of docked vacuole clusters (Figure 6). Vacuole membranes were visualized by the fluorescent probe PSS380, which binds to the bulk lipid phosphatidylserine (Kouvlov et al, 2003). Added purified actin, derivatized with the fluorophore Cy3, and GFP-tagged Vps33p (a subunit of HOPS) colocalize at the same vertices (Figure 6A–D). Quantitative measure of the enrichment of actin and Vps33p shows a strong correlation of their degree of enrichment at different vertices (Figure 6I), in accord with prior studies which suggest that actin has a direct role in vacuole fusion (Eitzen et al, 2002). In a similar experiment, we find that PI(3)P, assayed by binding of Cy3-labeled FYVE domain, a PI(3)P ligand, becomes enriched at the same vertices as GFP-tagged Vps33p (Figure 6E–H and J). We have also previously reported that Vps33p and Ypt7p become enriched at the same vertices in docked vacuole clusters (Wang et al, 2003). While we have thus far only examined a small subset of the many binary combinations of regulatory lipids and fusion proteins which are known to become vertex enriched, our data suggest that vertices are either enriched with many, or all, of the fusion proteins and lipids or none of them, in accord with interdependent and reversible pathways of enrichment.
Figure 6.
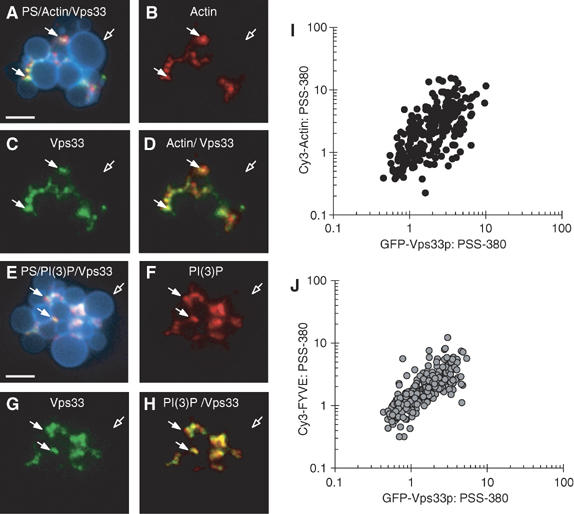
Regulatory lipids and fusion proteins are coenriched at identical vertices. Vacuoles isolated from yeast expressing Vps33p-GFP were incubated under docking conditions for 30 min at 27°C with fluorescent Cy3-labeled yeast actin or Cy3-labeled FYVE domain (see Materials and methods). After incubation, docking reactions were placed on ice and labeled with the fluorescent phosphatidylserine (PS) binding probe PSS-380. PS is evenly distributed on docked vacuoles when stained with 1 μM PSS-380 and appears blue (A, E). The localization of actin was monitored by adding 15 nM Cy3-Actin (B, D). Phosphatidylinositol 3-phosphate (PI(3)P) was specifically labeled with 0.2 μM Cy3-FYVE (F, H). Vps33p was visualized by its GFP tag (C, G). Panels A and D show merged images of PS, actin and Vps33p, or actin and Vps33p, respectively. Panels E and H show merged images of PS, PI(3)P and Vps33p, or PI(3)P and Vps33p, respectively. Solid arrows show examples of vertex sites enriched in Vps33p, PI(3)P, and actin. Hollow arrows specify outer membrane microdomains. Bars, 2 μm. Scatter plots depict the enrichment of each vertex for actin and for Vps33p (I) or PI(3)P and Vps33p (J). Intensities were measured in all three fluorescence channels at each subdomain and are expressed as a ratio of vertex concentration to PS label. Outer membrane ratios were normalized to a value of 1 and the vertex enrichment of actin, PI(3)P, and Vps33p were expressed relative to outer membrane intensities. Each data point represents a single vertex and is plotted according to its dual enrichment of vertex markers.
