

Abstract
Glucocorticoids are used to treat various inflammatory disorders, but the mechanisms underlying these actions are incompletely understood. The zinc finger protein tristetraprolin (TTP) destabilizes several proinflammatory cytokine mRNAs by binding to AU-rich elements within their 3′ untranslated regions, targeting them for degradation. Here we report that glucocorticoids induce the synthesis of TTP mRNA and protein in A549 lung epithelial cells and in rat tissues. Dexamethasone treatment leads to a sustained induction of TTP mRNA expression that is abrogated by RU486. Glucocorticoid induction of TTP mRNA is also blocked by actinomycin D but not by cycloheximide, suggesting a transcriptional mechanism which has been confirmed by transcription run-on experiments. The most widely characterized TTP-regulated gene is the AU-rich tumor necrosis factor alpha (TNF-α) gene. Dexamethasone represses TNF-α mRNA in A549 cells and decreases luciferase expression of a TNF-α 3′ untranslated region reporter plasmid in an orientation-dependent manner. Small interfering RNAs to TTP significantly prevent this effect, and a cell line stably expressing a short-hairpin RNA to TTP conclusively establishes that TTP is critical for dexamethasone inhibition of TNF-α mRNA expression. These studies provide the molecular evidence for glucocorticoid regulation of human TTP and reflect a novel inductive anti-inflammatory signaling pathway for glucocorticoids that acts via posttranscriptional mechanisms.
During inflammation, activated lymphocytes or macrophages secrete inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) or interleukin 1β (IL-1β). These cytokines in turn not only activate components of the inflammatory system but also exert profound excitatory effects on the hypothalamic-pituitary-adrenal axis, resulting in the synthesis and secretion of glucocorticoids. These adrenal steroids subsequently exert anti-inflammatory effects on many cell types, including T cells, macrophages, eosinophils, neutrophils, mast cells, and endothelial and epithelial cells, thereby creating a classical endocrine feedback loop (12). Glucocorticoids accomplish these actions in a glucocorticoid receptor (GR)-dependent manner through repression of proinflammatory signaling pathways, such as those activated by nuclear factor κB (NF-κB) or the mitogen-activated protein kinase (MAPK) pathway. GR-mediated abrogation of these proinflammatory signaling pathways leads to repression of the production of a number of cytokines, chemokines, and inflammatory enzymes that are relevant to inflammatory diseases, including TNF-α, granulocyte/macrophage colony-stimulating factor (GM-CSF), ILs (IL-1β, IL-2, IL-3, IL-6, IL-8, and IL-11), cyclooxygenase 2 (COX-2), and inducible nitric oxide synthase (1), by poorly understood mechanisms. Because of these profound anti-inflammatory effects, exogenous synthetic glucocorticoids are often prescribed for several immune and inflammatory diseases, including rheumatoid arthritis, inflammatory bowel disease, and asthma (4).
Transient inflammatory responses are often tightly controlled by both transcriptional and posttranscriptional mechanisms regulating proinflammatory gene expression. Posttranscriptional control, via regulation of mRNA turnover, is known to be conferred by adenylate-uridylate-rich elements (AREs) located in the 3′ untranslated regions (UTRs) of transcripts encoding various cytokines, chemokines, and other inflammatory proteins (3). These AREs, composed of multiple overlapping AUUUA motifs, promote the deadenylation of the cytokine mRNA and its subsequent degradation by the exosome (11). Therefore, once an adequate inflammatory response has been achieved, the ARE-dependent mRNA decay and subsequent inhibition of protein synthesis is vital for returning cytokines to basal levels. Prolonged expression of proinflammatory cytokines can lead to a variety of tissue-destructive pathologies. For example, transgenic mice expressing ARE-deleted TNF-α transcripts develop both chronic inflammatory arthritis and Crohn's disease-like inflammatory bowel disease as a result of prolonged overexpression of TNF-α (20). Therefore, the mRNA instability conferred by the AU-rich UTR is an important mechanism by which inflammatory responses are kept restrained.
The mechanism by which AREs regulate mRNA instability involves trans-acting factors which bind to the ARE and promote mRNA deadenylation and subsequent degradation. One such RNA binding protein is tristetraprolin (TTP) (23), also known as TIS11 (39), Nup 475 (14), or GOS24 (39). TTP is the prototype of a small family of tandem CCCH zinc finger proteins which bind to AREs through specific interactions with zinc finger motifs (21), promoting deadenylation through recruitment of a poly(A) RNase (22). TTP was initially described as being encoded by an immediate-early response gene (ZFP-36) which is rapidly and transiently induced in fibroblasts in response to insulin, phorbol esters, and serum (14, 23, 39). Since then, transient induction of TTP has been reported in T cells stimulated with transforming growth factor β (TGF-β) (30) and in macrophages treated with either TNF-α or lipopolysaccharide (LPS) (10). Importantly, in studies with myeloid cell types, TTP was found to be a component of a negative feedback loop which interferes with TNF-α production by destabilizing its mRNA. Model systems that characterize the effect of TTP on the TNF-α 3′ UTR have recently been developed. Human TTP reduces TNF-α 3′ UTR-dependent luciferase expression when cotransfected into HEK293 cells. Furthermore, mice deficient in TTP display increased TNF-α mRNA stability, overexpression of the cytokine, and a chronic inflammatory disease characterized by severe arthritis, autoimmunity, and myeloid hyperplasia (37). Overexpression of GM-CSF and COX-2 in cells derived from TTP-deficient mice has also been reported (9, 32), and less-direct evidence suggests that other targets of TTP include IL-2 (31, 33), IL-3 (35), and IL-8 (5).
