

Abstract
Mating pheromone represses synthesis of full-length PRY3 mRNA, and a new transcript appears simultaneously with its 5′ terminus 452 nucleotides inside the open reading frame (ORF). Synthesis of this shorter transcript results from activation of a promoter within the PRY3 locus, and its production is concomitant with the rapid disappearance of the full-length transcript. Evidence is consistent with the pheromone-induced transcription factor Ste12p binding two pheromone response elements within the PRY3 promoter, directly impeding transcription of the full-length mRNA while simultaneously inducing initiation of the short transcript. This process depends on a TATA box within the PRY3 ORF. Expression of full-length PRY3 inhibited mating, while no disadvantage was detectable for cells unable to make the short transcript. Therefore, Ste12p is utilized as a repressor of full-length PRY3 transcription, ensuring efficient mating. There is no evidence that production of the short PRY3 transcript is anything more than an adventitious by-product of this mechanism. It is possible that cryptic binding sites for transcriptional activators may occur frequently within genomes and have the potential of evolving for rapid, gene-specific repression by mechanisms analogous to PRY3. PRY3 regulation provides a model for the coordination of both inductive and repressive activities within a regulatory network.
Cells respond to changes in their environment through activation of genetic regulatory networks, groups of genes with mutual regulatory interactions. Coactivated or repressed genes and their specific regulators function together, altering the cellular composition to cope with a varying environment (18, 30, 43, 44). Upon reception of intrinsic or extrinsic cues, cellular needs are met both through production of new proteins and through silencing the expression of genes whose respective protein products are not needed or would be deleterious to the cell during a particular developmental program or environmental condition. Down-regulation of transcript levels can be mediated through multiple mechanisms of transcriptional repression (31, 35, 38) and though regulated degradation (37, 54). Furthermore, translation of transcripts can be silenced, either directly (29, 34, 37) or through alterations of transcript structures (29) resulting in either exposure of elements inhibitory to translation (26, 39, 55) or exclusion of translational start sites (29).
Our previous work revealed the frequent occurrence of altered 5′ termini among transcripts during cellular responses to new environmental conditions (29). In every case, one of the transcript forms was inefficiently translated, while the other was efficiently loaded with ribosomes. For some, the undertranslated transcript possessed a long 5′ leader originating from an upstream intergenic start site suggesting the presence of an independent promoter. However, several of the transcripts were shortened under the new conditions with their 5′ terminus residing within the open reading frame (ORF), in turn preventing the production of the full-length protein.
In order to better understand the processes by which these 5′ structural alterations occur, we analyzed the mechanism(s) responsible for producing, in response to Saccharomyces cerevisiae mating pheromone, an altered PRY3 transcript with its 5′ terminus residing within the ORF at +452 relative to the translational initiation codon of the full-length protein. Pry3p is a cell wall protein (56) of unknown function but whose transcript is cell cycle regulated with the “SIC1 cluster” (51). The promoter region of PRY3 contains putative binding sites for either the daughter cell-specific transcription factor Ace2p or the non-cell-specific activator Swi5p (11). The PRY3 promoter region directs reporter expression only in daughter cells (9), suggestive of a role specifically in their cell wall structure.
Here we report that the pheromone-responsive transcription factor Ste12p and its binding sites near the TATA box upstream of the PRY3 gene impede transcription from the full-length start site and in the process activate an intragenic promoter, which generates the short PRY3 transcript. The daughter-specific transcription factor Ace2p is required for active transcription from both full-length and internal start sites, and since full-length PRY3 expression is inhibitory to mating, repression by Ste12p must optimize the mating competence of newborn daughter cells. The multiple elements within the PRY3 locus are a clear example of local competition providing a platform for coordinating inductive and repressive activities within a regulatory network, in this instance the response to pheromone.
MATERIALS AND METHODS
Yeast strains and growth conditions.
Strains used in this study are listed in Table 1. All yeast strains were derived from strain BY2125 (W303 background; kindly provided by L. Breeden, Fred Hutchinson Cancer Research Center, Seattle, WA) by standard methods (22), except strain KB1132, which was derived from strain XWV01 followed by tetrad dissection. Yeast transformations were performed using the lithium acetate transformation method described previously (19), and gene replacements were introduced using the one-step gene disruption technique (45). pFA6a-13Myc-kanMX6 (33) was used as a PCR template for the kanamycin resistance marker used to replace the PRY3 locus (the PRY3 open reading frame together with 700 bp 5′ and 370 bp 3′; strain KB1132), and mutants were selected on G418 (Geneticin; Invitrogen). Media and culture conditions were essentially as described previously (49). Yeast cells were grown at 30°C in YPD (1% yeast extract, 2% peptone, and 2% glucose) or selective synthetic medium with 2% glucose (SD) supplemented with 50 μg/ml adenine, 100 μg/ml tryptophan, and 0.2% Casamino Acids and lacking uracil to select for URA3 plasmids. Unless otherwise noted, cells were grown to mid-log phase (approximately 1 × 107 cells/ml) before harvesting.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmida | Genotype or yeast marker(s) | Reference or source |
|---|---|---|
| Strains | ||
| BY2125 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 ssd1-1 | L. L. Breedenc |
| XWV01 | MATa/α ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 ssd1-1 | V. L. MacKayc |
| BY4742 | MATα his3-1 leu2-0 ura3-0 lys2-Δpep4 | 6 |
| VM1822 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 ssd1-1Δfar1::TRP1 | V. L. MacKayc |
| VM1718 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 ssd1-1Δste12::TRP1 | 29 |
| KB1127 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 ssd1-1Δpry3::HIS3 | This work |
| KB1132 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 ssd1-1Δpry3loc::Kanr | This work |
| KB1157 | MATa ade2-1 his3-11,15 leu2-3,112 trp1-1 ura3-1 can1-100 ssd1-1Δace2::URA3 | This work |
| Plasmids | ||
| pKB51 | PRY3 locus in pRS316 | This work |
| pKB52 | pKB51 NotI-to-BamHI fragment in pBSII SK(+) | This work |
| pKB232 | Mutated TATA box 2 in pKB51 | This work |
| pKB224 | Deleted PREs in pKB51 | This work |
| pKB122 | Deleted UAS1 in pKB51 | This work |
| pKB112 | Mutated TATA box 1 in pKB51 | This work |
| pRS316b | CEN6 ARSH4 URA3 f1 origin | 50 |
| pBSII SK(+) | f1 origin in positive orientation, SacI-to-KpnI polylinker orientation | Stratagene, La Jolla, CA |
| pFA6a-13Myc-kanMX6b | 13× Myc tag, KanMX6 selection | 33 |
All strains are W303 background, except BY4742 (S288C background).
Plasmids kindly provided by the Trisha Davis lab.
Strain kindly provided by this colleague.
Construction of CEN plasmids carrying the wild-type and mutant PRY3 locus.
The plasmids used in this study are listed in Table 1, and relevant primers are listed in Table 2. The PRY3 ORF plus 1,185 bp 5′ and 850 bp 3′ was cloned into the polylinker region of pRS316 at the NotI and HindIII sites to create pKB51. The BamHI site located near the HindIII site in the vector was destroyed by BamHI digestion, T4 DNA polymerase fill-in of the sticky ends, and blunt-end ligation to facilitate future cloning steps. This construct and future derivatives were sequenced to confirm accuracy. The 5′ region of the PRY3 locus was excised from pKB51 between NotI and an internal PRY3 BamHI site and ligated into pBSII SK(+) to generate pKB52. pKB52 was used as the template for site-directed mutagenesis of all promoter elements. After confirmation of the correct sequence, the NotI-BamHI fragment was excised using the same enzymes and religated back into the parent pKB51 plasmid to create the mutant PRY3 locus plasmids described below. Site-directed mutagenesis was performed with primers listed in Table 2. Briefly, PCR (15 cycles, 60°C annealing temperature) used Phusion Hi-Fi polymerase (2.5 U/reaction; Finnzymes, Espoo, Finland) and 125 ng each of forward and reverse primer-generated products. Reaction mixtures were DpnI digested, followed by phenol-chloroform (1:1) extraction. For linear products due to a short deletion, T4 polynucleotide kinase (20 U/reaction; New England Biolabs, Ipswich, MA) was used for addition of a terminal phosphate, followed by ligation. All plasmid PCR products were transformed into XL1-Blue competent cells (Stratagene, La Jolla, CA). For construction of pKB232, the TATA box at +385 was mutated from TATAAATAT to TACAAGTAT, which disrupts the consensus site (2) but codes for the identical amino acids. To generate pKB224, 22 bp from −175 to −154 (relative to the first coding AUG) was replaced by a 5-bp insertion (HindIII site), eliminating both pheromone response element (PRE) sequences. To generate pKB122, 42 bp from −500 to −459 (relative to the AUG) was replaced by a 6-bp insertion (NcoI site), eliminating all four Ace2p/Swi5p consensus sequences. For construction of pKB112, the TATA box at −188 was mutated from TATATA to ATCGAT.
