

Abstract
Splicing of exon 6B from the β-tropomyosin pre-mRNA is repressed in nonmuscle cells and myoblasts by a complex array of intronic elements surrounding the exon. In this study, we analyzed the proteins that mediate splicing repression of exon 6B through binding to the upstream element. We identified the polypyrimidine tract binding protein (PTB) as a component of complexes isolated from myoblasts that assemble onto the branch point region and the pyrimidine tract. In vitro splicing assays and PTB knockdown experiments by RNA interference demonstrated that PTB acts as a repressor of splicing of exon 6B. Using psoralen experiments, we showed that PTB acts at an early stage of spliceosome assembly by preventing the binding of U2 snRNA on the branch point. Using UV cross-linking and immunoprecipitation experiments with site-specific labeled RNA in PTB-depleted nuclear extracts, we found that the decrease in PTB was correlated with an increase in U2AF65. In addition, competition experiments showed that PTB is able to displace the binding of U2AF65 on the polypyrimidine tract. Our results strongly support a model whereby PTB competes with U2AF65 for binding to the polypyrimidine tract.
Alternative splicing is a widespread mechanism that increases protein diversity and regulates gene expression in higher eukaryotes. This process is particularly prominent in humans, as it has been estimated that at least 60% of the human genes are alternatively spliced. Alternative splicing generates several mRNAs from a single gene, leading to the synthesis of several proteins with distinct biological functions, different intracellular localizations, or different stabilities (reviewed in reference 47). Thus, alternative splicing plays a major role in defining the repertoire of proteins that are expressed in different cells. From numerous studies, it appears that the regulation of alternative splicing results from a complex interplay between multiple trans-acting factors and cis-acting sequences that facilitates or prevents the recruitment of splicing factors by the splicing machinery (reviewed in references 7 and 44).
The polypyrimidine tract binding protein (PTB), also known as hnRNP I, is one of the best-characterized splicing repressors. As demonstrated recently, PTB is an essential protein, needed for the development of Xenopus laevis embryos (28). Consistent with its widespread expression, PTB has been implicated in the repression of a large number of alternative splicing events (reviewed in references 7, 48, and 51). PTB recognizes short motifs, such as UCUU and UCUCU, located within a pyrimidine-rich context and often associated with the polypyrimidine tract upstream of the 3′ splice site of alternative exons (3, 8, 9, 21, 37). However, binding sites for PTB have also been found in exonic sequences and in introns downstream of regulated exons (13, 23, 27, 40). In most alternative splicing systems regulated by PTB, repression is achieved through the interaction of PTB with multiple PTB binding sites surrounding the alternative exon (3, 9-11, 21, 45, 46, 55). However, in a few cases, repression involves a single PTB binding site (23, 40). The mechanism by which PTB inhibits splicing is still poorly understood. Several models, depending on the position of PTB binding sites, have been proposed. In a model based on the presence of PTB binding sites within polypyrimidine tracts, splicing repression is proposed to occur by a direct competition between PTB and U2AF65, which in turn precludes the assembly of the U2 snRNP on the branch point (31, 35, 42). Another model, which involves PTB binding sites located on both sides of alternative exons, proposes that splicing repression would result from cooperative interactions between PTB molecules that would loop out the RNA, thereby making the splice sites inaccessible to the splicing machinery (2, 11). A third model proposes that the multimerization of PTB from a high-affinity binding site would create a repressive wave that covers the alternative exon and prevents its recognition (51). Recent studies of alternative splicing events in two different models, the c-src and Fas pre-mRNAs, have provided some clues about the mechanism of PTB repression. According to these studies, PTB represses splicing by preventing the communication between U1 snRNP and U2AF65, which are required for intron and exon definition (23, 39).
We are using the chicken β-tropomyosin (βTm) pre-mRNA as a model to investigate the regulation of alternative splicing. This pre-mRNA contains two mutually exclusive exons that are recognized differently according to myogenic differentiation. Exon 6A is present in nonmuscle cells and myoblasts, while exon 6B is present in skeletal muscle and myotubes. We and others have shown that mutations dispersed along the intron upstream of exon 6B activate splicing of exon 6B both in vitro and in vivo, suggesting that it contains several regulatory motifs involved in repression (22, 29, 30). This intron is characterized by a far upstream branch point and a high pyrimidine content. In the present study, we show that PTB binds to the intron upstream of exon 6B, at sites near the branch point and between the branch point and the 3′ splice site. In vitro splicing assays and PTB knockdown by RNA interference demonstrate that PTB is a repressor of exon 6B splicing. We provide evidence that PTB prevents the interaction of U2 snRNA with the branch point and antagonizes the binding of U2AF65.
MATERIALS AND METHODS
Plasmid constructions.
All βTm constructs were derived from a 1.7-kb chicken genomic clone spanning exons 4 to 7. The construct pSP65 5K6AΔ4-6B7 was derived from pSVK6AΔ4, in which the 5′ splice site of exon 6A has been mutated (4). The intron between exons 6B and 7 was deleted by reverse PCR, and the full exon 7 was restored by cloning a synthetically prepared hybrid oligonucleotide (5′ AGCTTCTAGGGGAGAAACTGAAGGAGGTAAGCCTCTAGA 3′). The template was linearized by XbaI for transcription. The construct glo-βTm, in which the 3′ region of the βTm upstream of exon 6B, extending from 10 nucleotides (nt) upstream of the branch site to the 3′ splice site, replaces the 3′ region upstream of globin exon 2 has been described previously (and named G1 in reference 18). The transcript pPmac-SexAI was obtained from pPmac by digestion with SexA1 (12). Mutation of the 5′ splice site of pPmac-SexAI was performed by PCR. Deletion of the 5′ splice site was performed by linearization with the BsmAI restriction enzyme, which also suppresses the last 16 nt of exon 6B. Transcription templates for BP, PY, 3′6B, BPmtPTB, and PYmtPTB were obtained by cloning of hybrid oligonucleotides flanked by XhoI and XbaI restriction sites into pSP72 (see the sequences in Fig. 1). Transcription templates for BP-PY and PY-3′6B were made by PCR with primer containing the SP6 RNA polymerase promoter. The transcription template for the high-affinity PTB binding site (Cs8 RNA) was obtained by cloning a hybrid oligonucleotide (5′ AATT[CTCTT]8A 3′) flanked by EcoRI and HindIII restriction sites into pSP65. pcDNA3 980 BPmtPTB was derived from the wild-type pcDNA3 980 (17) construct by reverse PCR using a sense oligonucleotide (5′ GTTCTCACACCAATAATCCTCCTTCCCTGTG 3′) and an antisense oligonucleotide (5′ AAGTTGTATTATTATTAAACTCAGAGTGG 3′). pcDNA3 980 PYmtPTB was derived from the wild-type pcDNA3 980 construct by reverse PCR using a sense oligonucleotide (5′ CCGCTCCCACCAATAACCCTTCATCA 3′) and an antisense oligonucleotide (5′ TTATTGCACAGGGTTATTGGAAAGAAGG 3′). The double mutant pcDNA3 980 db mtPTB was derived from pcDNA3 BPmtPTB with the sense oligonucleotide used to construct pcDNA3 980 PYmtPTB and an antisense oligonucleotide (5′ TTATTGCACAGGGTTATTGGATTATTGG 3′).
FIG. 1.

PTB is a component of myoblast protein complexes assembled onto BP and PY. (A) Schematic representation of the two mutually exclusive exons showing the regulatory elements upstream of exon 6B. PmlI and SexAI are restriction enzymes used to construct the plasmid pPmac SexAI. The sequences of the RNAs used for affinity purification of protein complexes are shown below. The A in bold type indicates the branch point upstream of exon 6B (22). (B) Two-dimensional gel electrophoresis of proteins from QM7 myoblast nuclear extracts assembled onto BP, PY, and 3′6B RNAs. Proteins were revealed by silver staining. The insets shown to the right of the BP and PY RNAs represent parts of the Western blot analysis performed on a two-dimensional gel with polyclonal antibodies against PTB. (C) Western blot analysis with anti-PTB of protein complexes assembled on wild-type or mutant RNAs. BPmtPTB and PYmtPTB are mutant BP and PY RNAs, respectively, in which putative PTB binding sites have been mutated (Fig. 1A). Odd and even lanes correspond to QM7 nuclear extracts from myoblasts (mb) and myotubes (mt) assembled onto RNAs, respectively.
In vitro transcription and splicing experiments.
Capped pre-mRNAs were synthesized in vitro from plasmids with SP6 RNA polymerase and [α-32P]UTP (26). RNAs for gel shift experiments and UV cross-linking experiments were transcribed with cap analogue and SP6 polymerase. RNAs for affinity selection were transcribed without capping by using an SP6 MEGAshortscript kit (Ambion) according to the manufacturer's recommendations. RNAs were then biotinylated as previously described (19).
Splicing reactions were performed with 20 fmol 32P pre-mRNA in 10-μl reactions with 40% HeLa nuclear extract, except as indicated. Competitor RNA and recombinant proteins were added to the splicing reaction on ice prior to addition of the labeled pre-mRNA. Reaction products were analyzed on 5% denaturing polyacrylamide gels except as indicated. RNA bands were quantified with a PhosphorImager (Molecular Dynamics). Splicing efficiency was calculated as the ratio between the mRNA value and the sum of the pre-mRNA value plus the mRNA value, with the number of uracil residues taken into account. HeLa cell nuclear extracts were purchased from A. Miller (Cilbiotech, Mons, Belgium). Myoblast and myotube nuclear extracts from QM7 were prepared as described previously (1, 15).
Spliceosome assembly analysis.
For spliceosome assembly analysis, 25 fmol of uniformly labeled RNA was incubated in a 25-μl reaction volume containing 40% HeLa nuclear extracts under splicing conditions at 30°C. Following incubation, the reaction was stopped by adding 0.2 mg/ml heparin. Splicing complexes were separated on a 2% agarose gel (SeaKem GTG agarose) and run at room temperature for 4 h at 100 V (14, 39).
Gel retardation experiments.