While GR repression of the proinflammatory transcription factors NF-κB and AP-1 is widely known as an important mechanism by which glucocorticoids exert their anti-inflammatory effects (13), glucocorticoids can also posttranscriptionally regulate genes of the inflammatory response. For example, they have been found to destabilize the mRNAs of some cytokines and inflammatory mediators (15, 17, 29); however, the mechanisms underlying these actions are unclear. Recently, GR-mediated induction of MAPK phosphatase 1 (MKP-1) and subsequent interruption of p38 MAPK signaling has been proposed as a new anti-inflammatory mechanism, and glucocorticoids have been shown to destabilize COX-2 mRNA through this mechanism (24, 25). However, the possibility that glucocorticoids may exert anti-inflammatory actions through other mechanisms clearly exists. We show here for the first time that glucocorticoids induce the synthesis of TTP mRNA and protein in A549 lung epithelial cells and in rat tissues. This induction appears to be through a direct, GR-mediated transcriptional mechanism, with downstream effects on TNF-α. These studies thus represent a novel signaling pathway by which glucocorticoids exert their anti-inflammatory effects.
MATERIALS AND METHODS
Cell culture and treatments.
A549 cells were purchased from ATCC and maintained as previously described (16) in Dulbecco's modified Eagle's medium/F-12 medium supplemented with 5% fetal calf serum, 100 IU of penicillin per ml, and 100 mg/ml of streptomycin. Dexamethasone and mifepristone (RU486) (Steraloids, Wilton, N.H.) were dissolved in water and ethanol, respectively, and used at the indicated concentrations. Treatments also included recombinant human TNF-α (Pierce), actinomycin D (Calbiochem), cycloheximide (Sigma), or LPS (Escherichia coli serotype O55:B5 [Sigma]) as indicated.
Animals and treatments.
One-month-old male Sprague-Dawley rats (Charles River) were bilaterally adrenalectomized at least 5 days before use. Dexamethasone was resuspended in phosphate-buffered saline by sonication and administered by intraperitoneal injection at a concentration of 5 mg/kg of body weight (about 1 mg). Animals were sacrificed by decapitation either 6 h (mRNA analysis) or 14 h (protein analysis) after injection, and tissues were surgically removed. All experimental protocols were approved by the animal review committee of the National Institute of Environmental Health Services (NIEHS) and were performed in accordance with the guidelines set forth in the NIH Guide for the Care and Use of Laboratory Animals.
Quantitative real-time RT-PCR.
Primer/probe sets for rat TTP, human TNF-α, human cyclophilin B, Renilla luciferase, and firefly luciferase were designed with PRIMER EXPRESS software, version 2.0 (Applied Biosystems). Predeveloped, validated primer/probe sets for human TTP and rat cyclophilin B were purchased (Applied Biosystems). Total RNA was extracted from cells and tissues with RNeasy kits (QIAGEN) according to the manufacturer's protocol. For tissue-extracted RNA, tissues were first placed in RNA stabilization reagent (RNAlater; QIAGEN) until the time of extraction and were homogenized with a Tissuemizer. For both cell culture and tissue-extracted RNA, DNase treatment with QIAGEN′s DNase Set was performed to remove residual DNA before reverse transcriptase (RT) PCR. RT-PCR mixtures were composed of the following: Universal PCR Master Mix, No AmpErase (Applied Biosystems), 200 nM concentrations of probe and primers (or an appropriate amount of predeveloped primer/probe sets, as described by the manufacturer), 0.5 μg RNA, 0.4 U/μl RNase inhibitor (Roche Diagnostics, Indianapolis, Ind.), and 0.4 U/μl murine leukemia virus RT (Roche). Negative controls, consisting of all components except for RT enzyme, gave no signal in every case. Reactions were run in duplicate on an ABI Prism 7900HT with thermal cycling parameters specific for one-step RT-PCR. The efficiency (slopes) of the target amplification and the efficiency of the reference endogenous control (cyclophilin B or Renilla luciferase) amplification were 100% (±10%). Target gene expression levels were normalized to endogenous controls in all experiments, and data were plotted relative to control (hormone-free) mRNA.
Western blot analysis.