Discussion
The vertex ring provides a useful model for studying the assembly of functional membrane microdomains. Such microdomains are commonly thought of as either fundamentally lipidic (e.g. lipid rafts, bearing proteins that have partitioned into the lipidic domain according to their lipid preferences) or basically proteinaceous (e.g. coated vesicles, assembling by a process driven by protein interactions). The vertex ring that surrounds the apposed membranes of docked yeast vacuoles shows enrichment of selected proteins (SNAREs, the Rab Ypt7p GTPase, and the HOPS complex) and chemically minor lipids, which are functionally vital for fusion (ergosterol, diacylglycerol, and phosphoinositides). These lipids and proteins are interdependent for their vertex enrichment (Fratti et al, 2004), confounding efforts to discover a simple linear pathway by which these components move to the vertex; indeed, it is unclear whether the pathway of vertex assembly is a linear, reversible pathway or entails more than one parallel pathway. In either case, we have shown that docking is not simply an ordered series of irreversible steps. We suggest that the reversible or concurrent steps of docking are the interdependent assembly of fusion factors into the vertex ring microdomain.
Analysis of protein and lipid function in fusion has rested on specific inhibitory ligands, and the utility of these ligands is enhanced by the ability to reverse their effects. Our current study extends the range of inhibitory ligands and establishes conditions for reversal or bypass of many inhibitors. Surprisingly, no docking inhibitor is bypassed during fusion reactions which are blocked by another docking inhibitor (Figure 4). It is noteworthy that U73122, an inhibitor of DAG formation, acts at a different step than C1b, a DAG-binding domain. It appears that the production of DAG by PLC on the vacuoles occurs rapidly and does not require docking, whereas the DAG ‘product' is needed throughout docking for the establishment and maintenance of a functional vertex ring. These data suggest that the enrichment of each component in the vertex ring is reversible, and reflects multiple cooperative interactions.
What kinds of cooperative interactions may occur during vacuole docking and vertex ring assembly? Several components (e.g. HOPS, PI3P, and Vam7p), which can each bind to the others with low pairwise affinity can, if present together, form a ternary complex. Each component would be stably bound to this ternary complex with far higher affinity, as its removal from the complex would require breaking two low-affinity associations. Although perhaps less likely, a second kind of cooperative affinity would be where the binding of two components activates their affinity for a third; an example of this is the binding of GTPγS to Ypt7p activating its affinity for HOPS (Seals et al, 2000). The only difference between these models of cooperative ternary complex formation is whether some binding associations are altering other binary association affinity constants. The availability of purified, active components allows these questions to be addressed quantitatively. Cooperativity may also suggest that high levels of certain components could bypass or reduce the need for others. This has certainly been seen in the vacuole fusion reaction; for example, high levels of Vam7p reduces the amount of Ypt7p needed for fusion (Thorngren et al, 2004).
The HOPS complex has direct interactions with Ypt7p (Seals et al, 2000; Wurmser et al, 2000), with SNAREs such as Vam3p and Vam7p (Dulubova et al, 2001; Stroupe et al, 2006), and with phosphoinositides (Stroupe et al, 2006). Since Vam7p binds directly to phosphoinositides through its PX domain (Cheever et al, 2001), we begin to see how a web of reversible interactions can drive vertex ring microdomain assembly. Three clusters of interactions have been described for vacuole fusion components: Ypt7p has direct affinity for HOPS (Seals et al, 2000; Wurmser et al, 2000), the four vacuole SNAREs bind each other (Ungermann et al, 1999; Collins et al, 2005), and the regulatory lipids may form a lipidic phase (Redfern and Gericke, 2004). Interactions between at least one member of each of these clusters—HOPS, Vam7p, and phosphoinositides—may link all the vertex constituents in a self-assembly network. It will be important to determine whether these interactions among Ypt7p:GTP, HOPS, SNAREs, and phosphoinositides are cooperative in a manner that enhances the specificity and rate of vertex assembly; for example, whether the affinity of HOPS for any one of its binding partners is enhanced by engagement with another. As expected from this working model, some vertex ring domains in a cluster of docked vacuoles are well advanced in assembly and others are not, but those that are well advanced in assembling are enriched for each of the tested proteins and lipids (Figure 6). These findings with yeast vacuoles may have parallels in other fusion reactions. Just as only a portion of the vacuole pairs in a large docked cluster have fully assembled vertices, ready for fusion, only a small fraction of the vesicles clustered at a synapse are in the readily releasable pool (Sudhof, 2004), although the molecular basis of the readily-releasable pool is not yet clear.