For gel retardation experiments, 25 fmol of uniformly labeled RNA was incubated in a 20-μl reaction volume containing 10 mM HEPES-KOH (pH 7.9), 100 mM KCl, 1 mM dithiothreitol, 10% glycerol, 0.02% Nonidet P-40, and 10 μg/ml Escherichia coli tRNA with his-PTB1 or GST-U2AF65, as indicated in the figures. Recombinant proteins were diluted in buffer D containing 0.5 mg/ml bovine serum albumin (15). The reaction was incubated for 15 min at 30°C, and the protein complexes were resolved by electrophoresis on a 6% nondenaturing polyacrylamide gel (39/1) with 0.5× Tris-borate-EDTA.
Purification of recombinant proteins.
Plasmids pET PTB1 and pQE30 PTB1 (gifts from C. Smith and J. Patton) were expressed in Escherichia coli BL21(DE3) and purified with Ni-nitrilotriacetic acid agarose (QIAGEN) according to the protocol described by the manufacturer. Recombinant GST-U2AF65 (a gift from J. Valcárcel) was purified according to standard procedures. Proteins were dialyzed against buffer D (15).
RNA affinity chromatography.
Protein complexes were purified by RNA affinity chromatography as described previously (16) (see the sequences in Fig. 1). Two-dimensional gel electrophoresis were performed with an 8% polyacrylamide gel in the second dimension (16). The selected proteins were visualized with a silver staining kit (Silverquest; Invitrogen). The identification of proteins by mass spectrometry was done by the Mass Spectrometry and Proteomic Technological Platform at Curie Institute, Paris, France.
Western blot analysis.
Western blotting was performed with the following antibodies: anti-PTB polyclonal antibodies (Ab-1; Calbiochem), anti-PTB monoclonal antibodies (Oncogene Research), anti-hnRNP A1 monoclonal antibodies (9H10; a gift from G. Dreyfuss), and anti-U2AF65 monoclonal antibodies (a gift from J. Valcárcel). The bands were detected with the SuperSignal West Pico detection kit (Pierce) and quantified with a Fuji LAS 3000.
Depletion of PTB from HeLa nuclear extracts.
The depletion of PTB with biotinylated RNA (Cs8) was performed with a ratio of 0.5 pmol RNA/μl of HeLa nuclear extracts. The RNA was first bound to streptavidin agarose (5 pmol RNA/μl of beads) and rotated with nuclear extracts for 1 h at 4°C. The beads were pelleted, and the supernatants were used as PTB-depleted extract. Mock-depleted nuclear extract was made under the same conditions but with the beads alone. Immunodepletion of PTB was done with a commercial mouse monoclonal anti-PTB antibody (Ab1; Oncogene Research). Then, 50 μl of antibodies (5 μg) was rocked with 20 μl of G plus agarose beads (Santa Cruz Biotechnology) overnight at 4°C. After washing of the beads with buffer D, 50 μl of nuclear extracts was added and rotated at 4°C for 3 h. The beads were pelleted, and the supernatants were used as PTB-depleted extract. Mock-depleted nuclear extract was made under the same conditions but with the beads alone.
Psoralen cross-linking experiments.
For psoralen cross-linking experiments, 25 fmol of [32P]UTP-labeled RNA was incubated in a 10-μl reaction volume containing 50% HeLa nuclear extract under splicing conditions with 25 ng/μl psoralen derivative AMT (4′-aminomethyl-4,5′,8-trimethylpsoralen) for 10 min at 30°C and irradiated on ice for 35 min at 365 nm. Cross-linked RNAs were analyzed on a 5% denaturing polyacrylamide gel. The identification of snRNA cross-linked products was performed by cleavage with RNase H in the presence of oligonucleotide complementary to snRNA U1 and U2 (38). Oligonucleotide U166-77 was 5′ TCCGGAGTGCAA 3′, U228-42 was 5′ CAGATACTACACTTG 3′, and U21-15 was 5′ AGGCCGAGAAGCGAT 3′.
The identification of the cross-link positions on the RNA was performed on the purified U2 and U1 cross-linked species by primer extension analysis with the 5′ end-labeled oligonucleotide primer I6A-6B rev (5′ GGGAGCGGAAGGAGCAC 3′), located 31 nt downstream of the branch point. Sequencing ladders were generated by using the un-cross-linked RNA pPmacSexAI as a template with the same primer. The identification of the cross-link position within U1 snRNA was performed by primer extension analysis with the U1 loop 2 primer (5′ TCCGGAGTGCAA 3′).
Site-specifically labeled pre-mRNA.
Labeling at specific positions was performed according to Yu and Steitz with 2′-O-methyl RNA-DNA chimera (54). pPmac-SexA1 was transcribed with SP6 RNA polymerase using an SP6 MEGAshortscript kit (Ambion) and was trace labeled with [α-32P]UTP. 5′ and 3′ half RNAs were derived by RNase H cleavage of pPmac-SexA1 directed by 2′-O-methyl RNA-DNA chimeras (position 21, 5′ GmAmGmdGdAdAdAGmAmAmGmGmAmGmAmGmAmGmAm 3′; position 34, 5′ GmAmGmdCdAdCdAGmGmGmAmAmGmGmAmGmGmAmAmAmGm 3′; position 58, 5′ AmAmGmdGdGdAdAGmAmAmGmGmUmGmGmGmAmGmCm 3′). RNAs were purified by polyacrylamide gel electrophoresis (PAGE). 3′ RNAs were dephosphorylated by calf intestinal phosphatase and 5′ labeled with polynucleotide kinase and [γ-32P]ATP. The 5′ and 3′ half RNAs (2.5 pmol) were ligated by bridging oligonucleotides (2.5 pmol) and T4 DNA ligase, as described previously (34). The bridging oligonucleotides for 2′-O-methyl RNA-DNA chimera positions 21, 34, and 58 were 5′ GCACAGGGAAGGAGGAAAGAAGGAGAGAGAAC 3′, 5′ GTGGGAGCGGAAGGAGCACAGGGAAGGAGGAAAG 3′, and 5′ GGAGGGGGTGATGAAGGGAAGAAGGTGGGAGCGGAA 3′, respectively. RNAs labeled at a specific position were then purified on polyacrylamide gels.
UV cross-linking and immunoprecipitation experiments.
Uniformly labeled RNA (100 fmol) or site-specifically labeled RNA (20 fmol) was incubated for 15 min at 30°C in 25 μl with 40% mock-depleted or PTB-depleted HeLa nuclear extracts under splicing conditions. Recombinant PTB1 (1.2 μg) that was added to the samples corresponded to the concentration (2 μM) of PTB1 in HeLa nuclear extracts. Samples were UV irradiated for 15 min and treated with RNase A and RNase T1 (17). Immunoprecipitation experiments were carried out as described previously (24). The UV cross-linking samples were rotated for 2 h at 4°C with 30 μl of anti-U2AF65 antiserum that had been prebound to protein A/G plus-agarose (Santa Cruz Biotechnology). The beads were washed three times with 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.05% Nonidet P-40 and boiled in loading buffer for 10 min. The cross-linked proteins were resolved on a 10% sodium dodecyl sulfate (SDS)-polyacrylamide gel and visualized by a PhosphorImager.
Cell culture and siRNA transfection.
QM7 quail myoblast cells were grown as described previously (17). For differentiation, cells were grown in Dulbecco's modified Eagle's medium with 4,500 mg/liter d-glucose supplemented with 1% fetal calf serum for 72 h. HeLa cells were grown in Dulbecco's modified Eagle's medium with 4,500 mg/liter d-glucose supplemented with 10% fetal bovine serum. For a typical small interfering RNA (siRNA) transfection, 12-well dishes were seeded with 105 HeLa cells 24 h prior to transfection. The following day (day 2), cells were transfected with 180 pmol of siRNA duplex (Eurogentec) by using Oligofectamine (Invitrogen). On day 3, cells were cotransfected with 0.5 μg plasmid carrying βTm minigenes, expression vector FLAG-PTB1 (a gift from C. Smith), and 180 pmol siRNA duplex by using Fugene 6 (Roche). Cells were harvested on day 5 for RNA and protein analysis. The siRNA directed against PTB was 5′ AACUUCCAUCAUUCCAGAGAA 3′ (P1 in reference 53), and the control luciferase siRNA was 5′ AACGUACGCGGAAUACUUCGA 3′. RNAs were prepared by the TriReagent method (Sigma), and splicing patterns were analyzed by reverse transcription (RT)-PCR, as previously described (17).
RESULTS
PTB binds to the intronic sequence upstream of exon 6B.
Previous works have shown that the intronic sequence upstream of exon 6B from the chicken βTm pre-mRNA was involved in the repression of exon 6B in nonmuscle cells and myoblasts (18, 22, 29). To identify factors that regulate splicing of exon 6B, we used an RNA affinity chromatography strategy to isolate protein complexes. To that purpose, the intronic sequence was arbitrarily divided into three contiguous RNA pieces (Fig. 1A). BP encompasses the branch point sequence, and it is 39 nt long. 3′6B contains the first nucleotide of exon 6B and extends 43 nt upstream of exon 6B. PY is located between BP and 3′6B, and it is 41 nt long (Fig. 1A). These RNAs were biotinylated, bound to streptavidin agarose beads, and incubated with QM7 nuclear extracts from myoblasts under splicing conditions. Proteins were then separated by two-dimensional gel electrophoresis with NEPHGE in the first dimension. Figure 1B shows that each RNA recruited numerous proteins, among which some were specifically associated with distinct RNA species. In particular, there was a group of proteins of around 60 kDa that were assembled on BP and PY but not on 3′6B and whose migration resembled that reported for PTB. Western blot analysis of two-dimensional gel electrophoresis with polyclonal antibodies against PTB (Fig. 1B, insert) and mass spectrometry analysis confirmed that this group of proteins was PTB and that they might represent different phosphorylation states of the same protein. Consistent with this result, BP and PY contained several UCUU and UCUCU motifs that were identified as optimal PTB binding sites (37). To test whether these sites were required for binding, RNA affinity chromatography was performed on RNAs carrying mutations in each of the putative binding motifs (Fig. 1A). The proteins were separated by SDS-PAGE and analyzed by Western blot experiments with polyclonal antibodies against PTB (Fig. 1C). In agreement with two-dimensional gel electrophoresis results, PTB interacted with wild-type BP and PY RNAs, albeit at a lower level with PY than with BP, whereas only a very faint interaction was detected with 3′6B (Fig. 1C, lanes 1, 5, and 9). In contrast, RNAs containing mutations that disrupt the putative PTB binding sites failed to assemble PTB into the complexes (Fig. 1C; compare lane 3 with lane 1 and lane 7 with lane 5). We also observed a decrease in the amount of PTB bound to BP and PY in complexes formed with myotube nuclear extracts compared to those formed with myoblasts, suggesting a reorganization of protein complexes during myogenic differentiation (Fig. 1C; compare lane 2 with lane 1 and lane 6 with lane 5).