Cells were detached from flasks with Versene (2.6 mM MgCl2, 1.5 mM KH2PO4, 136 mM NaCl, 0.5 mM EDTA, 8.1 mM Na2HPO4), pelleted, lysed in low-detergent buffer (20 mM Tris, 2 mM EDTA, 150 mM NaCl, 0.5% Triton X-100, 5% phosphatase inhibitors, 0.5% protease inhibitors), and sonicated with a cell disruptor (Branson Ultrasonic Co.). Tissue extracts were prepared by first disrupting tissue from organs in low-detergent buffer with a tissue grinder (Duall Type 22; Kontes Glass Co.) and then with a Tissuemizer, followed by centrifugation (18,000 × g, 1 min). Supernatants were then centrifuged again for 10 min (18,000 × g), and the resulting supernatants were used for Western blotting. Total protein for both cell and tissue extracts was determined with a protein assay reagent (Bio-Rad). Equivalent amounts of total protein were separated on sodium dodecyl sulfate (SDS)-polyacrylamide gels and transferred to nitrocellulose membranes. Antibodies for immunoblot analysis include anti-hTTP (C terminus) (6) (from W. Rigby, Dartmouth Medical School), anti-mTTP (8) (a gift of P. Blackshear, NIEHS, NIH), and anti-α-tubulin (Sigma). Densitometric analysis of immunoreactive bands was performed with NIH Image software, and TTP signals were normalized to those of α-tubulin for figures.
Nuclear run-on transcription analysis.
A549 cells were treated for 2 h with or without dexamethasone (100 nM). For preparation of nuclei, cells were washed with ice-cold phosphate-buffered saline and then harvested and lysed simultaneously with the Nuclei EZ Prep nucleus isolation kit (Sigma). Nuclei were immediately used in transcription reactions as described previously (40) by mixing them with an equal volume of reaction buffer containing the following components at final concentrations: 100 mM Tris-Cl (pH 7.9); 4 mM MgCl2; 200 mM NaCl; 0.4 mM EDTA (pH 8.0); 300 mM (NH4)2SO4; 4 mM MnCl2; 1.2 μM dithiothreitol; 1 mM (each) ATP, CTP, and GTP; 10 mM creatine phosphate; 240 U/ml RNasin (Promega); 29% glycerol; 0.1 mM phenylmethylsulfonyl fluoride; and 150 μCi [α-32P]UTP (800 Ci/mmol) (MP Biomedicals, Irvine, Calif.). Mixtures were then incubated at 30°C for 45 min. The reaction was terminated with a 4 M guanidine thiocyanate and 0.1 M β-mercaptoethanol solution. RNA was purified with an RNeasy Mini kit from QIAGEN and treated with DNase to remove contaminating DNA. The number of disintegrations per minute was determined with an LS 6500 scintillation counter (Beckman Coulter), and an equal number of counts were hybridized to target DNAs immobilized on Hybond N-plus nylon membranes (Amersham) for 48 h at 65°. Membranes were washed at room temperature twice with 5× SSPE (1× SSPE is 0.18 M NaCl, 10 mM NaH2PO4, and 1 mM EDTA [pH 7.7]) containing 0.1% SDS, followed by one wash with 2× SSPE containing 0.1% SDS, and were then placed in phosphorimaging cassettes for 1 to 5 days. Phosphorimaging was performed on a Typhoon 8600 variable-mode imager (Amersham), and relative quantification of specific run-on transcripts was determined with ImageQuant software, version 5.2. Specific hybridization was calculated by subtracting out the signal from vector DNA and then subtracting the sense signal from the antisense signal. The following DNAs (cloned into M13 vectors in antisense and sense orientations) were hybridized to slot blots: a 459-bp fragment from the 3′ region of human TTP, the 18S rRNA gene, and the actin gene.
ChIP assays.
The chromatin immunoprecipitation (ChIP) assay was performed with the ChIP assay kit from Upstate Biotechnologies (Lake Placid, N.Y.). A549 cells (1 × 107) were treated with 1 μM dexamethasone for 1 h and then fixed with 1% formaldehyde for 10 min at 37°C. Cross-linking was terminated with the addition of 125 mM glycine for 10 min at 37°C. Cells were harvested and lysed, and DNA was sheared by sonication (eight sets of 10-second pulses on a Branson Ultrasonic cell disruptor). An equal portion of the chromatin solution was saved for input from both control and dexamethasone-treated samples. For immunoprecipitations (IPs), a 1-μg amount of anti-GR antibody or rabbit immunoglobulin G (Upstate Biotechnologies) was added to the chromatin solution containing 50 μg DNA. After an overnight incubation, the antibody/GR complexes were captured by protein A agarose beads and subjected to serial washes. The chromatin fraction was eluted and reverse cross-linked at 65°C overnight in the presence of 200 nM NaCl. The DNA was then purified with the QIAGEN PCR purification kit according to the manufacturer's protocol, with an additional wash with buffer PE. DNA was eluted in 50-μl volumes for IP samples and 100-μl volumes for input samples. For PCR amplification of the TTP promoter region, −1314 to −901 (accession no. AY771351), which spans the STAT, Smad, and NF-κB consensus binding sites, forward and reverse primers (5′-CTCAGCCCAGGTGAGTTCTCCTTAAC-3′ and 5′-GGGCAACAGTGAGATCCCATGTCTAC-3′, respectively) were used to amplify the 413-bp region with 25 cycles of PCR, with 2 μl of DNA for IP reactions and 1 μl of DNA for input reactions. To amplify the 255-bp region of the TTP 3′ flanking region (+3283 to +3540), which spans a GRE half-site, forward and reverse primers (5′-GCGCCCGGAAGCCGCCCTGCGTCA-3′ and 5′-AAGTGGGAGAGGAGTAATGAGGGA-3′, respectively) were used with 10 μl DNA for IP reactions and 1 μl DNA for input reactions and subjected to 35 cycles of PCR. The 325-bp intronic region (+2023 to +2347) was amplified with forward and reverse primers (5′-AGACCAGCTTGGTGATTTGGAGGT-3′ and 5′-CAACTGGAGAAACCGGGCAAAGTT-3′, respectively) with 10 μl DNA for IP reactions and 1 μl DNA for input reactions and subjected to 35 cycles of PCR. PCR products were run on a 1% agarose gel with ethidium bromide and photographed.