Materials and methods
Plasmids, yeast strains, and reagents
The plasmid pYJ403-NYV1 for overexpression of Nyv1p under the constitutive ADH promoter was generated by introducing the 720 base-pair upstream region, the NYV1 open-reading frame, and its 300 base-pair downstream region into the XhoI/HindIII and BamHI/SacII sites of the pRS403 plasmid.
Yeast strains BJ3505 (Matα ura3-52 trp1-Δ101 his3-Δ200 lys2-801 gal2 (gal3) can1 prb1-Δ1.6R pep4::HIS3) (Jones, 2002) and DKY6281 (Mata ura3-52 leu2-3,112 trp1-Δ901 his3-Δ200 lys2-801 suc2-Δ9 pho8::TRP1) (Haas et al, 1994) were used for the vacuole fusion assay. Yeast strains BY4742 (Matα ura3Δ leu2Δ his3Δ lys2Δ) pep4Δ prb1Δ and BY4742 pho8Δ were used to generate strains overproducing Nyv1p by integrating pYJ403-NYV1 into the chromosome.
Recombinant his6-sVam3p, GST-sVam3p, Vam7p, GST-sNyv1p, and GST-sVti1 were generated as described (von Mollard et al, 1997; Dulubova et al, 2001; Merz and Wickner, 2004a; Thorngren et al, 2004). Affinity-purified anti-Nyv1p antibody was prepared as described (Thorngren et al, 2004).
D-Myo-phosphatidylinositol 4,5-bisphosphate (PIP2) was from Echelon Research, catalog P4516. It was dispersed at 3 mM in PS buffer by vortexing, and then 5 min bath sonication at 23°C prior to addition to fusion assays at 300 μM. It has a very high water solubility (Campbell et al, 2003) and thus will likely be as monomer/micelle in our reactions or partitioned into the bilayers.
Fusion assay
Standard assays of fusion (30 μl) contained 125 mM KCl, 6 mM MgCl2, 1 mM ATP, 1 mg/ml creatine kinase, 29 mM creatine phosphate, 20 mM PIPES-KOH, pH 6.8, 200 mM Sorbitol, 930 nM IB2, 10 μM CoA, and 3 μg each of BJ3505 vacuoles and DKY6281 vacuoles. After 90 min at 27°C, alkaline phosphatase activity was assayed.
Docking assay
Saccharomyces cerevisiae harboring Vps33p-GFP used in the docking assay was described previously (Wang et al, 2002, 2003). Docking reactions (30 μl) contained 6 μg of Vps33p-GFP vacuoles, docking reaction buffer (100 mM KCl, 0.5 mM MgCl2, 20 mM PIPES-KOH, pH 6.8, 200 mM sorbitol), ATP-regenerating system (0.3 mM ATP, 6 mM creatine phosphate, 0.7 mg/ml creatine kinase), 20 μM coenzyme A, 930 nM IB2, 8 nM his6-Sec18p, and either 0.2 μM Cy3-FYVE or 15 nM Cy3-labeled yeast actin. After 30 min at 27°C, vacuoles were placed on ice and stained with 1 μM PSS-380 to label phosphatidylserine (Fratti et al, 2004). Vacuoles were then mixed with 50 μl of 0.6% agarose in PS buffer at 42°C, and lightly vortexed (medium setting) for 3 s. Aliquots (15 μl) were mounted on slides and observed by fluorescence microscopy.