TABLE 2.
Primers relevant to this study
| Description | Primer name | Sequence (5′→3′) |
|---|---|---|
| S1 nuclease | P1 | GAGGACGAGGAAACAGTGCTTATAAGTGGTTCCACTTCCTCTGCAAACTCACCCAGGTAGTTTCCCTTCT |
| P2 | GGTTGTTGTTGTGGAAGAGGTGCCTTCGTTTGAGGGTTCG | |
| Clone PRY3 locus | PRY3loc-1 | ACTTATTTCATCGTGCTAGCTACTCTTC |
| PRY3loc-2 | CAAACAGAGGCTAAGTTCTTCAAAGC | |
| Site-directed mutagenesis | ||
| PRE deletion-F | PREdel-F | CTTCTCGGTCGAAGAATTCTCTAAAACATATTCTATACTTC |
| PRE deletion-R | PREdel-R | CTTCGGCCCTATATAGCAATTACATCCTTCCG |
| TATA box 2-F | TATAmut-F | GCCGAGATTGGATGTGGTTACAAGTATTGTGGTACGACATGG |
| TATA box 2-R | TATAmut-R | CCATGTCGTACCACAATACTTGTAACCACATCCAATCTCGGC |
| UAS deletion-F | FLpromD-F | TGGGTTTATAGTTACCGCAATGCAGGTCGC |
| UAS deletion-R | FLpromD-R | TGGCAATTTTTCGGCCAAACTTACCAACAATAGCG |
| TATA box 1-F | FLtataD-F | GATGGGCCGATGAAACAGTTCTCATGTTTCAAC |
| TATA box 1-R | FLtataD-R | GATGCAATTACATCCTTCCGTTGCTGGGTC |
| RLM-RACE | ||
| RNA adaptor | 5′Radap | GCUGAUGGCGAUGAAUGAACACUGCGUUUGCUGGCUUUGAUGAAA |
| RNA adaptor PCR forward 1 | 5ROPr | GCTGATGGCGATGAATGAACACTG |
| RNA adaptor PCR forward 2 | 5RIPr | CGCGGATCCGAACACTGCGTTTGCTGGCTTTGATG |
| PRY3 reverse transcribe | PRY3-RTL2 | CAGTGCTCGAGGTAGGGTCGGAAGAAG |
| PRY3 PCR reverse 1 | PRY3-GSL2a | CACTAGAGACGACGGTACTC |
| PRY3 PCR reverse 2 and sequence reverse | PRY3-LL77 | ATTACGGTAGAAGAGGACGCA |
| STE2 reverse transcribe | STE2-5′RT | CACAATCATACCTTTAACAGCGC |
| STE2 PCR reverse 1 | STE2-5′1 | CCTTTTGAAGTTGTCGCCTG |
| STE2 PCR reverse 2 | STE2-5′2 | GTTCCGCGGCTGCAGTTACCAGTGAAGTCTCAATAGAAG |
| STE2 sequence reverse | STE2-5′V1 | GGACTTGAATTATATTTGTAGCACC |
RNA analysis.
For total RNA isolation, cells were grown to mid-log phase, harvested, and lysed in 450 μl ice-cold RNA lysis buffer (50 mM Tris-HCl [pH 7.4], 100 mM NaCl, 10 mM EDTA) by glass bead bust (vigorous vortexing for 3 min at 4°C) followed by addition of 450 μl RNA buffer plus 1.3% sodium dodecyl sulfate. Samples were phenol, phenol-chloroform (1:1), and chloroform extracted. RNA was precipitated with NaCl and ethanol and resuspended with RNase-free water. For Northern blot analysis, aliquots (10 μg) of total RNA were run for 5 h at 96 V on 1% agarose gels containing 1.8 M formaldehyde in 40 mM morpholinepropanesulfonic acid (MOPS)-acetate buffer (pH 7.2), transferred to nylon membranes (GeneScreen Plus; Perkin-Elmer) for 72 h, and hybridized with [α-32P]dCTP-labeled probes specific for PRY3 (487 nucleotides from +1113 to +1600) and ACT1.
RNase protection assays were carried out as previously described (47) with a few modifications. The PRY3 RNase protection assay probe, containing 719 nucleotides of coding sequence and spanning both transcriptional start sites, was generated by linearization of pKB52 with SacII and synthesized from the T7 promoter incorporating [α-32P]UTP. The probe was isolated from a sequencing gel to ensure full-length integrity. The RNA probe was hybridized to 50 μg of total RNA for each sample at 65°C for 12 to 16 h, followed by RNase A (40 μg/ml) digestion at 4°C for 60 min. RNase-protected fragments were run on a 6% sequencing gel with an RNA Century Plus marker (Ambion).
S1 nuclease assays were performed as previously described (47) with gel-purified oligonucleotide probes (QIAGEN Corp.). Both S1 probes, P1 and P2, each contain five bases of nonhomologous sequence at the 5′ end (Table 2). Probes were individually end labeled with [γ-32P]ATP, mixed together in equal amounts, and hybridized at 65°C to 50 μg of total RNA (DNase I treated) for each sample. After hybridization, 60 U of S1 nuclease (Invitrogen) was used for digestion at 37°C for 30 min. S1 reactions and sequencing reactions, generated from pBSII SK(+), were run in parallel on 8% sequencing gels.
Hybridization or probe protection intensities for Northern blot, RNase protection, and S1 nuclease assays were quantified with a PhosphorImager and ImageQuant software (Molecular Dynamics, Sunnyvale, Calif.).
RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE) was carried out as previously described (32) with a synthesized RNA oligonucleotide adaptor (Invitrogen). The sequences for the RNA adaptor as well as specific primers used for reverse transcription and nested PCR can be found in Table 2. Enzymes used in this protocol were calf intestine alkaline phosphatase (CIP) (20 U/reaction; New England Biolabs), tobacco acid pyrophosphatase (TAP) (5 U/reaction; Epicenter Biotechnologies), RNaseOut (RNase inhibitor, 10 U/reaction; Invitrogen), T4 RNA ligase (2 U/reaction; New England Biolabs), and Thermo-X reverse transcriptase (200 U/reaction at 65°C; Invitrogen). Five micrograms of total RNA from untreated and α-factor-treated cells was analyzed by RNA adaptor ligation to non-CIP/non-TAP-treated aliquots (no cap) and to CIP/TAP-treated aliquots (capped). Some CIP-only-treated RNA aliquots were also ligated to the adaptor as a control for the CIP enzymatic reaction. PCR products were sequenced using gene-specific reverse primers (Table 2).
Yeast biological assays.
Cells were treated with α-factor (7 μM) dissolved in methanol as previously described (29, 34, 37). 1,10-Phenanthroline (Sigma-Aldrich) was dissolved in ethanol at a concentration of 100 mg/ml and added to cells to a final concentration of 100 μg/ml to inhibit RNA synthesis (20).