Plasmids, siRNAs, transfections, and dual-luciferase assay.
The pGL3-Control plasmid was purchased (Promega). pGL3-hRL (Renilla luciferase normalization plasmid) was generously provided by N. Lu (NIEHS). TNF-UTRLuc was constructed by inserting nucleotides 1261 to 1602 of the TNF-α 3′ UTR (accession number NM_000594) into the XbaI site of pGL3 control luciferase. The Rev-TNF-UTRLuc control was constructed similarly but in the opposite orientation from TNF-UTRLuc. The TNFLuc control was constructed by inserting a similar-size portion of the TNF-α 5′ region, which contains no AREs (nucleotides 4 to 342), into the XbaI site of pGL3 control luciferase. Small interfering RNAs (siRNAs) to human TTP and validated negative siRNA (scrambled RNA) were purchased (Ambion, Austin, Tex.) and used at a recommended final concentration of 100 nM. A549 cells (1.5 × 105) were transfected 24 h after plating. Initial experiments with the TNF-UTRLuc plasmid were performed with TransIT transfection reagent (Mirus Technologies, Madison, Wis.), and later experiments with TNF-UTRLuc and siRNAs were performed with Lipofectamine 2000 (Invitrogen). Twenty-four hours after transfection, cultures were treated with dexamethasone for 20 h and the dual-luciferase assay was subsequently performed according to the manufacturer's protocol (Promega). Firefly luciferase was normalized to Renilla luciferase in each sample. All luciferase assays reported here represent at least three independent experiments, each consisting of three wells per treatment with each well run in duplicate. Luciferase activity was measured with an MLX automated microtiter plate luminometer (Dynex). The transfection efficiency of the siRNAs is 93 to 98% in viable A549 cells, as determined by transfection with siGLO siRNA (Dharmacon Inc., Lafayette, Colo.) and analysis by flow cytometry on a FACS Vantage SE (data not shown). Transfection of TTP siRNAs into A549 cells did not affect cyclophilin B mRNA, as determined by real-time RT-PCR (not shown).
shRNAs and generation of stable cell lines.
Short-hairpin RNAs (shRNAs) cloned into the lentivirus vector pLKO.1-puro were chosen from the human library (MISSION TRC-Hs 1.0) (28) and purchased in either DNA or glycerol stock form from Sigma. The control shRNA (non-target shRNA vector, catalog no. SHC002; Sigma) contains a hairpin insert that will generate siRNAs but contains five base pair mismatches to any known human or mouse gene. A set of five shRNAs to TTP were tested for knockdown (SHGLY-NM_003407; Sigma); the shRNA to TTP containing the sequence CCGGGCTTCGCCAGAGCATCAGCTTCTCGAGAAGCTGATGCTCTGGCGAAGCTTTTT (TRCN0000005465; Sigma) was chosen for these experiments, since it was most efficient in knockdown of both TTP mRNA and protein. The control shRNA or shRNA to TTP lentivirus vectors was cotransfected with the packaging plasmids (Virapower System; Invitrogen) into 293 FT cells to generate lentivirus particles. These particles were then used to transduce A549 cells, and stably expressing cells were selected with puromycin at a concentration of 2 μg/ml.
Statistical analysis.
Statistical analysis was performed with one-way analysis of variance (ANOVA). Where ANOVA indicated statistical significance (P < 0.05), post hoc testing was applied. Statistical analyses of multiple comparisons to controls were performed with Dunnett's test to restrain the false-positive rate at 0.05. In comparisons involving only two groups, two-sample t tests were performed, unless the reference group was standardized to a constant with no variability. In this case, a one-sample t test was used. All data were considered statistically significant with a two-sided P value of <0.05. All analyses were carried out with JMP software (SAS Institute Inc., Cary, N.C.).
RESULTS
Glucocorticoids induce human TTP mRNA and protein in A549 lung epithelial cells.
TTP was first identified as a glucocorticoid-responsive gene in a microarray analysis of A549 (lung epithelial) cells treated with dexamethasone. To verify the array analysis, we evaluated the effect of dexamethasone on TTP mRNA expression by real-time RT-PCR. TTP mRNA expression levels were normalized to cyclophilin B, a constitutively expressed transcript unresponsive to dexamethasone (16). We observed a significant induction of TTP mRNA with concentrations as low as 10 nM and maximal effects on TTP mRNA levels with a 100 nM concentration (Fig. 1A). These concentrations of dexamethasone are in the physiologically relevant range for GR-mediated actions and well within therapeutic levels used for the treatment of inflammatory diseases. We next evaluated the kinetics of glucocorticoid treatment on the regulation of TTP mRNA expression. Dexamethasone treatment of A549 cells resulted in a sustained four- to fivefold induction of TTP mRNA, occurring within 2 h and stable through 8 h (Fig. 1B). These results are in contrast to TNF-α induction of TTP mRNA, which was transient, initially rising and then falling dramatically after 2 h. Combined treatment of TNF-α and dexamethasone also resulted in a sustained induction, with an initial spike in induction observed at 2 h, likely reflecting a response to TNF-α treatment. Thus, unlike other stimuli of TTP, dexamethasone treatment results in prolonged induction of TTP mRNA. Interestingly, TNF-α and dexamethasone do not have an antagonistic effect on TTP mRNA expression in A549 cells.