PTB represses splicing of exon 6B in vitro.
PTB has been involved in the repression of alternative splicing of numerous genes (reviewed in reference 7). To investigate whether PTB acts as a repressor in our model system, we first performed in vitro splicing assays in the presence of competitor RNAs that titrate PTB. To facilitate the analysis of splicing products, we used a pre-mRNA containing exon 5, exon 6A, and exon 6B fused to exon 7, where the 5′ splice site of exon 6A has been destroyed and the intronic enhancer S4 has been deleted (Fig. 2A). These mutations decrease the splicing efficiency of exon 6A and allow a partial derepression of exon 6B in HeLa cells (4, 19). We used as an RNA competitor Cs8, which contains eight copies of the high-affinity PTB binding site (52). In the absence of competitor RNA, one major splicing pathway was observed: exon 5 spliced to exon 6A (Fig. 2B, lane 2). Addition of increasing amounts of Cs8 in an in vitro splicing assay induced a switch toward exon 6B inclusion, with a 10-fold increase in exon 5 to exon 6B7 splicing and a strong decrease in exon 5 to exon 6A splicing (Fig. 2B; compare lanes 3 to 8 with lane 2). Adding back recombinant PTB1 reversed the splicing pattern toward the original one, suggesting that PTB is involved in the repression of exon 6B splicing (Fig. 2B; compare lanes 10 to 13 with lane 9). However, we could not totally exclude the possibility that Cs8 sequestered another factor in addition to PTB. Thus, a second set of in vitro splicing experiments was carried out in HeLa nuclear extracts that had been depleted of PTB by a commercial monoclonal PTB antibody. As shown in Fig. 7A, around 85 to 90% of the amount of PTB could be removed by the immunodepletion. The results in Fig. 2C clearly indicate that splicing of exon 6B was activated in PTB-immunodepleted nuclear extracts compared to mock-depleted nuclear extracts (fourfold increase). Addition of recombinant PTB1 at a concentration similar to that of the mock nuclear extract restores the splicing inhibition of exon 6B (Fig. 2C, lanes 7 to 9). Together, these results indicate that PTB acts as a repressor of exon 6B splicing in vitro. To get insight into the mechanism by which PTB represses splicing, we tested whether the 3′ intronic region upstream of exon 6B could make an heterologous exon responsive to PTB. To do so, we replaced the 3′ splice site region of the β-globin exon 2 with the 3′ splice site intronic region of exon 6B, which contains the BP, PY, and 3′6B fragments (construct glo-βTm [also named G1 in reference 18]) (Fig. 3A). As expected, the transfer of the 3′ intronic region of exon 6B upstream of the β-globin exon 2 represses the splicing of the glo-βTm transcript (Fig. 3B, lane 1). Moreover, the removal of PTB from nuclear extracts by immunodepletion relieved the repression and increased fivefold the splicing of the glo-βTm transcript, while the addition of recombinant PTB reestablished splicing inhibition (Fig. 3B, lanes 2 to 5). In contrast, neither PTB depletion nor the addition of PTB affected the splicing of the wild-type β-globin transcript (Fig. 3B). These results demonstrate that PTB, through its binding to the 3′ region upstream of exon 6B, represses splicing, and this repression is independent of the nature of the 5′ splice site and of putative PTB molecules bound elsewhere on the βTm transcript.
FIG. 2.

PTB represses in vitro splicing of exon 6B. (A) Schematic representation of the pre-mRNA used in in vitro splicing experiments. The X mark indicates the mutation of the 5′ splice site of exon 6A. The deletion of the intronic S4 enhancer sequence is indicated. (B) Addition of increasing amounts of Cs8 RNA competitor activates splicing of exon 6B, and adding back of recombinant PTB1 restores splicing inhibition. Labeled pre-mRNA 5-K6AΔ4-6B7 was incubated in 40% HeLa nuclear extracts for 90 min at 30°C in the absence (lane 2) or in the presence of 2, 4, 5, 6, and 8 pmol of Cs8 (lanes 3 to 8, respectively). Lane 1, pre-mRNA alone. In lanes 9 to 13, 4 pmol of Cs8 was added in the absence (lane 9) or in the presence of 0.25, 0.5, 0.75, or 1 μg of recombinant PTB (lanes 10 to 13, respectively). The products of the splicing reactions were separated by denaturing 5% PAGE. The identities of the splicing products are diagrammed to the left of the gel. (C) Immunodepletion of PTB from nuclear extracts activates splicing of exon 6B. Splicing experiments were carried out under the same conditions as for panel B, with mock-depleted (lanes 1 to 3) and PTB-immunodepleted (lanes 4 to 9) nuclear extracts in the presence of 0.2, 0.3, and 0.6 μg of recombinant PTB1 (lanes 7 to 9) for 60 min (lanes 1 and 4) or 90 min (lanes 2, 3, and 5 to 9). Note that the incubation of nuclear extracts with beads alone (mock-depleted nuclear extracts) resulted in a modification of the ratio between the splicing of exon 6A over the splicing of exon 6B (in panels B and C, compare lanes 2). We have no clear explanation for this finding except that the incubation of nuclear extracts, which decreases the splicing efficiency of the two splicing pathways, could allow a better competition between the splicing of exon 5 to exon 6B over the splicing of exon 5 to exon 6A.
FIG. 7.

PTB prevents the binding of U2AF65. (A) Western blot analysis of mock-depleted (M) HeLa nuclear extracts or extracts depleted with the Cs8 RNA (ΔR) or immunodepleted with a commercial monoclonal PTB antibody (Δα). The same blot was probed with the monoclonal anti-PTB antibody, then with the monoclonal anti-U2AF65 antibody. Note that the doublet below the U2AF65 signal corresponds to the remaining PTB signal. (B) Uniformly labeled pPmac-SexA1 RNA was incubated in mock-depleted (M) or PTB-depleted (ΔR and Δα) nuclear extracts under splicing conditions for 15 min at 30°C in the absence (lanes 1, 3, 5, and 7) or in the presence (lanes 2, 4, 6 and 8) of 1.2 μg of recombinant PTB1. After UV cross-linking, the samples were separated by SDS-PAGE (lanes 1 to 8, X-link) or immunoprecipitated with monoclonal anti U2AF65 antibodies (lanes 9 to 16, αU2AF65). The amount of UV cross-linked samples (lanes X-link) that were subjected to SDS-PAGE represents 1/10 of the volume of the immunoprecipitated samples. The positions of PTB and U2AF65 are indicated. (C) Uniformly labeled pPmac-SexA1 was incubated with 0.5, 1, 2, 4.3, 8.7, and 17.4 pmol of GST-U2AF65 (lanes 1 to 6, respectively) or with 0.44, 0.88, 1.76, 3.5, 7, and 14 pmol of his-PTB1 (lanes 7 to 12, respectively). (D) Uniformly labeled pPmac-SexA1 was incubated with 9 pmol of recombinant GST-U2AF65 (lanes 1 to 12) and with 0.22 pmol (lanes 2 and 8), 0.44 pmol (lanes 3 and 9), 0.88 pmol (lanes 4 and 10), 1.76 pmol (lanes 5 and 11), and 3.5 pmol (lanes 6 and 12) of recombinant PTB1 in the absence (lanes 1 to 6) or in the presence (lanes 7 to 12) of immunodepleted HeLa nuclear extracts. The samples were UV cross-linked, and the proteins were separated by SDS-PAGE.
FIG. 3.
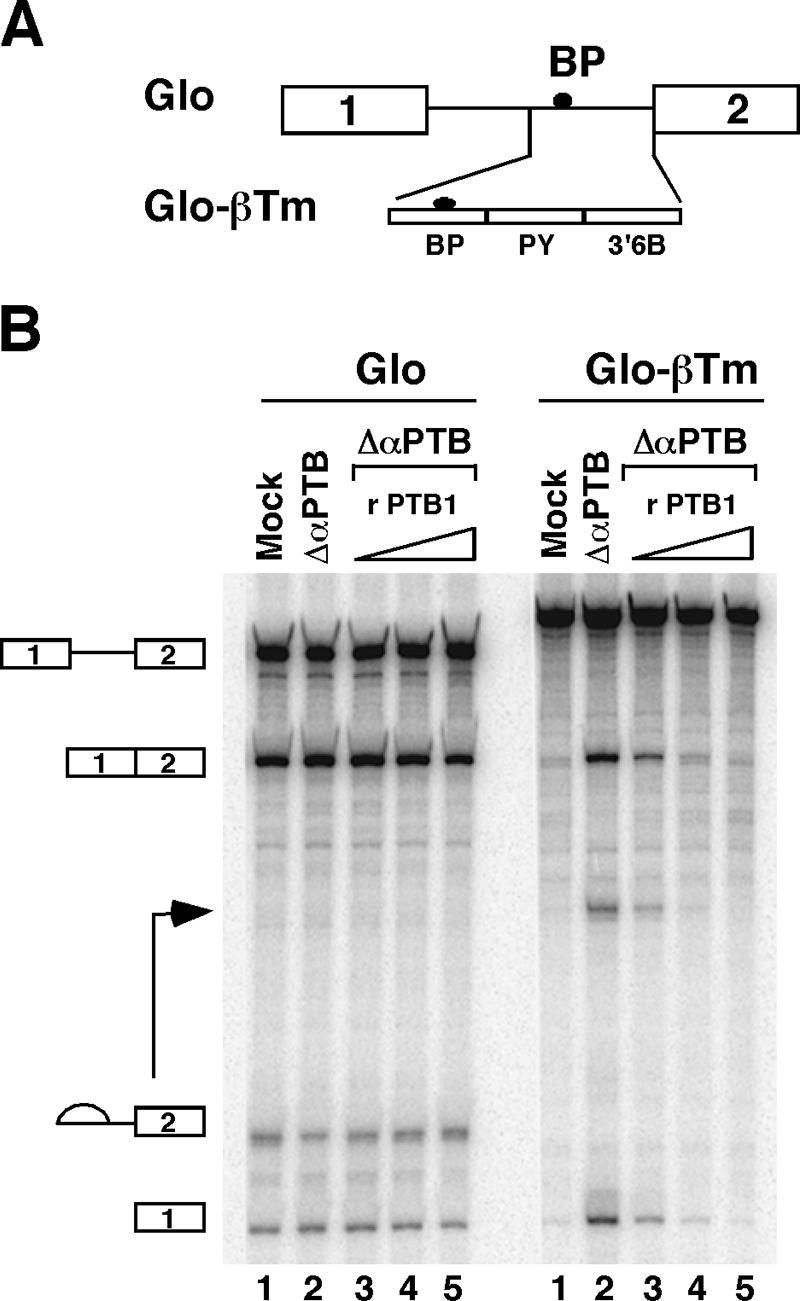
The 3′ region upstream of exon 6B makes pre-mRNA responsive to PTB. (A) Schematic representation of the Glo-βTm pre-mRNA used in in vitro splicing experiments. The intronic region upstream of exon 6B containing the BP, PY, and 3′ 6B fragments is shown. (B) In vitro splicing of the Glo-βTm pre-mRNA. Splicing experiments were performed as in Fig. 2B, with mock-depleted extracts (lane 1) and PTB-immunodepleted nuclear extracts (lanes 2 to 5) in the absence (lane 2) or in the presence of 0.05, 0.1, and 0.2 μg of recombinant PTB1 (lanes 3 to 5, respectively) for 90 min.