Quantitative mating assays were performed essentially as described previously (22). Cells (1 × 106) from a MATa experimental strain (KB1127) containing one URA3+ plasmid (pKB51, pRS316, or pKB224) grown in SD medium lacking uracil were mixed with 1 × 107 MATα wild-type cells (BY4742) grown in YPD and collected by vacuum filtration onto 0.45-μm-pore-size, 25-mm nitrocellulose filter disks (Millipore). Disks were incubated on top of YPD plates at 30°C for 3 h. Cells were resuspended in water, sonicated briefly, and diluted to titer a/α diploids (selected on SD plates lacking uracil and tryptophan) and MATa haploids plus a/α diploids (selected on SD plates lacking uracil). Mating efficiency was calculated as the number of a/α diploids divided by the total number of colonies on the SD plates lacking uracil (a/α diploids plus MATa haploids) and expressed as a percentage.
RESULTS
Mating pheromone induces an alteration in the 5′ terminus of the PRY3 transcript.
We previously reported an alteration in the 5′ structure of the PRY3 mRNA when cells are treated with α-factor (29). In growing cells, the 5′ terminus of PRY3 resides at −76 relative to the initiation AUG of the ORF, while following α-factor treatment, the PRY3 5′ terminus was found at +452 (29). The 3′ untranslated region of PRY3 is 357 nucleotides in length and did not change during pheromone treatment (L. Law, unpublished data). In Fig. 1A, we depict the established structure of the full-length PRY3 mRNA, including the sequence coding for the secretory signal for the full-length cell wall protein, and designate the start of the shorter +452 PRY3 form as well as the next in-frame AUG at +568. The results of 5′ RACE suggest that the transcript is contiguous with the gene sequence, eliminating the possibility of intron excision as an explanation for the decrease in transcript length (46). This was confirmed by RNase protection (Fig. 1B). RNase protection assays were carried out using a probe detecting both short and full-length transcriptional start sites. This probe was confirmed to be PRY3 specific by testing it on total RNA isolated from a Δpry3 strain (data not shown) and completely digestible with RNase A when incubated with tRNA (Fig. 1B, lane 4). RNase protection results in a prominent band consistent with the major 5′ terminus at −76 in growing cells (Fig. 1B, lane 5) and a band consistent with +452 in α-factor-treated cells (Fig. 1B, lanes 6 and 7).
FIG. 1.

Identification of an altered 5′ transcript structure of PRY3 in cells treated with α-factor. (A) Diagram of PRY3 transcript showing transcriptional and translational start sites for the full-length transcript. The terminus of the short 5′ transcript is marked by the line at +452. A possible translational start site at an in-frame AUG is highlighted at +568. (B) RNase protection assay of PRY3 performed on RNA isolated from untreated cells (lane 5) or cells treated with α-factor for 20 and 50 min (lanes 6 and 7). The RNase protection probe, containing 719 nucleotides of coding sequence and detecting both PRY3 transcriptional start sites, is shown in lanes 2 and 3 at two concentrations. An RNA ladder (lane 1) allows calculation of probe protection and confirms 5′ termini of the two PRY3 transcripts (marked with arrows). Lane 4 is probe mixed with tRNA (50 μg) as a digestion control. These data are representative of three independent experiments. (C) Northern analysis of PRY3 in wild-type cells treated with α-factor. RNA was isolated from cells that were treated or not for 20 and 50 min. The blot was reprobed for ACT1 as a loading control. This result was obtained in four independent experiments. The arrows mark the full-length and short PRY3-specific transcripts.
Northern blot assays also substantiate the previous results (Fig. 1C) and, together with RNase protection, demonstrate that the long form of the PRY3 transcript dissipates, while the shorter form of the transcript appears within 20 min and is retained for at least 50 min following addition of α-factor. Quantitation of Northern blots (four independent experiments) at 20 min after α-factor addition demonstrated a reproducible reduction in the level of PRY3 long form to 13% (±1%) of that of the untreated control. The short form, which is undetectable in untreated cells, accumulates to 66% (±2%) of the level of long form in untreated cells.
Kinetics of alteration in PRY3 transcript structure upon α-factor addition.
Northern blot analysis and RNase protection assays suggested that the switch between long and short form of the PRY3 transcript is complete within 20 min following α-factor exposure. To analyze further the kinetics of these structural changes, we developed an S1 nuclease assay using a radiolabeled oligonucleotide probe (P1), which crosses the +452 junction and differentiates between full-length and shorter PRY3 transcripts. A second radiolabeled oligonucleotide probe (P2) that detects a sequence common to both transcripts, located at the 3′ end of the PRY3 ORF, was added to each S1 nuclease reaction and used as a control for digestion and recovery (Fig. 2A). α-Factor was added to wild-type cells, and samples were harvested and frozen at the indicated times. Total RNA was isolated, and the PRY3 transcripts were analyzed quantitatively by S1 nuclease assay across the time course (Fig. 2B and C). The kinetics demonstrate that the decline in the level of the full-length form of PRY3 was nearly complete by 5 min after α-factor addition (Fig. 2B, lane 8; Fig. 2C), while the +452 form became detectable after 5 min and reached steady state by ∼15 min (Fig. 2C). The final levels of both transcript forms remained stable at least 90 min following pheromone addition (data not shown). These results were confirmed with an RNase protection assay over the same time course (data not shown). We conclude that these changes in PRY3 transcript structure are both rapid and sustained in the presence of α-factor.
FIG. 2.

Kinetics of alteration in PRY3 transcript structure during an α-factor time course. (A) Oligonucleotide probes for S1 nuclease assay were designed as shown, with an additional five bases of nonhomologous sequence at the 5′ end of the probe to control for S1 nuclease activity. (B) Total RNA was isolated from wild-type cells treated or not treated with α-factor for the indicated times. Probe hybridization was performed with a mix of both probes, followed by S1 nuclease digestion (see Materials and Methods). A sequencing reaction from pBSII SK(+) plasmid generated the base pair ladder (lanes 1 to 4). Lane 5 is the mixture of the two probes with no S1 added during the experiment. Lane 6 is the probe mixture hybridized to tRNA (50 μg) as a digestion control. These data are representative of five independent experiments. (C) Levels of full-length transcript (solid line with triangles) and +452 transcript (dashed line with squares) were quantified from the S1 nuclease assay in panel B. Values for both are shown as a percentage of the intensity of the full-length transcript at 0 min. Total PRY3 levels (dotted line with diamonds) are the P2 probe quantities for the α-factor time course plotted as percentage of the total transcript at 0 min.
The appearance of the PRY3 short transcript requires transcription.
The fact that the long PRY3 transcript disappeared more quickly than the short form accumulated is inconsistent with a simple processing mechanism that would convert the long to short forms. To address the possibility that the full-length PRY3 decay rate was accelerated in the presence of α-factor, we employed the drug 1,10-phenanthroline (20, 48) to inhibit RNA synthesis. 1,10-Phenanthroline was added to steady-state growing cells, and the full-length PRY3 transcript level (as assessed by probes P1 and P2) was quantified over a time course. The demonstrated half-life for full-length PRY3 was 6 to 10 min (Fig. 3A and B), similar to the rapid decline of full-length PRY3 observed in the presence of α-factor (Fig. 2). These results suggest no enhancement of degradation by pheromone.
FIG. 3.

Analysis of PRY3 transcript structure in the presence of RNA Pol II inhibitor. (A) Total RNA was isolated from wild-type cells treated or not treated with 1,10-phenanthroline (100 μg/ml) for the times indicated, followed by S1 nuclease assay as described in the Fig. 2 legend. (B) Levels of PRY3 transcript from panel A as detected by the 5′ oligonucleotide probe (P1; solid line with triangles) or as detected by the 3′ probe (P2; dotted line with diamonds) were individually quantified and shown as a fraction of the PRY3 transcript from cells at 0 min. Data are representative of four independent experiments. (C) Wild-type cells were treated with 1,10-phenanthroline (100 μg/ml) for 5 min prior to the addition of α-factor at 0 min. Total RNA was isolated from cells harvested and frozen at the times indicated, followed by S1 nuclease assay as described in the Fig. 2 legend. (D) Levels of full-length transcript (solid line with triangles) and +452 transcript (dashed line with squares) from panel C were quantified and shown as a percentage of the full-length transcript from cells at 0 min. Data are representative of three independent experiments.