FIG. 1.

Dexamethasone induction of hTTP mRNA and protein. (A) Real-time RT-PCR analysis (as described in Materials and Methods) of A549 cells treated for 8 h at the indicated concentrations of dexamethasone. Values are the means ± standard errors of the means for three experiments. *, P < 0.05 (relative to control). (B) Real-time RT-PCR analysis of A549 cells treated with TNF-α (100 ng/ml) and/or dexamethasone (100 nM). (C) A549 cells treated with dexamethasone (100 nM) for the indicated time points were processed for Western blot analysis with anti-hTTP antibody. The positive control (HEK cell extract overexpressing TTP) was used as a marker for identifying TTP; α-tubulin is shown as a normalization control.
The kinetics of TTP protein expression were next examined by Western blotting (Fig. 1C, upper panel). Multiple immunoreactive bands were observed at the later time points, indicative of the known phosphorylation of TTP protein. Densitometric analysis of immunoreactive bands revealed that dexamethasone treatment induced TTP protein to essentially the same level and kinetics as glucocorticoid induction of TTP mRNA, with protein enhancement also found after 24 h, reflective of the stable nature of this protein (8) (Fig. 1C, lower panel).
Dexamethasone induction of TTP mRNA and protein in vivo in rat tissues.
To determine whether glucocorticoids induce TTP mRNA in vivo, rats were adrenalectomized to deplete endogenous glucocorticoids and injected with dexamethasone, and tissues were analyzed for TTP mRNA (Fig. 2A). In agreement with Northern blot analyses of endogenous TTP mRNA in mouse tissues (14, 23), we find virtually undetectable levels in the brain, low to intermediate levels in the liver, and intermediate to high levels in the lungs, thymus, and spleen. Dexamethasone treatment resulted in a 3.1-fold induction of TTP mRNA in the lung, validating the use of A549 cells as a model system for study of the regulation of TTP. Glucocorticoids similarly caused a three- to fourfold induction of TTP mRNA expression in all tissues except for the spleen.
FIG. 2.

Dexamethasone induction of TTP mRNA and protein in rat tissues. Rats were injected with either vehicle (phosphate-buffered saline) or dexamethasone (5 mg/kg, intraperitoneally) for 6 h (RNA) or 14 h (protein). (A) RNA was extracted from a total of five control (−DEX) and five dexamethasone-treated (+DEX) rats and analyzed by real-time RT-PCR. TTP mRNA expression levels were first normalized to the rat cyclophilin B endogenous control, and the relative amount of TTP was determined by designating the control (−DEX) brain samples as the calibrator; this tissue had the lowest target expression level. *, P < 0.05 (versus vehicle-treated rats for all indicated). (B) Western blot analysis of rat tissues. While a rat-specific antibody to TTP was unavailable for these experiments, sequence comparison of the rat TTP protein with either the human or mouse TTP protein revealed a high level of homology to both, implicating probable cross-reactivity with these antibodies. (Upper panel) Lung tissue lysates from two control or two dexamethasone-injected rats probed with antibody to human TTP (lanes 1 to 4) or antibody to mouse TTP (lanes 6 to 9). The positive control (HEK cell extract overexpressing TTP) was visible with both antibodies. (Lower panel) A representative blot of rat brain, thymus, lung, spleen, and liver tissue extracts probed with antibody to human TTP. Positive control lysate (described above) and α-tubulin are again included for reference.
Western blotting was used to determine whether TTP protein was also induced by dexamethasone in vivo in these rat tissues. Administration of dexamethasone resulted in induction of TTP protein in the rat lung, as revealed by antibodies to both human and mouse TTP (Fig. 2B, upper panel). Analysis of other tissues indicated that dexamethasone induced TTP protein in the thymus, liver, and spleen, in addition to the lung (Fig. 2B, lower panel). Induction in the brain was difficult to ascertain given the low endogenous expression levels, an observation previously reported for the mouse (8, 26). Together our data indicate that dexamethasone induces TTP mRNA and protein in vivo, as well as in A549 cells, providing strong evidence for a physiological response of TTP to glucocorticoids.
RU486 abrogates dexamethasone induction of TTP.
To establish whether glucocorticoid induction of TTP is a GR-mediated event, A549 cells were treated with the GR antagonist RU486 at concentrations of 1 to 10 μM. Exposure of A549 cells to RU486 and dexamethasone resulted in inhibition of glucocorticoid-induced TTP mRNA (Fig. 3A, upper panel) and protein (Fig. 3A, lower panel). These results suggest that the glucocorticoid effect on TTP mRNA and protein requires binding of dexamethasone to its cognate receptor (GR).
FIG.3.