Silencing of PTB by RNA interference activates exon 6B splicing ex vivo.
In order to prove that PTB acts as a repressor of exon 6B splicing ex vivo, HeLa cells were deprived of PTB by RNA interference and then transfected with the pcDNA 980 minigene containing exons 5 to 7. Treatment of HeLa cells with an siRNA against PTB (named P1 in reference 53) led to a drastic reduction of the endogenous PTB, while a nonspecific siRNA (Luc siRNA) had no effect on the PTB protein level (Fig. 4A). In addition, the level of an unrelated protein, hnRNP A1, was not affected (Fig. 4A). RT-PCR analysis showed that in HeLa cells treated with the control Luc siRNA, the 980 minigene exhibited the expected splicing pattern, consisting of a predominant mRNA containing exon 5 spliced to exons 6A and 7 and only a small amount of mRNA containing exon 6B (Fig. 4C, lane 6). A similar pattern was observed in untreated cells (Fig. 4C, lane 11). In contrast, siRNA-mediated knockdown of PTB induced, on average, a sixfold increase in exon 6B inclusion and a concomitant decrease in exon 6A inclusion (Fig. 4C, lanes 1 and 6, and Fig. 4D, lanes 1 and 2). Cotransfection of HeLa cells with an expression vector coding for PTB1 reversed the splicing pattern to the original one (Fig. 4C, lanes 2 to 5). We have previously shown that PTB binds to the BP and PY elements. Thus, we tested whether these PTB binding motifs were involved in the response mediated by the knockdown of PTB. Mutations in the PTB binding sites were introduced into the 980 minigene to give rise to 980 BPmtPTB and 980 PYmtPTB. In cells transfected with the control siRNA, mutations of PTB binding sites within BP (980 BPmtPTB) resulted in a threefold increase in exon 6B inclusion compared to the wild-type minigene (Fig. 4D, lanes 1 and 3). Unexpectedly, mutation of PTB binding sites within PY (980 PYmtPTB) had a limited effect (Fig. 4D, lanes 1 and 5). In order to test whether mutations of PTB binding sites in PY had to be combined with those in BP to observe an effect, we constructed the double mutant. Unfortunately, RT-PCR analysis of the double mutant was not informative, because the level of exon 6B inclusion was similar to that in the wild-type minigene (data not shown). When the minigenes were cotransfected in cells deprived of PTB by RNA interference, different responses were observed. In agreement with a role for PTB binding sites within BP, the mutant 980 BPmtPTB had a reduced response to the knockdown of PTB. Only a threefold increase of exon 6B inclusion was observed, compared to the sixfold increase of exon 6B inclusion for the wild-type minigene (Fig. 4D; compare lanes 3 and 4 with lanes 1 and 2). In contrast, the 980 PYmtPTB mutant minigene exhibited a response to PTB knockdown which was intermediate between those of the wild-type minigene and of 980 BPmtPTB (Fig. 4D; compare lanes 5 and 6 with lanes 1 and 2). Taken together, in vitro and ex vivo results support a role for PTB in the repression of exon 6B splicing and indicate that PTB binding sites around the branch point mediate part of the PTB repression.
FIG. 4.

PTB knockdown activates splicing of exon 6B in HeLa cells. (A) Western blot analysis of PTB knockdown using anti-PTB antibodies. Twenty micrograms of total protein was loaded per lane. Lanes 1 and 5, cells not treated; lanes 2 to 4, cells treated with 20, 60, or 180 pmol PTB siRNA, respectively; lanes 6 to 8, cells treated with 20, 60, or 180 pmol luciferase siRNA, respectively. The blot was probed again with an anti-hnRNP A1 monoclonal antibody. (B) Schematic representation of the wild-type 980 βTm reporter minigene. The location of primers used for RT-PCR is indicated. Schematic diagrams of PCR-amplified splicing products are shown below. (C) HeLa cells treated with 180 pmol siRNA targeting PTB (lanes 1 to 5), with the same amount of a control luciferase siRNA (lanes 6 to 10) or without siRNA (lane 11), were transfected with 0.5 μg of the wild-type 980 minigene and with increasing amounts of expression vector coding for PTB1 (10, 30, 100, or 300 ng in lanes 2 to 5 and lanes 7 to 10). The splicing products were analyzed by RT-PCR and quantified with a PhosphorImager. (D) Wild-type and mutant minigenes were cotransfected with 180 pmol PTB siRNA or Luc siRNA in HeLa cells and analyzed by RT-PCR as for panel C. The percentage of exon 6B inclusion is shown as the mean ± standard deviation for at least four experiments.
PTB prevents the recruitment of U2 snRNA to the branch site upstream of exon 6B.
To investigate at which stage of spliceosome assembly PTB repression occurs, we examined the interactions of the snRNAs with the intron upstream of exon 6B by psoralen cross-linking experiments in PTB-depleted or mock-depleted HeLa nuclear extracts. PTB was depleted from HeLa nuclear extracts by the Cs8 biotinylated RNA bound to streptavidin agarose. The RNA used in the cross-link experiments (pPmac-SexA1) contained 150 nt of the intronic sequence upstream of exon 6B with all the regulatory sequences known to be involved in the repression, in addition to the full-length exon 6B and its downstream 5′ splice site. Incubation of pPmac-SexA1 with PTB-depleted or mock-depleted nuclear extracts revealed several cross-linked species that migrated more slowly than the free RNA (Fig. 5A). Band 1, which appeared as a doublet, dramatically increased in PTB-depleted nuclear extracts, about six- to sevenfold compared to the mock-depleted nuclear extract (Fig. 5A; compare lanes 1 and 2 with lanes 6 and 7). This cross-link disappeared upon addition of recombinant PTB1, suggesting that it could correspond to a specific snRNA interaction impaired by PTB repression (Fig. 5A, lanes 3 to 5). Band 2 also increased following PTB depletion, but only twofold compared to the mock-depleted nuclear extract, and diminished following the addition of high concentrations of recombinant PTB1 (Fig. 5A). In contrast, band 3 was affected neither by the removal of PTB nor by the addition of recombinant PTB1 (Fig. 5A, insert showing a low exposure). To identify the snRNA that was cross-linked to the RNA, cross-linking reactions were treated with RNase H in the presence of specific oligonucleotides directed against U2 or U1 snRNA. The cross-linked doublet (band 1) disappeared upon treatment with RNase H and an oligonucleotide complementary to the U2 region that is involved in base pairing with the branch point (Fig. 5B, lanes 2 and 3). Treatment with an oligonucleotide complementary to the 5′ end of U2 shortened the size of the cross-linked doublet (Fig. 5B, lanes 4 and 5). Together, these data demonstrated that the doublet corresponded to cross-linked products with U2 snRNA. Primer extension analysis performed on purified cross-linked species 1 revealed that U2 snRNA contacted the RNA on the U residues present on both sides of the branch point (Fig. 5C, black arrows). When psoralen cross-linking assays were treated with RNase H and an oligonucleotide complementary to nucleotides 66 to 77 of U1, both cross-linked species 2 and 3 were strongly decreased, suggesting that in addition to the binding of U1 snRNA on the 5′ splice site, another U1 snRNA molecule bound elsewhere on the RNA (Fig. 5B, lanes 6 and 7). RNase H-mediated cleavage on purified cross-linked band 3 with oligonucleotides directed at the end of exon 6B indicated that band 3 corresponded to the cross-linking of U1 snRNA on the 5′ splice site (data not shown). Primer extension analysis on purified cross-linked species 2 identified a binding of U1 snRNA on a U residue, 17 nt upstream of the branch point, in a region that does not exhibit any putative 5′ splice site (Fig. 5C, lane 2). To characterize the region of U1 snRNA that contacted the RNA, a primer extension analysis was performed with the U1-BP cross-linked species and an oligonucleotide complementary to nt 66 to 77 of U1. Several major reverse transcriptase stops were observed that were not present when the primer extension was performed with snRNA from HeLa cells or with an in vitro-transcribed U1 snRNA (Fig. 5D; compare lane 1 with lanes 2 to 4). Two reverse transcriptase stops mapped to cytosine 9 and uridine 10 of the U1 snRNA, and two others mapped to uridines 42 and 43 of the U1 snRNA (Fig. 5D, lane 1). Interactions of positions 8 to 10 of U1 snRNA with an intronic G-rich enhancer have already been observed, suggesting that these positions might represent a novel type of interaction involved in regulation (32).