To determine if RNA polymerase II (Pol II) activity is required for the appearance of the short form, 1,10-phenanthroline was added to growing wild-type cells to block transcription 5 min prior to the addition of α-factor to the culture. Total RNA was isolated from cell samples frozen at the indicated times, and PRY3 transcripts were analyzed by S1 nuclease assay as previously described. As expected, following α-factor addition, the level of the full-length form of PRY3 declined while the +452 form of PRY3 did not accumulate significantly, consistent with transcription being required for this event (Fig. 3C and D). The small amount of +452 transcript formed and the remaining long form were most likely due to residual RNA Pol II activity in the presence of 100 μg/ml of 1,10-phenanthroline (48). These data imply that while the α-factor-dependent decline in full-length PRY3 is consistent with a block in transcription followed by normal PRY3 decay, the short form is produced by new transcription in response to pheromone treatment. However, the requirement for transcription could be indirect.
If the +452 form of PRY3 were the result of cleavage or degradation of the full-length transcript, it should not contain a cap at its 5′ terminus. Alternatively, if the PRY3 +452 transcript were produced from of a new promoter within the PRY3 locus, it should possess a 7-methyl guanosine cap (21). To differentiate between these possibilities, we used RLM-RACE to selectively amplify gene-specific PCR products from pools of capped and uncapped total RNA isolated from cells treated and not treated with α-factor (Fig. 4). Briefly, an RNA adaptor was ligated to termini of RNAs containing an exposed 5′ phosphate (most uncapped RNAs). To examine pools of uncapped RNAs, we ligated the RNA adaptor to transcripts from total RNA isolations and then carried out gene-specific reverse transcription and amplification (Fig. 4, “no cap”). Pools of capped RNAs were examined by a three-step process: removal of 5′ phosphates from uncapped RNAs using CIP and removal of 7-methyl guanosine caps (leaving exposed 5′ phosphates) using TAP, followed by ligation of the RNA adaptor to this pool of RNAs (Fig. 4, “cap”). To confirm that we were detecting only capped RNAs in this pool, we ligated the RNA adaptor to a pool of RNAs treated only with CIP as a test for incomplete phosphatase reactions (Fig. 4, “control”). Following ligation of the RNA adaptor selectively to pools of capped and uncapped RNAs, RNAs were reverse transcribed with PRY3 and STE2 gene-specific primers. Gene-specific RACE products, from two independent primer sets, were derived from PCRs using an adaptor-specific forward primer and gene-specific reverse primer.
FIG. 4.

PRY3 5′ cap analysis by RNA ligase-mediated RACE. Transcript-specific RACE PCR products were analyzed by agarose gel electrophoresis. Data shown are representative of three independent experiments each using two independent, gene-specific primer sets demonstrating transcript specificity. For both PRY3- and STE2-specific RACE reactions, amplification products in the “no cap” lanes indicate the RNA adaptor ligated to the exposed 5′ phosphates of uncapped transcripts. “Control” lanes indicate completeness of the CIP reaction. Amplification products in the “cap” lanes indicate the transcripts in which the RNA adaptor ligated to the newly exposed 5′ phosphate groups of capped transcripts following the removal of the 5′ cap by TAP. “NT” represents no-template control for PCRs. Lanes 1, 16, 17, and 25 are loaded with a 100-bp ladder. PCR products were isolated from lanes 3, 6, 7, 10, 13, 14, 22, and 23 and sequenced. (A) PRY3 RLM-RACE results for both primer sets used. Sequencing results from PCR products from lanes 3, 7, 10, and 14 demonstrated 5′ termini at +452 relative to the AUG translation initiation codon. Sequencing results from lanes 6 and 13 demonstrated 5′ termini at −76. (B) STE2, an α-factor-induced transcript which has a known 5′ transcriptional start site of −31 relative to the initiating AUG, was used as a control (results shown for primer set 1).
PRY3-specific long and short RLM-RACE products from untreated and α-factor-treated (30-min) cell RNA extracts were found in the capped pools of mRNA (Fig. 4A, lanes 6, 7, 13, and 14). We subsequently sequenced these amplification products to verify that the 5′ termini were at −76 (untreated) and +452 (α-factor treated) relative to the first coding AUG as was found previously (29). Neither of these PRY3 amplification products was found in the “control” lanes (Fig. 4A, lanes 4, 5, 11, and 12). Interestingly, the +452 RLM-RACE products were amplified in the “no-cap” (+α-factor) condition (Fig. 4A, lanes 3 and 10), demonstrating that some uncapped +452 PRY3 transcript exists. The uncapped population of +452 transcripts could result from processing the long PRY3 transcript; however, because a capped population of short transcript exists, it is most likely due to decapping of the newly transcribed short mRNA. The RLM-RACE and sequencing results with STE2 serve as a control for the fidelity of this experimental approach as its 5′ terminus is known to remain at −31 relative to the translation AUG initiation codon in both treated and untreated cells (Fig. 4B, lanes 22 and 23) (29). From both the RLM-RACE and the 1,10-phenanthroline results, we conclude that the +452 PRY3 transcript is the result of new transcription at the PRY3 locus following exposure to α-factor.
Influence of ACE2 on PRY3 transcript expression.
Given that PRY3 is expressed during G1 (51), and the full-length promoter region drives expression of a reporter gene specifically in daughter cells (9), we tested the influence of the ACE2 gene, which regulates daughter cell-specific transcription during G1, on the structure and quantity of the PRY3 transcripts. Total RNA was isolated from Δace2 cells with and without α-factor treatment for 30 min. Northern blot analysis demonstrated that ACE2 was required for wild-type levels of PRY3 expression (compare lanes 1 and 5 in Fig. 5), confirming daughter cell-specific expression of this gene. The small amount of PRY3 transcript detected (6% of wild-type levels) was possibly due to activation by Swi5p in the vicinity of Ace2p binding sites or possibly through other transcriptional regulators; both have been demonstrated for other genes in the absence of Ace2p (13). The peak of PRY3 transcription during early G1 and the presence of possible Ace2p binding sites in the region of the PRY3 promoter are consistent with a direct role of Ace2p. Production of the short form of PRY3 was also dependent on ACE2 (Fig. 5, lane 6) and suggests that it, like the long form, may be restricted to daughter cells.
FIG. 5.

PRY3 expression is FAR1 independent but ACE2 dependent. Northern analysis of PRY3 in total RNA isolated from wild-type (WT), Δfar1, and Δace2 cells untreated or treated with α-factor for 30 min. The blot was reprobed for ACT1 as a loading control. Data are representative of two independent experiments.
Pheromone regulates PRY3 transcript structure via the Ste12p transcription factor.
Acute pheromone treatment of wild-type cells drastically changes the transcriptome through the combined action of the mitogen-activated protein kinase-activated transcription factor Ste12p and cell cycle arrest, which is dependent on Far1p. Thus, either or both of these pheromone-induced pathways could be responsible for the appearance of the short PRY3 transcript (15, 44). It seems unlikely that PRY3 short form is regulated by the cell cycle, since no short form was detected by either RNase protection assay or Northern blotting in steady-state, cycling cells (Fig. 1B, lane 5, and 1C, lane 1). To determine if the short PRY3 transcript is dependent on cell cycle arrest in G1 phase, we disrupted the FAR1 gene. When treated with α-factor, Δfar1 cells continue to cycle but can still mate, albeit with severely reduced efficiency (41). Northern blot analysis showed that the short form of PRY3 accumulated in Δfar1 cells, to a level similar to that of wild type, when treated with α-factor for 30 min (Fig. 5, compare lanes 2 and 4). The Δfar1 strain did not cell cycle arrest since 75% of Δfar1 cells had buds (after 90 min of α-factor treatment), while only 6% of wild-type cells were budded after the same treatment. This result implies that pheromone-induced events other than cell cycle arrest are required for the alteration in PRY3 transcript structure.