(A) RU486 abrogates dexamethasone induction of TTP. (Upper panel) Real-time RT-PCR analysis of A549 cells treated for 2 h with 1 μM RU486 (or vehicle) and/or 100 nM dexamethasone. Values are the means ± standard errors of the means for three experiments. *, P < 0.001. (Lower panel) Western analysis of A549 cells treated for 8 h with 10 μM RU486 (or vehicle) and/or dexamethasone (100 nM) as indicated. (B) Dexamethasone induction of TTP mRNA is abrogated by actinomycin D but not by cycloheximide. Representative results of experiments of real-time RT-PCR (three wells per treatment), run in duplicate, are shown. Subsequent experiments gave similar results. (Left panel) Real-time RT-PCR analysis of A549 cells pretreated with vehicle or actinomycin D (ACT.D, 10 μg/ml) for 30 min and then treated for 2 h with 100 nM dexamethasone as indicated. *, P < 0.001 (relative to control). (Right panel) Real-time RT-PCR analysis of A549 cells pretreated with vehicle or cycloheximide (CHX, 35 μM) for 1 h and then treated for 2 h with 100 nM dexamethasone as indicated. *, P < 0.001 (relative to control or versus CHX). (C) Dexamethasone enhances hTTP transcription. Nuclear run-on transcription analysis of A549 cells treated for 2 h with or without dexamethasone (100 nM) is shown. The value obtained for TTP mRNA is normalized to each internal control (18S or actin) and is depicted as the level relative to treated control cells. The results represent the mean values (± standard deviations) from two independent experiments, each run in duplicate. (D) GR occupancy of the TTP promoter and 3′ flanking region. In the ChIP assay, A549 cells were treated for 1 h with 1 μM dexamethasone, after which they were lysed, sonicated, and immunoprecipitated with either nonspecific (NS) antibody to rabbit immunoglobulin G or specific GR antibody. The immunoprecipitated 5′ flanking region (top gel) and 3′ flanking region of TTP (center gel) were identified by PCR. (Lower gel) Serving as a negative control, an intronic region of TTP was also analyzed with the same DNA as that in the experiments described above. Shown are representative results of three experiments.
Dexamethasone induction of TTP mRNA is abrogated by actinomycin D but not by cycloheximide.
To elucidate the mechanism by which glucocorticoids induce TTP mRNA, we analyzed TTP expression in the presence of actinomycin D, which prevented glucocorticoid induction of TTP mRNA (Fig. 3B, left panel), suggesting that dexamethasone induction of TTP is a transcriptional event. To determine whether de novo protein synthesis was required for glucocorticoid induction of TTP mRNA, A549 cells were treated with cycloheximide and TTP mRNA levels were analyzed (Fig. 3B, right panel). Exposure of A549 cells to cycloheximide resulted in superinduced levels of TTP mRNA, a common phenomenon associated with protein synthesis inhibitors (19). Nevertheless, treatment with dexamethasone for 2 h in the presence of cycloheximide resulted in a fourfold induction of TTP mRNA, similar to induction of TTP mRNA by dexamethasone alone (fivefold).
Dexamethasone enhances TTP gene transcription.
While all of our data suggested a direct GR-mediated transcriptional effect on TTP, a promoter-luciferase reporter containing bp −2682 to +56 of the hTTP gene that was successfully used to analyze TGF-β-induced transcription (30) was consistently unresponsive to glucocorticoid treatment following transfection into A549 cells (data not shown). Thus, nuclear run-on transcription assays were undertaken to confirm that dexamethasone induces endogenous TTP expression through a transcriptional process. Nascent RNA chains in nuclei isolated from control or dexamethasone-treated A549 cells were allowed to elongate in the presence of [32P]UTP and nucleotide triphosphates. Actin and 18S gene transcription remained relatively unchanged during the 2 h of dexamethasone treatment. TTP gene transcription, however, was increased five- to sevenfold after normalization to actin or 18S, respectively (Fig. 3C). These data demonstrated that the number of active transcription complexes for the hTTP gene was greater in dexamethasone-treated A549 cells than in control cells. To determine whether dexamethasone may also be acting posttranscriptionally to stabilize TTP mRNA, A549 cells were treated with actinomycin D, RNA was harvested at 10-min intervals, and TTP transcript levels were assayed by real-time RT-PCR. The half-life of hTTP mRNA was found to be comparable in control and dexamethasone-treated A549 cells (37.4 min [96% confidence interval] and 38.8 min [97% confidence interval], respectively), indicating that the effect of dexamethasone on TTP is at a primarily transcriptional level.
GR occupancy of the promoter and 3′ flanking region of TTP.