FIG. 5.

Depletion of PTB from nuclear extracts promotes the interaction of U2 snRNP. (A) Psoralen cross-linking reactions using 32P-labeled pPmac-sexA1 were carried out with PTB-depleted (lanes 1 to 5) or mock-depleted (lanes 6 to 10) nuclear extracts in the presence of 50 ng (lanes 3 and 8), 100 ng (lanes 4 and 9), or 150 ng (lanes 5 and 10) of recombinant PTB1. RNAs were recovered and analyzed on a 5% polyacrylamide denaturing gel. Cross-linked species are indicated on the left. (B) Identification of cross-linked products. 32P-labeled pPmac-sexA1 was incubated with PTB-depleted nuclear extracts. Purified RNA from cross-linked reactions was mock-treated (lane 1) or treated with RNase H and oligonucleotides complementary to snRNA, as indicated at the top of each lane. Lanes 2 and 3, 20 and 200 pmol oligonucleotide complementary to nucleotides 29 to 42 of U2 snRNA, respectively; lanes 4 and 5, 10 and 100 pmol oligonucleotide complementary to nucleotides 1 to 15 of U2 snRNA, respectively; lanes 6 and 7, 10 and 100 pmol oligonucleotide complementary to nucleotides 66 to 77 of U1 snRNA, respectively. (C) Mapping of cross-linking sites on the RNA. Primer extension of U2 and U1-BP gel-purified cross-linked species was performed with an oligonucleotide complementary to the intron between exons 6A and 6B. Lanes 1 and 2, primer extension from U2 and U1-BP cross-linked species, respectively; lane 3 (Ex), primer extension from pPmac-SexA1; lanes 4 to 7, dideoxy sequencing reaction performed on pPmac-SexA1. Black and white arrows indicate transcription stops on pPmac-SexA1 with U2 and U1-BP cross-linked species, respectively. The positions of psoralen cross-links are indicated by circles on the sequence of pPmac-SexA1, shown on the right. (D) The cross-linked sites on U1-BP snRNA were mapped with an oligonucleotide complementary to nt 66 to 77 of U1 snRNA on the purified U1-BP species. Lane 1, primer extension of U1-BP snRNA cross-linked species; lanes 2 and 3, primer extension of snRNA U1 from HeLa nuclear extracts; lane 4 (Ex), primer extension of snRNA U1 transcribed from pBSU1 (25); lanes 5 to 8, dideoxy sequencing of snRNA U1 transcribed from pBSU1. Black arrows indicate specific transcription stops on U1 snRNA. The positions of the psoralen cross-links are indicated by circles on the sequence of U1 snRNA, shown on the right.
The stable interaction of U2 snRNA on the branch point that defines the A complex was shown to be dependent on both ATP and the presence of U1 snRNA (6). We next tested whether the interaction of U2 snRNP with the intron upstream of exon 6B would be relevant to the A complex formed onto an entire precursor. We found that the absence of ATP abolished the cross-linking of U2 snRNA on pPmac-SexAI RNA (Fig. 6A). In addition, the deletion of the downstream 5′ splice site or the mutation of the 5′ splice site (GTATGA to GACCCG), which precludes the binding of U1 snRNA, resulted in a significant reduction in the binding of U2 snRNA (Fig. 6B). These results indicate that the complex assembled onto the 3′ splice site has all the hallmarks of a 3′ functional complex. In agreement with that proposal, spliceosome analysis showed that the 6A-6B precursor proceeded through the assembly of A, B, and C complexes in PTB-titrated nuclear extracts, while only the heterogeneous H complex was observed in mock nuclear extracts (Fig. 6C). Combined with previous experiments, these data give strong support to a model in which the repression by PTB occurs at an early stage of spliceosome assembly before or at the interaction of U2 snRNA with the branch point upstream of exon 6B.
FIG. 6.

(A) The interaction of U2 snRNP on the branch point requires the presence of ATP. Psoralen cross-linking experiments were performed as described in the legend to Fig. 5A, with or without ATP (lanes 1 and 2, respectively). The bracket on the right indicates intramolecular cross-linked species. (B) The interaction of U2 snRNP on the branch point requires a downstream 5′ splice site. Psoralen cross-linking experiments were performed as for panel A with wild-type pPmac-SexA1 (lanes 1 to 4) or with 5′ splice site mutated RNA (lanes 5 to 8) or with deleted 5′ splice site (lanes 9 to 12) in the absence (lanes, 1, 2, 5, 6, 9, and 10) or in the presence (lanes, 3, 4, 7, 8, 11 and 12) of 0.5 pmol of Cs8/μl of nuclear extracts. For each RNA, odd and even lanes are duplicated samples. The star represents intramolecular cross-linked species. (C) PTB prevents spliceosome complex formation. The pre-mRNA containing exon 6A, intron, and exon 6B was incubated under splicing conditions without (lanes 1 to 5) or with (lanes 6 to 10) 0.5 pmol of Cs8/μl of nuclear extracts for 0 min (lanes 1 and 6), 15 min (lanes 2 and 7), 30 min (lanes 3 and 8), 60 min (lanes 4 and 9), and 90 min (lanes 5 and 10) at 30°C. H and SP represent heterogeneous and spliceosome complexes, respectively. Lanes 11 to 15, control pre-mRNAs.
PTB prevents the binding of the large U2AF65 subunit on the pyrimidine tract.
One of the first steps that is needed for the recruitment of U2 snRNP onto the branch point is the binding of U2AF65 to the polypyrimidine tract. Thus, we investigated whether PTB could affect the binding of the large U2AF65 subunit. To test this hypothesis, we performed UV cross-linking and immunoprecipitation experiments with anti-U2AF65 monoclonal antibodies in mock and PTB-depleted HeLa nuclear extracts. The depletion of PTB from nuclear extracts was performed either with Cs8 RNA bound to streptavidin agarose or by immunodepletion. The extent of PTB depletion was about 85% with nuclear extracts that have been depleted with Cs8 and 90% with the immunodepleted nuclear extract (Fig. 7A, ΔR and Δα, respectively). The same blot probed with antibodies against U2AF65 indicated that the level of U2AF65 was unaffected by PTB depletion (Fig. 7A). Uniformly labeled pPmac-SexA1 RNA was incubated under splicing conditions in mock and PTB-depleted HeLa nuclear extracts (Fig. 7B). A protein of around 60 kDa, migrating as a doublet, was predominantly cross-linked to the RNA in mock-depleted nuclear extracts (Fig. 7B, lanes 1 and 5). This protein was identified as PTB by immunoprecipitation experiments with anti-PTB antibodies (data not shown and Fig. 8E). As expected, cross-linking of PTB was reduced in PTB-depleted nuclear extracts compared to mock-depleted nuclear extracts (Fig. 7B; compare lanes 3 and 7 with lanes 1 and 5, respectively). However, whereas the level of PTB depletion was estimated to be around 80%, we repeatedly observed a reduction of cross-linked PTB of only 40 to 60%. The most likely explanation is that the high affinity of PTB for the pyrimidine-rich sequences upstream of exon 6B and the high concentration of PTB in HeLa nuclear extracts before the depletion (2 μM) could allow the cross-linking of the residual PTB. As shown by cross-linking/immunoprecipitation experiments with anti-U2AF65 monoclonal antibodies, the reduction of PTB cross-linking was associated with a 1.75-fold increase in U2AF65 cross-linking in nuclear extracts that have been depleted with the Cs8 RNA (two independent experiments) or a 2.3-fold increase in U2AF65 cross-linking in immunodepleted nuclear extracts (four independent experiments) (Fig. 7B; compare lanes 11 and 9 and lanes 15 and 13). Furthermore, addition of recombinant PTB1 at a concentration that reestablished in vitro splicing inhibition of exon 6B also rerepressed the binding of U2AF65 (Fig. 7B, lanes 12 and 16). Together, these results indicate that PTB prevents the binding of U2AF65. PTB binding sites were found in the polypyrimidine tract just downstream of the branch point, raising the possibility that PTB could compete with U2AF65 binding. To test this possibility, UV cross-linking/competition assays were performed with recombinant PTB and U2AF65 proteins in the absence or in the presence of nuclear extracts that have been PTB-immunodepleted. As expected from previous results, recombinant PTB binds avidly to the labeled pPmac-SexA1 RNA, PTB cross-linking being observed at the lowest concentration used (Fig. 7C, lanes 7 to 12). In contrast, U2AF65 cross-linked more weakly to pPmac-SexA1 than did PTB (Fig. 7C, lanes 1 to 6). Competition experiments performed with increasing amounts of recombinant PTB showed that PTB was able to displace not only the recombinant U2AF65 protein alone but also the recombinant U2AF65 incubated in PTB-immunodepleted nuclear extracts (Fig. 7D). Thus, these results suggest that PTB competes with U2AF65 for binding to the polypyrimidine tract.
FIG. 8.

PTB competes with U2AF65 for binding. (A) A truncated part of pPmac-SexA1 is shown with the intron upstream of exon 6B indicated in lowercase letters and the beginning of exon 6B in capital letters. The positions of site-specific labeling are indicated. The branch point is shown in bold. The positions of the different RNA fragments are indicated with their lengths. (B) U2AF65 interacts mainly with BP-PY. Gel shift experiments were performed without (lanes 1) or with (lanes 2 to 5, respectively) 1.7 nM, 5.4 nM, 17 nM, and 54 nM concentrations of recombinant GST-U2AF65. The Kd(app) was estimated by the concentration of protein that gives 50% of the RNA bound to the protein. (C) Binding of PTB to the intronic sequences upstream of exon 6B. Gel retardation assays were performed as for panel B without (lanes 1) or with (lanes 2 to 5, respectively) 2.8, 8.8, 28, and 88 nM recombinant his-PTB1. Note that higher-order complexes were detected at high PTB concentrations, as observed by others (2, 23). (D) UV cross-linking experiments on site-specifically labeled pPmac-SexA1. The reactions were performed as described for Fig. 7B with mock-depleted (M) or PTB-depleted (ΔR and Δα) nuclear extracts. After UV cross-linking, the samples were separated by SDS-PAGE (lanes 1 to 4, 9 to 12, and 17 to 20 [X-link]) or immunoprecipitated with anti U2AF65 antibodies (lanes 5 to 8, 13 to 16, and 21 to 24 [αU2AF65]). X denotes unknown cross-linked proteins with the RNA labeled at position 34 downstream of the branch point. The lower panel shows same samples as the upper panel, but for each position, different times of exposure on the PhosphorImager screen are presented. (E) Immunoprecipitation with anti-PTB antibodies. UV cross-linking experiments were performed under the same conditions as described for panel D and immunoprecipitated with anti-PTB antibodies.