The primary transcription factor responsible for induction of pheromone-regulated genes is Ste12p (12, 44). We tested whether the transcription factor was required for the change in PRY3 transcript structure by treating wild-type cells and cells lacking STE12 with pheromone for 30 min. Northern analysis revealed no change in transcript structure in Δste12 cells treated with α-factor (Fig. 6A). We confirmed these results by inducing the Δste12 mutant with α-factor over a 60-min time course, isolating total RNA, and analyzing PRY3 transcript structure by S1 nuclease assay (Fig. 6B). In this experiment, the +452 transcript was undetectable over the 60-min time course. In addition, the full-length PRY3 transcript did not decrease in the Δste12 mutant in the presence of pheromone. These results suggest a model in which appearance of the +452 PRY3 transcript and repression of full-length PRY3 transcription both require the Ste12p transcription factor. However, the involvement of Ste12p is not necessarily directly at the PRY3 locus.
FIG. 6.

Pheromone-induced PRY3 transcript regulation is STE12 dependent. (A) Northern analysis of PRY3 in total RNA isolated from wild-type (WT) and Δste12 strains untreated and treated with α-factor for 30 min. The blot was reprobed for ACT1 as a loading control. (B) S1 nuclease assay was performed on total RNA extracts from Δste12 cells isolated during an α-factor time course assay as described in the Fig. 2 legend.
Regulatory elements within the PRY3 locus.
The PRY3 locus (the PRY3 ORF plus 1,185 nucleotides upstream and 850 nucleotides downstream of the full-length ORF) was cloned into a yeast CEN plasmid carrying URA3 for selection. This wild-type construct was transformed into a MATa haploid strain in which the chromosomal PRY3 ORF plus 700 nucleotides upstream and 370 nucleotides downstream was replaced with the kanamycin resistance marker (Δpry3loc). Both the full-length and short PRY3 mRNAs were transcribed from the wild-type construct in quantities similar to those of a wild-type strain (Fig. 7B and 7C; compare lanes 1 to 3 with Fig. 1C, lanes 1 to 3). An S1 nuclease assay over a 60-min α-factor time course confirmed that the plasmid-derived short form of PRY3 starts at +452 (data not shown). Additionally, the change in PRY3 transcript structure derived from the wild-type construct occurs at the same rate as in wild-type cells carrying an empty vector under the same growth conditions (data not shown). Therefore, all the features necessary for initiating a new transcriptional start site at +452 are found within the PRY3 ORF and the associated 5′ and 3′ flanking sequences.
FIG. 7.

Analysis of PRY3 promoter elements. (A) Diagram depicting promoter elements for the full-length and α-factor-induced transcripts. (B) Northern analysis of PRY3 transcript in total RNA isolated from Δpry3 strains carrying the following plasmids: wild-type (WT) construct, mutated TATA box 2 at +385, deleted PREs, and empty vector. RNA was isolated from cells untreated and treated with α-factor for 20 and 50 min. The blot was reprobed for ACT1 as a loading control. Data shown are representative of two biologically independent experiments from Δpry3loc and Δpry3 strains carrying the plasmids. (C) Northern analysis of PRY3 in total RNA isolated from Δpry3 strains carrying the following plasmids: wild-type (WT) construct, deleted UAS 1, mutated TATA box 1 at −188, and empty vector. RNA was isolated from cells untreated and treated with α-factor for 20 and 50 min. The blot was reprobed for ACT1 as a loading control. Data shown are representative of two biologically independent experiments from Δpry3loc and Δpry3 strains carrying the plasmids.
Sequence inspection revealed the presence of sequences that could potentially regulate a change in promoter utilization at the PRY3 locus upon pheromone treatment. Two putative PREs, which are Ste12p binding sites (12), were discovered juxtaposed between putative Ace2p/Swi5p binding sites and the transcriptional start site of the full-length transcript at −76 (Fig. 7A). Additionally, a potential TATA box was found at +385, which is 5′ of the +452 transcriptional start site (Fig. 7A). To determine if these putative promoter elements are important for PRY3 transcript regulation, we tested two mutant constructs: a double point mutation of the putative TATA box at +385 (“ΔTATA box 2”), which altered it from TATAAATAT to TACAAGTAT, and a deletion of both Ste12p binding sites (“ΔPREs”). By Northern analysis we observed that in cells containing ΔTATA box 2, the level of full-length PRY3 was decreased after treatment with α-factor similarly to the wild-type construct (10% of the level of long transcript in untreated cells). The short transcript starting at +452 appeared as only a faint, diffuse band (Fig. 7B; compare lanes 4 to 6 with 1 to 3 and Fig. 1C), possibly due to weak cryptic transcriptional start sites as revealed previously in yeast by removing a functional TATA box (40). When cells carrying the ΔPRE construct were treated with α-factor, the level of the full-length transcript did not dissipate as observed in wild-type cells and the short transcript was never detected (Fig. 7B, lanes 7 to 9). This result is identical to that obtained with the Δste12 mutant in Fig. 6A and consistent with the Ste12p transcription factor acting directly at the PRY3 locus. These results suggest that there is a promoter located within the PRY3 locus specific for the short form and that during pheromone induction for mating, the short transcript is actively produced at the expense of the full-length transcript.
To further define the nature of the apparent switch in promoter usage, we sought to confirm the sites of the promoter elements for the full-length transcript. A possible upstream activation site (UAS 1) for the full-length transcript consists of a series of four putative Ace2p/Swi5p binding sites residing between −485 and −460 relative to the translation AUG initiation codon (Fig. 7A). Additionally there is a putative TATA box at −188, located 5′ of and in close proximity to the two PREs (Fig. 7A). To investigate a potential role for these putative elements, two additional mutant constructs were made: a deletion of the putative Ace2p/Swi5p binding sites (“ΔUAS 1”) and a disruption of the TATA box associated with the upstream promoter (“ΔTATA box 1”), changing it from TATATA to ATCGAT. Like the Δace2 mutant strain, the full-length PRY3 transcript was greatly reduced in the ΔUAS 1 construct (6% of that of wild-type levels), consistent with the Ace2p transcription factor acting directly at the PRY3 promoter (Fig. 7C, compare lanes 1 and 4). In the absence of the UAS 1 region, the +452 PRY3 transcript failed to accumulate to wild-type levels following pheromone treatment (5% of the level of full-length transcript in untreated cells), suggesting that the UAS 1 region is also required for full activation of the α-factor-induced transcript (Fig. 7C, compare lanes 5 and 6 with lanes 2 and 3). In untreated cells carrying the ΔTATA box 1 construct, expression of the full-length transcript was greatly diminished (11% of that of wild-type levels) and a small amount of short PRY3 transcript was detectable (Fig. 7C, lane 7). In contrast to the results from the ΔUAS 1 construct, α-factor-treated cells carrying the ΔTATA box 1 construct were able to produce wild-type levels of +452 PRY3 transcript (Fig. 7C, lanes 8 and 9). These results indicate that the Ace2p binding region is required for wild-type levels of full-length PRY3 transcription in growing cells, as well as +452 PRY3 transcription in the presence of α-factor. Additionally, the TATA box found at −188 is required for induction of full-length PRY3 transcription and also contributes to preventing transcription from the +452 start site in the absence of α-factor.
Expression of full-length PRY3 suppresses mating.
It has been previously reported that strains deficient in PRY3 have increased mating efficiency (16). To test if mating is influenced by long or short PRY3 transcript expression, a quantitative mating assay was used to establish mating efficiencies of a Δpry3 MATa yeast strain carrying the following three plasmids: (i) wild-type PRY3, (ii) empty vector (no PRY3 expression), and (iii) a construct that expresses only full-length PRY3 from the daughter cell-specific promoter with the PREs deleted (ΔPREs). These three strains were mated with a wild-type MATα strain for 3 h and screened for diploid formation. The Δpry3 strain mated with an efficiency at least equal to that of wild type in four independent experiments (Fig. 8). In contrast, strains expressing only full-length transcript, due to the PRE deletions, reproducibly mated 40 to 50% less efficiently than wild type (Fig. 8).