The presence of a bona fide consensus GRE sequence was not predicted from the 5.4 kb of available hTTP sequence (accession no. AY771351), containing 1.8 kb of 5′ flanking region, intron, mRNA coding region and UTRs and 1.1 kb of 3′ flanking region. Only a single hexameric half-GRE sequence was apparent in the TTP 3′ flanking region. However, there were numerous STAT, Smad, and NF-κB consensus binding sites in the promoter region; importantly, each of these transcription factors has been shown to function cooperatively with GR to regulate other genes, such as beta-casein (34), mouse mammary tumor virus (2), or TLR2 (16). Thus, we employed the ChIP assay to examine the ability of GR to interact with the endogenous TTP 5′ and 3′ flanking regions. Primers which span putative binding sites for STAT, Smad, and NF-κB in the promoter region (5′ flanking region) and the GRE-like sequence (3′ flanking region) were synthesized. Minimal occupancy of either the 5′ or 3′ flanking region was detected in the absence of hormone; however, treatment with dexamethasone resulted in a substantial increase of GR recruitment to the TTP 5′ flanking region (Fig. 3D, top gel) and the 3′flanking region (center gel). In contrast, no increase of GR recruitment to the intron region of TTP was observed with dexamethasone treatment (lower gel). While it remains unclear whether this GR binding is functional, these results suggest that there are at least two regions of the TTP gene that interact with GR: a portion of the promoter containing STAT, Smad, and NF-κB binding sites and a portion of the 3′ flanking region which contains the half-GRE sequence.
Dexamethasone decreases TNF-α mRNA through its 3′ UTR.
To investigate whether glucocorticoid-induced TTP has functional downstream consequences on TNF-α mRNA levels (10, 20), we analyzed TNF-α mRNA expression levels in response to dexamethasone and a proinflammatory stimulus, LPS (Fig. 4A). After 2 h of treatment with dexamethasone, TNF-α mRNA expression decreased to 59% of control (untreated) levels. Treatment with LPS (0.1 μg/ml) for 2 h induced TNF-α mRNA by 1,293%, but cotreatment with dexamethasone and LPS resulted in only a 456% increase. Dexamethasone continued to affect both endogenously produced and LPS-induced TNF-α mRNA levels at 4, 6, and 8 h. These results indicate that glucocorticoids repress the ARE-containing TNF-α gene and can antagonize proinflammatory stimulus (LPS)-induced TNF-α in A549 cells. However, since glucocorticoids are also known to repress TNF-α mRNA through its promoter region as well as through other posttranscriptional mechanisms (1, 15), it was unclear whether dexamethasone also impedes expression of TNF-α via its 3′ UTR and through TTP. To assess whether dexamethasone inhibits TNF-α through its 3′ UTR, we generated a TNF-3′ UTR reporter (6) by inserting a portion of the human TNF-3′ UTR into a pGL3-Control vector downstream from the luciferase coding region and upstream of the poly(A) tail (Fig. 4B, upper panel). In addition to the empty control vector, two other controls were included for determination of specificity: Rev-TNF-UTRLuc was constructed with the same insert but in the opposite orientation from TNF-UTRLuc, and TNFLuc was generated by inserting a similar-size portion of the TNF-α promoter into the same region of the pGL3-Control vector. Transfection of A549 cells with these constructs in the absence of hormone revealed that pGL3-Control and TNFLuc were expressed to a higher level than Rev-TNF-UTRLuc, and TNF-UTRLuc activity was even lower (Fig. 4B, lower panel), suggesting that expression of the TNF-α UTR in either orientation may be reduced by endogenous factors which bind to AU-rich mRNAs in A549 cells. No significant decrease was observed in pGL3-Control, TNFLuc, or Rev-TNF-UTRLuc activity after treatment with glucocorticoids. In marked contrast, dexamethasone treatment significantly decreased (50%) TNF-UTRLuc activity. Since all of the reporters contain identical promoter and enhancer sequences, and the TNF-α 3′ UTR regulates gene expression at the level of mRNA turnover and/or translation (6, 20), these results indicate that dexamethasone acts posttranscriptionally to suppress TNF-α expression through its 3′ UTR. The absence of a dexamethasone-mediated reduction in Rev-TNF-UTRLuc activity further indicates that the orientation of the sequence, in addition to the sequence itself, is imperative for this response.
FIG. 4.

Dexamethasone inhibits TNF-α through its 3′ UTR. (A) Dexamethasone represses TNF-α mRNA in A549 cells. Real-time RT-PCR analysis of A549 cells treated with 0.1 μg/ml LPS and/or 100 nM dexamethasone for the indicated times is shown. Values are the means ± standard errors of the means for three experiments. *, P < 0.05 (relative to control levels at the 0-h time point); #, P < 0.05 (versus LPS levels). (B) Dexamethasone decreases the luciferase activity of a TNF-UTR reporter construct. Construction of TNF-UTRLuc and from pGL3-Control is shown at the top. The Rev-TNF-UTRLuc control was generated similarly but inserted in the opposite orientation, and the TNFLuc control was constructed by insertion of 338 nucleotides of the TNF-α promoter into the same site (XbaI) of pGL3-Control. For the dual luciferase assay, A549 cells transfected with pGL3-Control, TNFLuc, or TNF-UTRLuc and a Renilla luciferase normalization plasmid were treated with 100 nM dexamethasone as indicated. Data were plotted as percentages of hormone-free pGL3-Control. Values are the means ± standard errors of the means for three experiments; *, P < 0.0001 (relative to hormone-free TNF-UTRLuc levels).
siRNAs to TTP abrogate the dexamethasone-mediated decrease in TNF-UTRLuc activity.