To test this hypothesis more directly, UV cross-linking/immunoprecipitation experiments were performed on pPmac-SexA1 labeled at specific positions (Fig. 8A). First, we performed gel shift experiments. Gel retardation assays showed that recombinant U2AF65 bound mainly to the BP-PY RNA segment {apparent affinity [Kd(app)], 13.5 nM} and with a lower affinity [Kd(app), 54 nM] to the PY-3′6B segment (Fig. 8B). No binding was observed on BP, PY, and 3′6B. This suggests that U2AF65 interacts with two regions within the polypyrimidine tract, approximately 20 and 60 nt downstream of the branch point. As expected from previous results, PTB bound to all of the RNA segments except 3′6B, but with a higher affinity for BP and BP-PY [Kd(app), 6.5 nM] than for PY-3′6B [Kd(app), 17 nM] and PY [Kd(app), 75 nM] (Fig. 8C). From these results, we conclude that U2AF65 and PTB bind to the same regions and that the binding sites for these two proteins are in close proximity. Therefore, UV cross-linking/immunoprecipitation experiments were carried out on pPmac-SexA1 containing a single labeled phosphate incorporated 21, 34, or 58 nt downstream of the branch point (Fig. 8A). PTB was more efficiently cross-linked to the RNA labeled at position 21 than to the RNA labeled at position 58, whereas only a trace of cross-linking was observed for the RNA labeled at position 34 (Fig. 8D; compare lanes 1 and 3 with lanes 17 and 19, upper panel). The depletion of PTB from nuclear extracts either with the Cs8 RNA or the anti-PTB antibodies resulted in the reduction of PTB cross-linking (Fig. 8D, lanes 2 and 4 and lanes 18 and 20). As already observed with the uniformly labeled RNA, the reduction of PTB cross-linking was limited (around 30 to 40%), which was confirmed by UV cross-linking/immunoprecipitation experiments using monoclonal anti-PTB antibodies (Fig. 8E). In this case, a 35% decrease in PTB was observed at position 21 in immunodepleted nuclear extracts compared to mock-depleted nuclear extracts (Fig. 8E, lanes 1 and 2). More interestingly, at each position 21 and 58 that showed a decrease in PTB cross-linking, there was an increase in U2AF65 binding (about twofold) (Fig. 8D, lanes 6 and 8 and lanes 22 and 24, respectively). No detectable binding of U2AF65 could be observed at position 34. Altogether, these results strongly support the conclusion that PTB prevents the binding of U2AF65 by directly interfering with the binding of U2AF65 on the polypyrimidine tract upstream of exon 6B, which in turn prevents the recruitment of U2 snRNP on the branch site.
DISCUSSION
PTB is a well-known splicing repressor involved in the regulation of multiple pre-mRNAs. In this study, using the chicken βTm pre-mRNA as a model, we have shown that PTB inhibits exon 6B splicing by competing with the splicing factor U2AF65.
Based on an RNA affinity chromatography, we identified PTB as a potential regulator of exon 6B splicing. Using several approaches that combined in vitro splicing experiments and RNAi-mediated knockdown of PTB, we demonstrated that PTB blocks the splicing of exon 6B. It is noteworthy that exon 7 of the rat βTm, which is the equivalent of exon 6B, is the first example for which a repressive role for PTB has been proposed (35). Arbitrary division of the intronic sequence upstream of exon 6B into small RNA pieces allows us to find that PTB binds to the region encompassing the branch point (BP) and the PY region, which is located between the BP and 3′6B RNAs (Fig. 1 and 8). However, the two regions do not seem to be equivalent in mediating splicing repression. While mutations of the PTB binding sites in BP activated splicing of exon 6B in vivo, mutations in PY had only a limited effect (Fig. 4D). Similarly, whereas PTB depletion had a reduced impact on the minigene mutated in the BP region compared to the wild-type minigene, it still had an impact on the minigene mutated in the PY region (Fig. 4D). Several hypotheses might explain these results. Since the binding of PTB is significantly stronger to BP than to PY, with a sevenfold-increased affinity (Fig. 8C), one can suggest that PTB binding to PY plays a minor role in the repression of exon 6B splicing. Similar observations have been reported for the regulation of immunoglobulin M (IgM) pre-mRNA splicing, whereby of the two PTB binding sites located on exon M2 of the IgM pre-mRNA, only one seems to be needed for the repression (40). Another possibility is that PTB binding sites within PY are not sufficient by themselves but are needed in cooperation with those located on BP to regulate exon 6B splicing. The requirement for multiple PTB binding sites with different affinities is well exemplified in the case of the N1 exon from c-src. In this case, the efficient repression of the N1 exon requires, in addition to a high-affinity PTB binding site, an additional weaker binding site, which alone has little effect on splicing (2). Finally, one cannot totally exclude the possibility that the mutations have disrupted an enhancer element in PY. Transfection in HeLa cells of the 980 minigene containing the double mutation in BP and PY PTB binding sites did not allow us to test this hypothesis (data not shown). We found that the percentage of exon 6B inclusion in the double mutant was similar to that in the wild-type minigene, suggesting that the combination of mutations in both PTB binding sites was detrimental to exon 6B splicing. Indeed, we noticed a 2.5-fold decrease in U2AF65 binding to the RNA containing the double mutation compared to the wild-type RNA (data not shown). This could contribute to the reduced activity of exon 6B splicing. However, it is also possible that the double mutation has disrupted an enhancer element or generated a silencer element. The most convincing demonstration of an interplay between PTB binding sites within PY and BP would be to disrupt PTB binding without interfering with U2AF65 binding. As of now, we have not succeeded in constructing mutants in which the activities of PTB and U2AF65 could be separated (data not shown).
Despite the fact that PTB is a potent regulator of alternative splicing for numerous genes, the mechanism by which this protein represses splicing has remained elusive until recently. Using psoralen experiments, we showed that PTB prevented the binding of U2 snRNP to the branch point, which indicates that PTB acts at an early stage of spliceosome assembly. By UV cross-linking experiments performed on uniformly labeled RNA or RNA labeled at specific positions, we found that the removal of PTB correlated with an increase in U2AF65 binding, suggesting strongly that PTB competes with U2AF65 for the binding to the pyrimidine tract. In favor of this conclusion, we found that PTB was able to displace the binding of U2AF65. This finding was observed with the recombinant proteins alone but also in the presence of the proteins from nuclear extracts, arguing that PTB bound to the polypyrimidine tract blocks the access of U2AF65 in vivo (Fig. 7D). The second argument was provided by experiments with labeled RNA at site-specific positions. The observation that the decrease in PTB was associated with an increase in U2AF65 binding also argued that binding sites for PTB and U2AF65 were overlapping. Of the three positions that have been site-specifically labeled, two were present in intronic sequences that interacted with both U2AF65 and PTB (Fig. 8). A strong signal for U2AF65 was observed 21 nt downstream of the branch point, and a second, weaker signal was observed 58 nt downstream. This finding is in agreement with gel shift experiments showing that U2AF65 bound mainly to BP-PY [Kd(app), 13.5 nM], which covers position 21, and more weakly to PY-3′6B [Kd(app), 54 nM], which covers position 58 (Fig. 8B). Even if we have not entirely covered the polypyrimidine tract by site-specific labeling, we think that the interaction of U2AF65 at these positions is biologically relevant. Position 21 is within the polypyrimidine tract, which is adjacent to the branch point, in a configuration that has been shown to be required for splicing of introns with a far upstream branch point (43). In addition, the increase in U2AF65 binding after PTB removal was approximately the same whether the RNA was uniformly labeled or site-specifically labeled, suggesting that there are no additional U2AF65 binding sites elsewhere on the RNA. According to a recent model for U2AF65 interaction, we propose that the binding of U2AF65 at positions 21 and 58 could reflect the sliding of its RRM domains, RRM2 and RRM1, around these positions (5). The weaker binding observed at position 58 could be due to a lower affinity of the register or to an intrinsically less efficient cross-linking at this position than at position 21. Finally, the last argument in favor of a direct competition between PTB and U2AF65 was provided by the transfer of the 3′ region upstream of exon 6B in a heterologous context. We showed that this region, when transferred upstream of the β-globin exon 2, made the pre-mRNA responsive to PTB (Fig. 3). This demonstrates that the repression by PTB is independent of the nature of the 5′ splice site and of other sequences located elsewhere on the βTm transcript. Thus, our results support a model according to which PTB represses splicing of exon 6B by directly interfering with the binding of U2AF65. This model is in marked contrast with recent models proposing that PTB would disrupt the communication between U1 snRNP bound to the 5′ splice site and U2AF65 bound to the polypyrimidine tract across the exon or the intron (23, 39). On the contrary, our results validate a competition model that was proposed earlier but was not definitively proven (3, 37, 42). The competition between a negative regulator and a splicing factor for the binding to splicing signals has been well exemplified by the regulation of the TRA mRNA, whereby the binding of the female-specific factor SXL on the male-specific 3′ splice site blocks the binding of U2AF65, thus repressing the use of the male-specific exon (49).