FIG. 8.
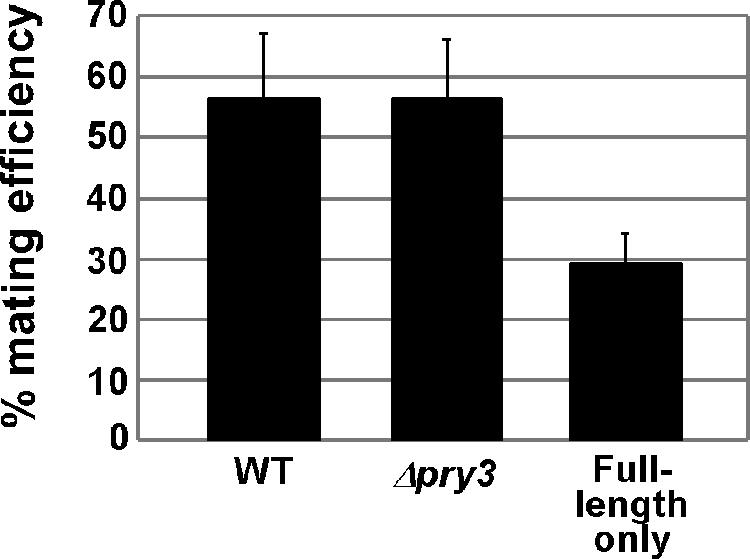
Production of full-length PRY3 is inhibitory to mating. The figure shows MATa Δpry3 strains carrying the PRY3 wild-type (WT) construct, empty vector, or the PRY3 PRE deletion construct (full length only). Results are averages of four independent experiments. Wild-type cells carrying an empty vector mated with the same efficiency as did Δpry3 cells carrying a wild-type construct in two experiments (data not shown).
DISCUSSION
Start site selection is dictated by the architecture of the PRY3 regulatory region.
There are multiple elements within the PRY3 locus that control two promoters. An upstream activation sequence containing as many as four putative Ace2p/Swi5p binding sites is found at −485 to −460, and as expected from the presence of these binding sites (9), wild-type transcription is dependent on the ACE2 gene. This upstream activation region is necessary to support transcription from both PRY3 start sites. Expression of full-length (mapped to −76) PRY3 requires a TATA box at −188 (TATA box 1). Juxtaposed between the −76 start site and TATA box 1 are two PREs which are necessary to invoke a rapid repression of full-length PRY3 transcription upon α-factor exposure. In addition to repression of the long transcript, these putative Ste12p binding sites are required for induction of the short form initiating at +452, and both repression and induction through the PREs require the STE12 gene. Finally, the +452 PRY3 transcript requires the TATA box positioned at +385 (TATA box 2) for full induction. Sequence alignment of four closely related Saccharomyces species demonstrates that all of the promoter elements 5′ of the full-length ORF are perfectly conserved (8, 24). However, in only two species, Saccharomyces mikatae and Saccharomyces paradoxus, are the intragenic TATA box 2 at +385 and the in-frame ATG at +568 conserved. This evidence suggests that the mechanism for down-regulating the long PRY3 transcript is likely conserved; however, it also alludes to the possibility that the act of transcription of the short form of PRY3 may not be an essential property of this regulatory mechanism.
Does the short PRY3 transcript have an independent biological role?
The full-length PRY3 transcript codes for a cell wall protein (56) containing a conserved sperm-coating glycoprotein (SCP) domain from amino acids 24 through 152. The function of SCP domains remains elusive; however, these proteins have been implicated in pathogen resistance in plants (25) and immune responses in humans (14), suggesting an intriguing functional link (52). It is possible that a truncated form of Pry3p with biological function is translated from the pheromone-induced short form of the PRY3 transcript, as has been reported previously for a number of genes in yeast, including SUC2, MOD5, LEU4, and KAR4 among others (3, 5, 17, 53). The fact that ribosomes load on the short PRY3 transcript after α-factor treatment, albeit inefficiently (approximately 2 ribosomes per 1,000 nucleotides compared to a transcriptome average in yeast of 4.4 ribosomes per 1,000 nucleotides), leaves open the possibility that the +452 form of PRY3 may produce a protein product (34). There is an in-frame AUG at +568 that could be utilized for translational initiation. This putative short Pry3 protein would lack a secretory signal and therefore, if made, would likely be cytoplasmic. In addition, the shorter protein would lack the SCP domain and have no other recognizable function. If a short form of Pry3p is made, it is not required for mating, since strains with a complete deletion of PRY3 mate as efficiently as (this paper) or better than (16) wild-type strains; however, this does not preclude the possibility that a putative short Pry3p could have an undiscovered function that is not required for efficient mating. These results, together with the lack of an established function for the short transcript, point to an alternative class of interpretations in which the regulated shift in promoter usage is the by-product of a repression mechanism directed towards limiting expression of inhibitory full-length PRY3 in daughter cells during mating.
In contrast to the Δpry3 strain, removal of the two PREs, which prevented down-regulation of PRY3 in addition to blocking induction of the short transcript, resulted in a 40 to 50% reduction in mating efficiency of the total population compared to wild type. This result is consistent with continued production of full-length Pry3p completely inhibiting mating in newborn daughter cells, which compose 40% of a growing population of cells (42).
Regulation of PRY3 in daughter cells.
During their extended G1 phase (9, 28), daughter cells express PRY3 from the −76 transcriptional start site in an Ace2p- and TATA box-dependent fashion. Upon exposure to pheromone, expression of the short form of PRY3 also relies on Ace2p-dependent activity at the UAS 1 promoter region, despite the fact that Ste12p is a robust transcriptional activator. This observation suggests models where the induction of full-length PRY3 transcript in newborn daughter cells and the Ste12p-dependent induction of the short form depend on Ace2p-dependent chromatin remodeling for exposure of TATA box 1 and the two PREs. Another possibility is that Ace2p and Ste12p might both be required to recruit TATA binding protein (TBP), RNA Pol II transcriptional machinery, or other cofactors to the downstream start site to generate the short form of PRY3 following pheromone exposure. Based on the results reported here, Ste12p would not be expected to activate synthesis of the +452 PRY3 transcripts in mother cells, because of the dependence on Ace2p.
Ste12p functions as a transcriptional repressor.
Ste12p is a potent transcriptional activator. Induction of the pheromone response pathway leads to activation of more than 200 genes in a STE12-dependent manner (43, 44). In addition, microarray studies have also shown that approximately 200 genes are repressed by pheromone, albeit the vast majority of these are cell cycle regulated and their repression is FAR1 dependent (44). From studies reported here, binding of Ste12p to the PREs in the promoter of PRY3 appears to provide a mechanism for rapid, gene-specific repression. To our knowledge this is the first report of Ste12p acting directly as a repressor of gene expression through any mechanism. The concomitant activation of the intragenic PRY3 promoter by Ste12p, producing the short transcript, could be due to the unmasking of cryptic promoter elements as the result of the proximate binding of this strong transcriptional activator. It seems quite possible that activation of cryptic intragenic promoters may often occur when transcription activators are utilized as repressors and in the absence of any selection against it. Cryptic intragenic TATA boxes and transcriptional start sites have previously been found, although experimentally induced aberrations in transcriptional elongation were required to reveal these cryptic start sites (7, 23).
Given the location of the PREs only 12 nucleotides 3′ of TATA box 1, pheromone-activated Ste12p could compete for occupancy of this region (57), either displacing TBP or inhibiting assembly of an RNA Pol II preinitiation complex (27). Transcriptional interference through promoter occlusion has been demonstrated previously; however, this typically requires activation of an upstream promoter followed by transcriptional elongation through the downstream promoter, in the process disrupting assembly of functional transcription complexes (1, 10, 36). The results with PRY3 suggest a model where Ste12p acts directly as a repressor of transcription by possibly preventing nucleation of the RNA Pol II transcriptional machinery at the full-length core promoter region or through another chromatin-modifying mechanism. A similar mechanism has been suggested for repression of ZRT2 during severe zinc limitation, through binding of the transcriptional activator Zap1p at a zinc response element located 3′ of the TATA box and overlapping the transcriptional start site (4).