Microscopy
Images were acquired using an Olympus BX51 microscope equipped with a 100 W mercury arc lamp, Plan Apochromat × 60 objective (1.4 NA) and a Sensicam QE CCD camera (Cooke). Images were acquired at 23°C without pixel binning. Vps33p-GFP images were acquired using an Endow GFP filter set (Chroma Technologies). Cy3-FYVE and Cy3-Actin images were acquired using a TRITC/DiI filter set (Chroma Technologies). An XF06 filter set (Omega Optical Inc.) was used to acquire PSS-380 images. Filter sets were housed in a motorized turret. PSS-380 was used to focus fields and photobleaching was performed for 20 s between channels. Bleaching was normalized by automated image acquisition using IP Lab software (Scanalytics), ensuring that bleaching is uniform and negating it as a factor in calculating intensity ratios. Images were processed and analyzed using Image/J software (NIH). Vertex enrichment was calculated by ratiometric quantitation. Maximum pixel values for vertex, boundary, and outer membrane were measured in each fluorescence channel and every vertex and outer membrane within a vacuole cluster was measured. Vertex ratios were normalized by dividing the mean ratio values of outside edges. Clusters (15–20) with 100–300 vertex sites were analyzed from multiple independent experiments.
Preparation of yeast actin
Yeast actin was prepared as described (Goode, 2002) with modification. S. cerevisiae BY4742 grown in YPD broth to stationary phase were washed in G-buffer (10 mM Tris–Cl, pH 8, 0.2 mM CaCl2, 2 mM DTT, and 0.5 mM ATP) and frozen dropwise in liquid N2. Frozen cell pellets were lysed in a Waring blender in liquid N2. Lysate was thawed in G-buffer with protease inhibitors and centrifuged in a JA-20 rotor (13 400 g, 4°C, 30 min) followed by a high-speed centrifugation (45Ti, 100 000 g, 4°C, 120 min). The high-speed supernatant was filtered through cheesecloth and loaded onto an immobilized DNAse-I column. The column was washed with G-buffer containing protease inhibitors and 10% deionized formamide, followed by one column volume of G-buffer containing protease inhibitors and 10% deionized formamide and 0.2 M NH4Cl, and then G-buffer alone. Actin was eluted with G-buffer containing protease inhibitors and 50% deionized formamide and dialyzed against PS buffer with 125 mM KCl and 5 mM MgCl2.
Cloning, expression, and purification of untagged Rdi1p
The RDI1 gene from S. cerevisiae was PCR amplified from strain DKY6281, using 5′-GGTGGTCATATGGCCGAAGAAAGTACCGAC-3′ and 5′-GGTGGTCCCGGGTTTTTTGACAATTTCGACC-3′ primers to introduce NdeI and SmaI restriction sites (underlined). The product and the vector pTYB2 (New England Biolabs) were digested with NdeI and SmaI and ligated. The resultant vector (pVJS2) allowed production of Rdi1p with a C-terminal chitin-binding domain (CBD). Removal of the CBD from the C-terminus via intein cleavage would result in an Rdi1p protein containing a single C-terminal glycyl residue.
E. coli Rosetta2 (λDE3) (Novagen) harboring pVJS2 was grown in TB plus ampicillin (100 μg/ml) and chloramphenicol (12.5 μg/ml) to OD600=1.0 at 30°C. The culture was placed on ice for 15 min. Isopropyl-β-D-thiogalactopyranoside was added to 0.3 mM, and the culture was shaken overnight at 18°C. After cell harvest, Rdi1p-CBD was purified by chitin-affinity chromatography (New England Biolabs), according to modified manufacturer's instructions. The cell lysis/binding buffer contained 0.02 M HEPES–KOH, pH 8.0, 0.5 M KCl, 1 mM EDTA, and 0.1% Triton X-100. After binding Rdi1p-CBD, the column resin was washed with 15 volumes of binding buffer without Triton X-100. Overnight cleavage of the intein was in wash buffer plus 50 mM DTT at 4°C. Eluted Rdi1p was concentrated, dialyzed into PS buffer with 125 mM KCl, frozen in liquid nitrogen, and stored at −80°C.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Acknowledgments
This work was supported by grants from the NIH. RAF was supported by a postdoctoral fellowship from the Helen Hay Whitney Foundation. VS was supported by a Damon Runyon Cancer Research Foundation Fellowship (DRG-1837). KMC received predoctoral support from the NIH (T32 AR97576, NIAMS).