Recently, the structure of PTB has been solved, leading to models of how PTB functions as a repressor. In particular, it has been proposed that the structural features of RRM3 and RRM4, which are tightly associated, allow the looping out of the RNA around an alternative exon or a branch site (36, 50). We noticed that in BP there is a 13-nt pyrimidine-rich segment with the CUCUCU motif just upstream of the branch point (Fig. 1A). Thus, the clustering of PTB binding sites on each side of the branch point could allow the looping out of the branch point, precluding the stabilization of the U2 snRNA-branch point interaction. Another possibility is that this particular organization of PTB binding sites around the branch point could also prevent the binding of a factor that is required for U2 snRNA stabilization. In favor of this hypothesis, we noticed that after PTB removal, the increase in U2 snRNA bound to the RNA was higher than that of U2AF65 (about six- to sevenfold for the former compared to two- to threefold for the latter). However, we cannot totally exclude the possibility that the increased level of U2AF65 cross-linking has been underestimated due to the fact that PTB is a very abundant protein, which avidly binds to the polypyrimidine tract. In this regard, it would be interesting to dissect the function of the different PTB motifs around the branch point and to test the function of different variants of PTB truncated in RRM4 in the repression of exon 6B splicing.
During this study, we observed that U1 snRNA could be cross-linked to an element upstream of the branch site (Fig. 5). The functional significance of this U1 snRNA interaction in the regulation of exon 6B remains obscure. In other models in which U1 snRNP is involved in the regulation, it has been shown to repress or activate splicing of alternative exons. As a repressor, U1 binding to regulatory elements functions as a decoy that diverts U1 snRNP from binding to the accurate 5′ splice site (20, 33, 41). As an activator, the binding of U1 snRNA to the G-rich sequence has been proposed to increase the informational content of the 5′ splice site and reinforce the selection of the authentic 5′ splice site (32). Here we show that in contrast to U2 snRNA binding, U1 snRNA interacted with the RNA even in the presence of PTB (Fig. 5). However, psoralen cross-linking experiments also showed that the amount of bound U1 snRNA increased in PTB-depleted nuclear extracts and is decreased by adding recombinant PTB, although the amplitude of the response to PTB was much less pronounced than for U2 snRNA (Fig. 5). We also noticed that U1-BP interacts with the RNA in the absence of U2 snRNP and that this interaction does not increase when U2 snRNP cannot bind, suggesting that the binding of U1 and U2 snRNP is not mutually exclusive (Fig. 5 and data not shown). If the interaction of U1 snRNA with an element upstream of the branch point is involved in the regulation, what could be its function? In the βTm alternative splicing model, the mutually exclusive character of exons 6A and 6B implies that they are never spliced together and that the inclusion of exon 6B in myotubes is associated with a downregulation of exon 6A. It could be possible that U1 snRNP bound in close proximity to U2 snRNP increases the informative content of the 3′ splice site upstream of exon 6B in such a way that in myotubes, splicing of exon 5 to exon 6B wins the competition over splicing of exon 5 to exon 6A. Another possibility is that U1 binding upstream of the branch site interrupts the interaction between U1 snRNA bound to the 5′ splice site of exon 6A and U2 snRNA bound to the branch point, thus preventing splicing of exons 6A and 6B. Clearly, further experiments are needed to understand the function of U1 snRNP in the splicing of exon 6B and how U1 binding intervenes in this regulation.
In conclusion, we have shown that PTB inhibits the splicing of the muscle-specific exon from the βTm pre-mRNA at an early stage of spliceosome assembly, before U2 snRNA interaction on the branch site. Is PTB the only determinant of the repression? The answer is probably no. In HeLa cells, PTB knockdown led to an increase of exon 6B inclusion from 4 to about 22% (Fig. 4). The partial derepression of exon 6B splicing when PTB has been removed suggests that other factors could be involved in the repression of exon 6B splicing in nonmuscle cells and myoblasts. In favor of this hypothesis, previous works have shown that mutations in the 3′6B fragment, which is not a target for PTB binding, also derepress splicing of exon 6B both in vitro and ex vivo (12, 29). During this study, we identified hnRNP K in the complex assembled onto the 3′6B fragment (data not shown). Interestingly, addition of poly(C), which is known to bind hnRNP K, to psoralen reactions increased the interaction of U2 snRNA with the branch site, suggesting that hnRNP K could be involved in the repression of exon 6B (data not shown). It has also been shown that the repression of exon 6B involves the S4 sequence that lies downstream of exon 6A and is a binding target for hnRNP K, PTB, and the SR proteins ASF/SF2 and SC35 (4, 16, 18, 19). It is noteworthy that the architecture of the intron separating exons 6A and 6B is striking, with the 5′ and the 3′ end of the intron containing contiguous binding sites for hnRNP K and PTB (this study and reference 16). The function (if any) of this architecture remains obscure. However, it must also be recalled that contrary to the α-tropomyosin model, for which the mutually exclusive behavior of exons 2 and 3 is easily explained by a steric hindrance between the 5′ splice site and the branch point, the mechanism by which exons 6A and 6B are mutually exclusive remains to be established. One can suggest that some of the regulatory elements and the proteins through which the regulation is mediated are involved in maintaining the mutually exclusive character of exons 6A and 6B. In addition to regulatory elements lying upstream of exon 6B, our recent studies have shown that a G-rich element located downstream of exon 6B is involved in regulation (17). This element, through the binding of hnRNP A1, represses the splicing of exon 6B (17). Thus, it is clear from these data that the repression of exon 6B splicing in nonmuscle cells results from the combination of multiple elements and factors that bind on both sides of exon 6B and which are probably required to fully block the recognition of exon 6B in this cellular context. In addition, one can also suggest that the partial derepression of exon 6B induced by the removal of PTB may indicate that activating factors needed for the recognition of exon 6B in myotubes are missing or are in a concentration too low to counteract the activity of repressive factors. Preliminary data suggest that this could be the case (data not shown). Future experiments are required to understand how these factors work together and what is the hierarchy among these factors in regulating splicing of exon 6B during myogenic differentiation.
Acknowledgments
We gratefully acknowledge G. Dreyfuss and J. Valcárcel for the kind gifts of antibodies. Many thanks go to J. Patton, J. Valcárcel, and C. Smith for providing plasmids. We thank D. Libri and S. Camier for fruitful discussions and advice during the course of preparation of the manuscript. Special thanks go to K. Tanner for carefully reading the manuscript.
This work was supported by CNRS and Ligue Nationale Contre le Cancer. J. Saulière is supported by a fellowship from Association Française contre les Myopathies.
Footnotes
Published ahead of print on 18 September 2006.
REFERENCES
- 1.Abmayr, S. M., R. Reed, and T. Maniatis. 1988. Identification of a functional mammalian spliceosome containing unspliced pre-mRNA. Proc. Natl. Acad. Sci. USA 85:7216-7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amir-Ahmady, B., P. L. Boutz, V. Markovtsov, M. L. Phillips, and D. L. Black. 2005. Exon repression by polypyrimidine tract binding protein. RNA 11:699-716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ashiya, M., and P. J. Grabowski. 1997. A neuron-specific splicing switch mediated by an array of pre-mRNA repressor sites: evidence of a regulatory role for the polypyrimidine tract binding protein and a brain-specific PTB counterpart. RNA 3:996-1015. [PMC free article] [PubMed] [Google Scholar]
- 4.Balvay, L., D. Libri, M. Gallego, and M. Y. Fiszman. 1992. Intronic sequence with both negative and positive effects on the regulation of alternative transcripts of the chicken beta tropomyosin transcripts. Nucleic Acids Res. 20:3987-3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banerjee, H., A. Rahn, W. Davis, and R. Singh. 2003. Sex lethal and U2 small nuclear ribonucleoprotein auxiliary factor (U2AF65) recognize polypyrimidine tracts using multiple modes of binding. RNA 9:88-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barabino, S. M., B. J. Blencowe, U. Ryder, B. S. Sproat, and A. I. Lamond. 1990. Targeted snRNP depletion reveals an additional role for mammalian U1 snRNP in spliceosome assembly. Cell 63:293-302. [DOI] [PubMed] [Google Scholar]
- 7.Black, D. L. 2003. Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 72:291-336. [DOI] [PubMed] [Google Scholar]
- 8.Carstens, R. P., E. J. Wagner, and M. A. Garcia-Blanco. 2000. An intronic splicing silencer causes skipping of the IIIb exon of fibroblast growth factor receptor 2 through involvement of polypyrimidine tract binding protein. Mol. Cell. Biol. 20:7388-7400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chan, R. C., and D. L. Black. 1997. The polypyrimidine tract binding protein binds upstream of neural cell-specific c-src exon N1 to repress the splicing of the intron downstream. Mol. Cell. Biol. 17:4667-4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charlet, B. N., P. Logan, G. Singh, and T. A. Cooper. 2002. Dynamic antagonism between ETR-3 and PTB regulates cell type-specific alternative splicing. Mol. Cell 9:649-658. [DOI] [PubMed] [Google Scholar]
- 11.Chou, M. Y., J. G. Underwood, J. Nikolic, M. H. Luu, and D. L. Black. 2000. Multisite RNA binding and release of polypyrimidine tract binding protein during the regulation of c-src neural-specific splicing. Mol. Cell 5:949-957. [DOI] [PubMed] [Google Scholar]