The architecture of the PRY3 promoter region is one solution to actively repressing expression of a gene in response to an environmental signal. PRY3 regulation may be representative of a general strategy for transcriptional repression, since other genes in our original screen for altered 5′ transcript leaders (29) have similar promoter architectures (our unpublished observation), including another cell wall protein, CRH1. It seems likely that other instances of this regulatory strategy will be found to coordinate induction and repression within a regulatory network under conditions where transcription factors are activated in response to environmental cues.
Acknowledgments
We are grateful to Linda Breeden, Vivian MacKay, Trisha Davis, and Per Widlund for their generous gifts of plasmids, strains, and technical advice. We thank Lynn Law, Linda Breeden, and Adam Geballe for suggestions throughout this work and for comments on the manuscript.
This study was supported by research grants from the National Institutes of Health (CA89807 and CA71453). K.S.B. was supported under a National Science Foundation Graduate Research Fellowship and in part by PHS NRSA T32 GM07270 from NIGMS.
Footnotes
Published ahead of print on 28 August 2006.
REFERENCES
- 1.Adhya, S., and M. Gottesman. 1982. Promoter occlusion: transcription through a promoter may inhibit its activity. Cell 29:939-944. [DOI] [PubMed] [Google Scholar]
- 2.Basehoar, A. D., S. J. Zanton, and B. F. Pugh. 2004. Identification and distinct regulation of yeast TATA box-containing genes. Cell 116:699-709. [DOI] [PubMed] [Google Scholar]
- 3.Beltzer, J. P., S. R. Morris, and G. B. Kohlhaw. 1988. Yeast LEU4 encodes mitochondrial and nonmitochondrial forms of alpha-isopropylmalate synthase. J. Biol. Chem. 263:368-374. [PubMed] [Google Scholar]
- 4.Bird, A. J., E. Blankman, D. J. Stillman, D. J. Eide, and D. R. Winge. 2004. The Zap1 transcriptional activator also acts as a repressor by binding downstream of the TATA box in ZRT2. EMBO J. 23:1123-1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boguta, M., L. A. Hunter, W. C. Shen, E. C. Gillman, N. C. Martin, and A. K. Hopper. 1994. Subcellular locations of MOD5 proteins: mapping of sequences sufficient for targeting to mitochondria and demonstration that mitochondrial and nuclear isoforms commingle in the cytosol. Mol. Cell. Biol. 14:2298-2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brachmann, C. B., A. Davies, G. J. Cost, E. Caputo, J. Li, P. Hieter, and J. D. Boeke. 1998. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 14:115-132. [DOI] [PubMed] [Google Scholar]
- 7.Carrozza, M. J., B. Li, L. Florens, T. Suganuma, S. K. Swanson, K. K. Lee, W. J. Shia, S. Anderson, J. Yates, M. P. Washburn, and J. L. Workman. 2005. Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123:581-592. [DOI] [PubMed] [Google Scholar]
- 8.Cliften, P., P. Sudarsanam, A. Desikan, L. Fulton, B. Fulton, J. Majors, R. Waterston, B. A. Cohen, and M. Johnston. 2003. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science 301:71-76. [DOI] [PubMed] [Google Scholar]
- 9.Colman-Lerner, A., T. E. Chin, and R. Brent. 2001. Yeast Cbk1 and Mob2 activate daughter-specific genetic programs to induce asymmetric cell fates. Cell 107:739-750. [DOI] [PubMed] [Google Scholar]
- 10.Corbin, V., and T. Maniatis. 1989. Role of transcriptional interference in the Drosophila melanogaster ADH promoter switch. Nature 337:279-282. [DOI] [PubMed] [Google Scholar]
- 11.Dohrmann, P. R., W. P. Voth, and D. J. Stillman. 1996. Role of negative regulation in promoter specificity of the homologous transcriptional activators Ace2p and Swi5p. Mol. Cell. Biol. 16:1746-1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dolan, J. W., C. Kirkman, and S. Fields. 1989. The yeast STE12 protein binds to the DNA sequence mediating pheromone induction. Proc. Natl. Acad. Sci. USA 86:5703-5707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doolin, M. T., A. L. Johnson, L. H. Johnston, and G. Butler. 2001. Overlapping and distinct roles of the duplicated yeast transcription factors Ace2p and Swi5p. Mol. Microbiol. 40:422-432. [DOI] [PubMed] [Google Scholar]
- 14.Eberle, H. B., R. L. Serrano, J. Fullekrug, A. Schlosser, W. D. Lehmann, F. Lottspeich, D. Kaloyanova, F. T. Wieland, and J. B. Helms. 2002. Identification and characterization of a novel human plant pathogenesis-related protein that localizes to lipid-enriched microdomains in the Golgi complex. J. Cell Sci. 115:827-838. [DOI] [PubMed] [Google Scholar]
- 15.Elion, E. A. 2000. Pheromone response, mating and cell biology. Curr. Opin. Microbiol. 3:573-581. [DOI] [PubMed] [Google Scholar]
- 16.Entian, K. D., T. Schuster, J. H. Hegemann, D. Becher, H. Feldmann, U. Guldener, R. Gotz, M. Hansen, C. P. Hollenberg, G. Jansen, W. Kramer, S. Klein, P. Kotter, J. Kricke, H. Launhardt, G. Mannhaupt, A. Maierl, P. Meyer, W. Mewes, T. Munder, R. K. Niedenthal, R. M. Ramezani, A. Rohmer, A. Romer, A. Hinnen, et al. 1999. Functional analysis of 150 deletion mutants in Saccharomyces cerevisiae by a systematic approach. Mol. Gen. Genet. 262:683-702. [DOI] [PubMed] [Google Scholar]
- 17.Gammie, A. E., B. G. Stewart, C. F. Scott, and M. D. Rose. 1999. The two forms of karyogamy transcription factor Kar4p are regulated by differential initiation of transcription, translation, and protein turnover. Mol. Cell. Biol. 19:817-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gasch, A. P., P. T. Spellman, C. M. Kao, O. Carmel-Harel, M. B. Eisen, G. Storz, D. Botstein, and P. O. Brown. 2000. Genomic expression programs in the response of yeast cells to environmental changes. Mol. Biol. Cell 11:4241-4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gietz, R. D., and R. A. Woods. 2002. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 350:87-96. [DOI] [PubMed] [Google Scholar]
- 20.Grigull, J., S. Mnaimneh, J. Pootoolal, M. D. Robinson, and T. R. Hughes. 2004. Genome-wide analysis of mRNA stability using transcription inhibitors and microarrays reveals posttranscriptional control of ribosome biogenesis factors. Mol. Cell. Biol. 24:5534-5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gu, M., and C. D. Lima. 2005. Processing the message: structural insights into capping and decapping mRNA. Curr. Opin. Struct. Biol. 15:99-106. [DOI] [PubMed] [Google Scholar]
- 22.Guthrie, C., and G. R. Fink. 1991. Guide to yeast genetics and molecular biology. Academic Press, San Diego, Calif.