References
- Brandhorst D, Zwilling D, Rizzoli SO, Lippert U, Lang T, Jahn R (2006) Homotypic fusion of early endosomes: SNAREs do not determine fusion specificity. Proc Natl Acad Sci USA 103: 2701–2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell RB, Liu F, Ross AH (2003) Allosteric activation of PTEN phosphatase by phosphatidylinositol 4,5-bisphosphate. J Biol Chem 278: 33617–33627 [DOI] [PubMed] [Google Scholar]
- Cheever ML, Sato TK, de Beer T, Kutateladze TG, Emr SD, Overduin M (2001) Phox domain interaction with PtdIns(3)P targets the Vam7 t-SNARE to vacuole membranes. Nat Cell Biol 3: 613–618 [DOI] [PubMed] [Google Scholar]
- Collins KM, Thorngren NL, Fratti RA, Wickner WT (2005) Sec17p and HOPS, in distinct SNARE complexes, mediate SNARE complex disruption or assembly for fusion. EMBO J 24: 1775–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova I, Yamaguchi T, Wang Y, Sudhof TC, Rizo J (2001) Vam3p structure reveals conserved and divergent properties of syntaxins. Nat Struct Biol 8: 258–264 [DOI] [PubMed] [Google Scholar]
- Eitzen G, Thorngren N, Wickner W (2001) Rho1p and Cdc42p act after Ypt7p to regulate vacuole docking. EMBO J 20: 5650–5656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eitzen G, Wang L, Thorngren L, Wickner W (2002) Remodeling of organelle-bound actin is required for yeast vacuole fusion. J Cell Biol 158: 669–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford MG, Mills IG, Peter BJ, Vallis Y, Praefcke GJK, Evans PR, McMahon HT (2002) Curvature of clathrin-coated pits driven by epsin. Nature 419: 361–366 [DOI] [PubMed] [Google Scholar]
- Fratti RA, Jun Y, Merz AJ, Margolis N, Wickner W (2004) Interdependent assembly of specific regulatory lipids and membrane fusion proteins into the vertex ring domain of docked vacuoles. J Cell Biol 167: 1087–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode BL (2002) Purification of yeast actin and actin-associated proteins. Methods Enzymol 351: 433–441 [DOI] [PubMed] [Google Scholar]
- Haas A, Conradt B, Wickner W (1994) G-protein ligands inhibit in vitro reactions of vacuole inheritance. J Cell Biol 126: 87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JE, Giorgione J, Newton AC (2000) The C1 and C2 domains of protein kinase C are independent membrane targeting modules, with specificity for phosphatidylserine conferred by the C1 domain. Biochem 39: 11360–11369 [DOI] [PubMed] [Google Scholar]
- Jones EW (2002) Vacuolar proteases and proteolytic artifacts in Saccharomyces cerevisiae. Methods Enzymol 351: 127–150 [DOI] [PubMed] [Google Scholar]
- Jun Y, Fratti RA, Wickner W (2004) Diacylglycerol and its formation by phospholipase C regulate Rab- and SNARE-dependent yeast vacuole fusion. J Biol Chem 279: 53186–53195 [DOI] [PubMed] [Google Scholar]
- Kouvlov AV, Stucker KA, Lakshmi C, Robinson JP, Smith BD (2003) Detection of apoptotic cells using a synthetic fluorescent sensor for membrane surfaces that contain phosphatidylserine. Cell Death Differ 10: 1357–1359 [DOI] [PubMed] [Google Scholar]
- Mayer A, Wickner W, Haas A (1996) Sec18p (NSF)-driven release of Sec17p (α-SNAP) can precede docking and fusion of yeast vacuoles. Cell 85: 83–94 [DOI] [PubMed] [Google Scholar]
- Mayer A, Wickner W (1997) Docking of yeast vacuoles is catalyzed by the Ras-like GTPase Ypt7p after symmetric priming by Sec18p (NSF). J Cell Biol 136: 307–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz A, Wickner W (2004a) Trans-SNARE interactions elicit Ca2+ efflux from the yeast vacuole lumen. J Cell Biol 164: 195–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merz A, Wickner W (2004b) Reconstitution of organelle docking and fusion kinetics in a cell-free assay. Proc Natl Acad Sci USA 101: 11548–11553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller O, Johnson DI, Mayer A (2001) Cdc42p functions at the docking stage of yeast vacuole membrane fusion. EMBO J 20: 5657–5665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeffer SR (2005) Structural clues to Rab GTPase functional diversity. J Biol Chem 280: 15485–15488 [DOI] [PubMed] [Google Scholar]