- 12.Clouet d'Orval, B., Y. d'Aubenton-Carafa, J. M. Brody, and E. Brody. 1991. Determination of an RNA structure involved in splicing inhibition of a muscle-specific exon. J. Mol. Biol. 221:837-856. [DOI] [PubMed] [Google Scholar]
- 13.Cote, J., S. Dupuis, and J. Y. Wu. 2001. Polypyrimidine track-binding protein binding downstream of caspase-2 alternative exon 9 represses its inclusion. J. Biol. Chem. 276:8535-8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Das, R., and R. Reed. 1999. Resolution of the mammalian E complex and the ATP-dependent spliceosomal complexes on native agarose mini-gels. RNA 5:1504-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dignam, J. D., R. M. Lebovitz, and R. G. Roeder. 1983. Accurate transcription initiation by RNA polymerase II in a soluble extract from isolated mammalian nuclei. Nucleic Acids Res. 11:1475-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Expert-Bezancon, A., J. P. Le Caer, and J. Marie. 2002. Heterogeneous nuclear ribonucleoprotein (hnRNP) K is a component of an intronic splicing enhancer complex that activates the splicing of the alternative exon 6A from chicken beta-tropomyosin pre-mRNA. J. Biol. Chem. 277:16614-16623. [DOI] [PubMed] [Google Scholar]
- 17.Expert-Bezancon, A., A. Sureau, P. Durosay, R. Salesse, H. Groeneveld, J. P. Lecaer, and J. Marie. 2004. hnRNP A1 and the SR proteins ASF/SF2 and SC35 have antagonistic functions in splicing of beta-tropomyosin exon 6B. J. Biol. Chem. 279:38249-38259. [DOI] [PubMed] [Google Scholar]
- 18.Gallego, M. E., L. Balvay, and E. Brody. 1992. cis-acting sequences involved in exon selection in the chicken β-tropomyosin gene. Mol. Cell. Biol. 12:5415-5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallego, M. E., R. Gattoni, J. Stevenin, J. Marie, and A. Expert-Bezancon. 1997. The SR splicing factors ASF/SF2 and SC35 have antagonistic effects on intronic enhancer-dependent splicing of the beta-tropomyosin alternative exon 6A. EMBO J. 16:1772-1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giles, K. E., and K. L. Beemon. 2005. Retroviral splicing suppressor sequesters a 3′ splice site in a 50S aberrant splicing complex. Mol. Cell. Biol. 25:4397-4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gooding, C., G. C. Roberts, and C. W. Smith. 1998. Role of an inhibitory pyrimidine element and polypyrimidine tract binding protein in repression of a regulated alpha-tropomyosin exon. RNA 4:85-100. [PMC free article] [PubMed] [Google Scholar]
- 22.Goux-Pelletan, M., D. Libri, Y. d'Aubenton-Carafa, M. Fiszman, E. Brody, and J. Marie. 1990. In vitro splicing of mutually exclusive exons from the chicken beta-tropomyosin gene: role of the branch point location and very long pyrimidine stretch. EMBO J. 9:241-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Izquierdo, J. M., N. Majos, S. Bonnal, C. Martinez, R. Castelo, R. Guigo, D. Bilbao, and J. Valcarcel. 2005. Regulation of Fas alternative splicing by antagonistic effects of TIA-1 and PTB on exon definition. Mol. Cell 19:475-484. [DOI] [PubMed] [Google Scholar]
- 24.Kan, J. L., and M. R. Green. 1999. Pre-mRNA splicing of IgM exons M1 and M2 is directed by a juxtaposed splicing enhancer and inhibitor. Genes Dev. 13:462-471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Konarska, M. M., and P. A. Sharp. 1987. Interactions between small nuclear ribonucleoprotein particles in formation of spliceosomes. Cell 49:763-774. [DOI] [PubMed] [Google Scholar]
- 26.Krainer, A. R., T. Maniatis, B. Ruskin, and M. R. Green. 1984. Normal and mutant human beta-globin pre-mRNAs are faithfully and efficiently spliced in vitro. Cell 36:993-1005. [DOI] [PubMed] [Google Scholar]
- 27.Le Guiner, C., A. Plet, D. Galiana, M. C. Gesnel, F. Del Gatto-Konczak, and R. Breathnach. 2001. Polypyrimidine tract-binding protein represses splicing of a fibroblast growth factor receptor-2 gene alternative exon through exon sequences. J. Biol. Chem. 276:43677-43687. [DOI] [PubMed] [Google Scholar]
- 28.Le Sommer, C., M. Lesimple, A. Mereau, S. Menoret, M. R. Allo, and S. Hardy. 2005. PTB regulates the processing of a 3′-terminal exon by repressing both splicing and polyadenylation. Mol. Cell. Biol. 25:9595-9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Libri, D., L. Balvay, and M. Y. Fiszman. 1992. In vivo splicing of the beta tropomyosin pre-mRNA: a role for branch point and donor site competition. Mol. Cell. Biol. 12:3204-3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Libri, D., M. Goux-Pelletan, E. Brody, and M. Y. Fiszman. 1990. Exon as well as intron sequences are cis-regulating elements for the mutually exclusive alternative splicing of the β-tropomyosin gene. Mol. Cell. Biol. 10:5036-5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lin, C. H., and J. G. Patton. 1995. Regulation of alternative 3′ splice site selection by constitutive splicing factors. RNA 1:234-245. [PMC free article] [PubMed] [Google Scholar]
- 32.McCullough, A. J., and S. M. Berget. 2000. An intronic splicing enhancer binds U1 snRNPs to enhance splicing and select 5′ splice sites. Mol. Cell. Biol. 20:9225-9235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McNally, L. M., and M. T. McNally. 1999. U1 small nuclear ribonucleoprotein and splicing inhibition by the Rous sarcoma virus negative regulator of splicing element. J. Virol. 73:2385-2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moore, M. J., and P. A. Sharp. 1992. Site-specific modification of pre-mRNA: the 2′-hydroxyl groups at the splice sites. Science 256:992-997. [DOI] [PubMed] [Google Scholar]
- 35.Mulligan, G. J., W. Guo, S. Wormsley, and D. M. Helfman. 1992. Polypyrimidine tract binding protein interacts with sequences involved in alternative splicing of beta-tropomyosin pre-mRNA. J. Biol. Chem. 267:25480-25487. [PubMed] [Google Scholar]
- 36.Oberstrass, F. C., S. D. Auweter, M. Erat, Y. Hargous, A. Henning, P. Wenter, L. Reymond, B. Amir-Ahmady, S. Pitsch, D. L. Black, and F. H. Allain. 2005. Structure of PTB bound to RNA: specific binding and implications for splicing regulation. Science 309:2054-2057. [DOI] [PubMed] [Google Scholar]
- 37.Perez, I., C. H. Lin, J. G. McAfee, and J. G. Patton. 1997. Mutation of PTB binding sites causes misregulation of alternative 3′ splice site selection in vivo. RNA 3:764-778. [PMC free article] [PubMed] [Google Scholar]
- 38.Sawa, H., and Y. Shimura. 1992. Association of U6 snRNA with the 5′-splice site region of pre-mRNA in the spliceosome. Genes Dev. 6:244-254. [DOI] [PubMed] [Google Scholar]
- 39.Sharma, S., A. M. Falick, and D. L. Black. 2005. Polypyrimidine tract binding protein blocks the 5′ splice site-dependent assembly of U2AF and the prespliceosomal E complex. Mol. Cell 19:485-496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen, H., J. L. Kan, C. Ghigna, G. Biamonti, and M. R. Green. 2004. A single polypyrimidine tract binding protein (PTB) binding site mediates splicing inhibition at mouse IgM exons M1 and M2. RNA 10:787-794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Siebel, C. W., L. D. Fresco, and D. C. Rio. 1992. The mechanism of somatic inhibition of Drosophila P-element pre-mRNA splicing: multiprotein complexes at an exon pseudo-5′ splice site control U1 snRNP binding. Genes Dev. 6:1386-1401. [DOI] [PubMed] [Google Scholar]
- 42.Singh, R., J. Valcarcel, and M. R. Green. 1995. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science 268:1173-1176. [DOI] [PubMed] [Google Scholar]
- 43.Smith, C. W., E. B. Porro, J. G. Patton, and B. Nadal-Ginard. 1989. Scanning from an independently specified branch point defines the 3′ splice site of mammalian introns. Nature 342:243-247. [DOI] [PubMed] [Google Scholar]
- 44.Smith, C. W., and J. Valcarcel. 2000. Alternative pre-mRNA splicing: the logic of combinatorial control. Trends Biochem. Sci. 25:381-388. [DOI] [PubMed] [Google Scholar]
- 45.Southby, J., C. Gooding, and C. W. Smith. 1999. Polypyrimidine tract binding protein functions as a repressor to regulate alternative splicing of α-actinin mutually exclusive exons. Mol. Cell. Biol. 19:2699-2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Spellman, R., A. Rideau, A. Matlin, C. Gooding, F. Robinson, N. McGlincy, S. N. Grellscheid, J. Southby, M. Wollerton, and C. W. Smith. 2005. Regulation of alternative splicing by PTB and associated factors. Biochem. Soc. Trans. 33:457-460. [DOI] [PubMed] [Google Scholar]
- 47.Stamm, S., S. Ben-Ari, I. Rafalska, Y. Tang, Z. Zhang, D. Toiber, T. A. Thanaraj, and H. Soreq. 2005. Function of alternative splicing. Gene 344:1-20. [DOI] [PubMed] [Google Scholar]
- 48.Valcarcel, J., and F. Gebauer. 1997. Post-transcriptional regulation: the dawn of PTB. Curr. Biol. 7:R705-R708. [DOI] [PubMed] [Google Scholar]
- 49.Valcarcel, J., R. Singh, P. D. Zamore, and M. R. Green. 1993. The protein Sex-lethal antagonizes the splicing factor U2AF to regulate alternative splicing of transformer pre-mRNA. Nature 362:171-175. [DOI] [PubMed] [Google Scholar]
- 50.Vitali, F., A. Henning, F. C. Oberstrass, Y. Hargous, S. D. Auweter, M. Erat, and F. H. Allain. 2006. Structure of the two most C-terminal RNA recognition motifs of PTB using segmental isotope labeling. EMBO J. 25:150-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wagner, E. J., and M. A. Garcia-Blanco. 2001. Polypyrimidine tract binding protein antagonizes exon definition. Mol. Cell. Biol. 21:3281-3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wollerton, M. C., C. Gooding, F. Robinson, E. C. Brown, R. J. Jackson, and C. W. Smith. 2001. Differential alternative splicing activity of isoforms of polypyrimidine tract binding protein (PTB). RNA 7:819-832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wollerton, M. C., C. Gooding, E. J. Wagner, M. A. Garcia-Blanco, and C. W. Smith. 2004. Autoregulation of polypyrimidine tract binding protein by alternative splicing leading to nonsense-mediated decay. Mol. Cell 13:91-100. [DOI] [PubMed] [Google Scholar]
- 54.Yu, Y. T., and J. A. Steitz. 1997. A new strategy for introducing photoactivatable 4-thiouridine ((4S)U) into specific positions in a long RNA molecule. RNA 3:807-810. [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang, L., W. Liu, and P. J. Grabowski. 1999. Coordinate repression of a trio of neuron-specific splicing events by the splicing regulator PTB. RNA 5:117-130. [DOI] [PMC free article] [PubMed] [Google Scholar]