- 23.Kaplan, C. D., L. Laprade, and F. Winston. 2003. Transcription elongation factors repress transcription initiation from cryptic sites. Science 301:1096-1099. [DOI] [PubMed] [Google Scholar]
- 24.Kellis, M., N. Patterson, M. Endrizzi, B. Birren, and E. S. Lander. 2003. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature 423:241-254. [DOI] [PubMed] [Google Scholar]
- 25.Kitajima, S., and F. Sato. 1999. Plant pathogenesis-related proteins: molecular mechanisms of gene expression and protein function. J. Biochem. (Tokyo) 125:1-8. [DOI] [PubMed] [Google Scholar]
- 26.Kozak, M. 1991. Structural features in eukaryotic messenger RNAs that modulate the initiation of translation. J. Biol. Chem. 266:19867-19870. [PubMed] [Google Scholar]
- 27.Kuras, L., and K. Struhl. 1999. Binding of TBP to promoters in vivo is stimulated by activators and requires Pol II holoenzyme. Nature 399:609-613. [DOI] [PubMed] [Google Scholar]
- 28.Laabs, T. L., D. D. Markwardt, M. G. Slattery, L. L. Newcomb, D. J. Stillman, and W. Heideman. 2003. ACE2 is required for daughter cell-specific G1 delay in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 100:10275-10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Law, G. L., K. S. Bickel, V. L. MacKay, and D. R. Morris. 2005. The under-translated transcriptome reveals widespread translational silencing by alternative 5′ transcript leaders. Genome Biol. 6:R111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee, T. I., N. J. Rinaldi, F. Robert, D. T. Odom, Z. Bar-Joseph, G. K. Gerber, N. M. Hannett, C. T. Harbison, C. M. Thompson, I. Simon, J. Zeitlinger, E. G. Jennings, H. L. Murray, D. B. Gordon, B. Ren, J. J. Wyrick, J. B. Tagne, T. L. Volkert, E. Fraenkel, D. K. Gifford, and R. A. Young. 2002. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science 298:799-804. [DOI] [PubMed] [Google Scholar]
- 31.Li, B., and J. C. Reese. 2001. Ssn6-Tup1 regulates RNR3 by positioning nucleosomes and affecting the chromatin structure at the upstream repression sequence. J. Biol. Chem. 276:33788-33797. [DOI] [PubMed] [Google Scholar]
- 32.Liu, X., and M. A. Gorovsky. 1993. Mapping the 5′ and 3′ ends of Tetrahymena thermophila mRNAs using RNA ligase mediated amplification of cDNA ends (RLM-RACE). Nucleic Acids Res. 21:4954-4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Longtine, M. S., A. McKenzie III, D. J. Demarini, N. G. Shah, A. Wach, A. Brachat, P. Philippsen, and J. R. Pringle. 1998. Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14:953-961. [DOI] [PubMed] [Google Scholar]
- 34.MacKay, V. L., X. Li, M. R. Flory, E. Turcott, G. L. Law, K. A. Serikawa, X. L. Xu, H. Lee, D. R. Goodlett, R. Aebersold, L. P. Zhao, and D. R. Morris. 2004. Gene expression analyzed by high-resolution state array analysis and quantitative proteomics: response of yeast to mating pheromone. Mol. Cell. Proteomics 3:478-489. [DOI] [PubMed] [Google Scholar]
- 35.Maldonado, E., M. Hampsey, and D. Reinberg. 1999. Repression: targeting the heart of the matter. Cell 99:455-458. [DOI] [PubMed] [Google Scholar]
- 36.Martens, J. A., P. Y. Wu, and F. Winston. 2005. Regulation of an intergenic transcript controls adjacent gene transcription in Saccharomyces cerevisiae. Genes Dev. 19:2695-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mata, J., S. Marguerat, and J. Bahler. 2005. Post-transcriptional control of gene expression: a genome-wide perspective. Trends Biochem. Sci. 30:506-514. [DOI] [PubMed] [Google Scholar]
- 38.Mennella, T. A., L. G. Klinkenberg, and R. S. Zitomer. 2003. Recruitment of Tup1-Ssn6 by yeast hypoxic genes and chromatin-independent exclusion of TATA binding protein. Eukaryot. Cell 2:1288-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Morris, D. R., and A. P. Geballe. 2000. Upstream open reading frames as regulators of mRNA translation. Mol. Cell. Biol. 20:8635-8642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagawa, F., and G. R. Fink. 1985. The relationship between the “TATA” sequence and transcription initiation sites at the HIS4 gene of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 82:8557-8561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nern, A., and R. A. Arkowitz. 1999. A Cdc24p-Far1p-Gbetagamma protein complex required for yeast orientation during mating. J. Cell Biol. 144:1187-1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Porro, D., E. Martegani, B. M. Ranzi, and L. Alberghina. 1997. Identification of different daughter and parent subpopulations in an asynchronously growing Saccharomyces cerevisiae population. Res. Microbiol. 148:205-215. [DOI] [PubMed] [Google Scholar]
- 43.Ren, B., F. Robert, J. J. Wyrick, O. Aparicio, E. G. Jennings, I. Simon, J. Zeitlinger, J. Schreiber, N. Hannett, E. Kanin, T. L. Volkert, C. J. Wilson, S. P. Bell, and R. A. Young. 2000. Genome-wide location and function of DNA binding proteins. Science 290:2306-2309. [DOI] [PubMed] [Google Scholar]
- 44.Roberts, C. J., B. Nelson, M. J. Marton, R. Stoughton, M. R. Meyer, H. A. Bennett, Y. D. He, H. Dai, W. L. Walker, T. R. Hughes, M. Tyers, C. Boone, and S. H. Friend. 2000. Signaling and circuitry of multiple MAPK pathways revealed by a matrix of global gene expression profiles. Science 287:873-880. [DOI] [PubMed] [Google Scholar]
- 45.Rothstein, R. 1991. Targeting, disruption, replacement, and allele rescue: integrative DNA transformation in yeast. Methods Enzymol. 194:281-301. [DOI] [PubMed] [Google Scholar]
- 46.Ruegsegger, U., J. H. Leber, and P. Walter. 2001. Block of HAC1 mRNA translation by long-range base pairing is released by cytoplasmic splicing upon induction of the unfolded protein response. Cell 107:103-114. [DOI] [PubMed] [Google Scholar]
- 47.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 48.Santiago, T. C., I. J. Purvis, A. J. Bettany, and A. J. Brown. 1986. The relationship between mRNA stability and length in Saccharomyces cerevisiae. Nucleic Acids Res. 14:8347-8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sherman, F. 1991. Getting started with yeast. Methods Enzymol. 194:3-21. [DOI] [PubMed] [Google Scholar]
- 50.Sikorski, R. S., and P. Hieter. 1989. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae 554. Genetics 122:19-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Spellman, P. T., G. Sherlock, M. Q. Zhang, V. R. Iyer, K. Anders, M. B. Eisen, P. O. Brown, D. Botstein, and B. Futcher. 1998. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell 9:3273-3297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Szyperski, T., C. Fernandez, C. Mumenthaler, and K. Wuthrich. 1998. Structure comparison of human glioma pathogenesis-related protein GliPR and the plant pathogenesis-related protein P14a indicates a functional link between the human immune system and a plant defense system. Proc. Natl. Acad. Sci. USA 95:2262-2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Taussig, R., and M. Carlson. 1983. Nucleotide sequence of the yeast SUC2 gene for invertase. Nucleic Acids Res. 11:1943-1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valencia-Sanchez, M. A., J. Liu, G. J. Hannon, and R. Parker. 2006. Control of translation and mRNA degradation by miRNAs and siRNAs. Genes Dev. 20:515-524. [DOI] [PubMed] [Google Scholar]
- 55.Wilkie, G. S., K. S. Dickson, and N. K. Gray. 2003. Regulation of mRNA translation by 5′- and 3′-UTR-binding factors. Trends Biochem. Sci. 28:182-188. [DOI] [PubMed] [Google Scholar]
- 56.Yin, Q. Y., P. W. de Groot, H. L. Dekker, L. de Jong, F. M. Klis, and C. G. de Koster. 2005. Comprehensive proteomic analysis of Saccharomyces cerevisiae cell walls: identification of proteins covalently attached via glycosylphosphatidylinositol remnants or mild alkali-sensitive linkages. J. Biol. Chem. 280:20894-20901. [DOI] [PubMed] [Google Scholar]
- 57.Yuan, Y. L., and S. Fields. 1991. Properties of the DNA-binding domain of the Saccharomyces cerevisiae STE12 protein. Mol. Cell. Biol. 11:5910-5918. [DOI] [PMC free article] [PubMed] [Google Scholar]