- Redfern DA, Gericke A (2004) Domain formation in phosphatidylinositol monophosphate/phosphatidylcholine mixed vesicles. Biophys J 86: 2980–2992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seals DF, Eitzen G, Margolis N, Wickner WT, Price A (2000) A Ypt/Rab effector complex containing the Sec1 homolog Vps33p is required for homotypic vacuole fusion. Proc Natl Acad Sci USA 97: 9402–9407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel DP, Banschbach J, Alford D, Ellens H, Lis LJ, Quinn RJ, Yeagle PL, Bentz J (1989) Physiological levels of diacylglycerols in phospholipid membranes induce membrane fusion and stabilize inverted phases. Biochemistry 28: 3703–3709 [DOI] [PubMed] [Google Scholar]
- Stroupe C, Collins KM, Fratti RA, Wickner WT (2006) Purification of active HOPS complex reveals its affinities for phosphoinositides and the SNARE Vam7p. EMBO J 25: 1579–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudhof T (2004) The synaptic vesicle cycle. Ann Rev Neurosci 27: 509–547 [DOI] [PubMed] [Google Scholar]
- Thorngren N, Collins KN, Fratti RA, Wickner W, Merz AJ (2004) A soluble SNARE drives rapid docking, bypassing ATP and Sec17/18p for vacuole fusion. EMBO J 23: 2765–2776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungermann C, Nichols BJ, Pelham HRB, Wickner W (1998) A vacuolar v-t-SNARE complex, the predominant form in vivo and on isolated vacuoles, is disassembled and activated for docking and fusion. J Cell Biol 140: 61–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungermann C, Price A, Wickner W (2000) A new role for a SNARE protein as a regulator of the Ypt7/Rab-dependent stage of docking. Proc Natl Acad Sci USA 97: 8889–8891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungermann C, von Mollard GF, Jensen ON, Margolis N, Stevens TH, Wickner W. (1999) Three v-SNAREs and two t-SNAREs, present in a pentameric cis-SNARE complex on isolated vacuoles, are essential for homotypic fusion. J Cell Biol 145: 1435–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- vonMollard GF, Nothwehr SF, Stevens TH (1997) The yeast v-SNARE Vti1p mediates two vesicle transport pathways through interactions with the t-SNAREs Sed5p and Pep12p. J Cell Biol 137: 1511–1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdez-Taubas J, Pelham HRB (2003) Slow diffusion of proteins in the yeast plasma membrane allows polarity to be maintained by endocytic cycling. Curr Biol 13: 1636–1640 [DOI] [PubMed] [Google Scholar]
- Wada Y, Ohsumi Y, Anraku Y (1992) Isolation and characterization of two classes of vam mutants. J Biol Chem 267: 18665–18670 [PubMed] [Google Scholar]
- Wang J, Arbuzova A, Hangyas-Mihalyne G, McLaughlin S (2001) The effector domain of myristoylated alanine-rich C kinase substrate binds strongly to phosphatidylinositol 4,5-bisphosphate. J Biol Chem 276: 5012–5019 [DOI] [PubMed] [Google Scholar]
- Wang L, Merz AJ, Collins KM, Wickner W (2003) Hierarchy of protein assembly at the vertex ring domain for yeast vacuole docking and fusion. J Cell Biol 160: 365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Seeley ES, Wickner W, Merz AJ (2002) Vacuole fusion at a ring of vertex docking sites leaves membrane fragments within the organelle. Cell 108: 357–369 [DOI] [PubMed] [Google Scholar]
- Wickner W (2002) Yeast vacuoles and membrane fusion pathways. EMBO J 21: 1241–1247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurmser AE, Sato TK, Emr SD (2000) New component of the vacuolar class C-Vps complex couples nucleotide exchange on the Ypt7 GTPase to SNARE-dependent docking and fusion. J Cell Biol 151: 551–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2