Using this model system, we investigated whether glucocorticoid-induced TTP specifically mediates this inhibitory effect on the TNF-α 3′ UTR. For these experiments, siRNAs to TTP or control siRNA (scrambled RNA) were transfected into A549 cells. Two different siRNAs to TTP were found to substantially reduce, but not completely eliminate, endogenously expressed and dexamethasone-induced TTP mRNA (Fig. 5A, upper panel) and protein (Fig. 5A, center and lower panels). Cotransfection of the siRNAs with TNF-UTRLuc and Renilla normalization plasmid into A549 cells resulted in TNF-UTRLuc activity which was elevated in samples transfected with siRNA A and elevated to a statistically significant level with siRNA B compared with control siRNA. siRNAs A and B were both also effective in significantly elevating TNF-UTRLuc activity in the presence of dexamethasone (Fig. 5B, upper panel). To more clearly represent the effect of the TTP siRNAs on dexamethasone inhibition of the TNF-α UTR, these data were plotted as the percent decrease of TNF-UTRLuc activity for each of the siRNAs (Fig. 5B, lower panel). The dexamethasone-mediated decrease in luciferase activity for control siRNA was 50.9% (similar to previous experiments) but was only 36.9% for cells transfected with siRNA A and 36.3% for siRNA B. This rescue, resulting from TTP siRNAs, suggests that dexamethasone-induced TTP has a significant effect on TNF-α half-life and/or translation. To delineate whether these observed effects were occurring at the mRNA or protein level, luciferase mRNA was quantitated (Fig. 5C). A549 cells were transfected as above and treated with dexamethasone for 8 h, after which RNA was extracted and subjected to real-time RT-PCR. The firefly luciferase signal was normalized to the Renilla luciferase signal for these analyses. As expected, dexamethasone had no effect on firefly luciferase mRNA in cells transfected with the Rev TNF-UTRLuc negative control. Dexamethasone also did not regulate firefly luciferase mRNA in cells transfected with pGL3-Control vector (data not shown). In contrast, cells transfected with TNF-UTRLuc exhibited a 38% decrease in firefly luciferase mRNA after treatment with dexamethasone, whereas cells transfected with TNF-UTRLuc and siRNAs to TTP failed to show any significant decrease in firefly luciferase mRNA with glucocorticoid treatment. These results indicate that one component of the dexamethasone-mediated anti-inflammatory action occurs at the 3′ UTR of TNF-α at the level of the mRNA and is mediated by induction of TTP synthesis.
FIG. 5.

Dexamethasone inhibition of the TNF-α 3′ UTR is mediated by TTP. (A) Real-time RT-PCR analysis of A549 cells transfected with control (scrambled) siRNA or siRNAs to TTP for 48 h and treated with dexamethasone (100 nM, 4 h) as indicated (upper panel). Similar results with real-time RT-PCR were observed after 24 h of transfection. Western blot analysis of A549 cells transfected with control siRNA or siRNAs to TTP for 24 h and treated with dexamethasone (100 nM, 4 h), as indicated is shown in the center panel. Densitometric analysis of immunoreactive bands is shown in the lower panel. (B) Dual luciferase assay. A549 cells were transfected with control siRNA or siRNAs to TTP, TNF-UTRLuc, and a Renilla normalization plasmid and treated with 100 nM dexamethasone (or left hormone-free). Data were plotted as percentages of TNF-UTRLuc activity from hormone-free control siRNA. Values are means ± standard errors of the means for five independent experiments. *, P < 0.01 (relative to hormone-free control siRNA); #, P < 0.01 (versus dexamethasone treated control siRNA). (Lower panel) Data from the upper panel replotted to show the average dexamethasone-mediated decrease in luciferase activity. Significant rescue was observed with both siRNA A and siRNA B. *, P < 0.005 (relative to control siRNA). (C) Real-time RT-PCR analysis. A549 cells were cotransfected with control siRNA or siRNAs to TTP, Renilla normalization plasmid, and either Rev-TNF-UTRLuc or TNF-UTRLuc and treated with dexamethasone (100 nM, 8 h) as indicated. The firefly luciferase mRNA signal was normalized to that of Renilla luciferase, and data were plotted as percentages of pGL3-Control luciferase mRNA expression (not shown). Values are the means ± standard errors of the means for four independent experiments. *, P < 0.05 (relative to hormone-free TNF-UTRLuc mRNA expressing control siRNA).
Dexamethasone-induced TTP is critical for inhibition of TNF-α mRNA expression.
To determine whether dexamethasone induction of TTP and subsequent suppression of the TNF-α 3′ UTR has an overall impeding effect on TNF-α mRNA expression, we generated an A549 cell line stably expressing a shRNA to TTP. A549 cells expressing this shRNA to TTP inhibited TTP mRNA by 65% (Fig. 6A, upper panel) and TTP protein by approximately 60% (Fig. 6A, middle and lower panels) compared to A549 cells expressing the nontarget control shRNA. Using this TTP-depleted cell model, TNF-α mRNA was then analyzed by real-time RT-PCR (Fig. 6B). Treatment with dexamethasone decreased TNF-α mRNA expression 64% in the A549 cells stably expressing the nontarget control shRNA but only 28% in A549 cells stably expressing the shRNA to TTP. These results indicate that TTP is critical for glucocorticoid-mediated inhibition of TNF-α mRNA expression.
FIG. 6.
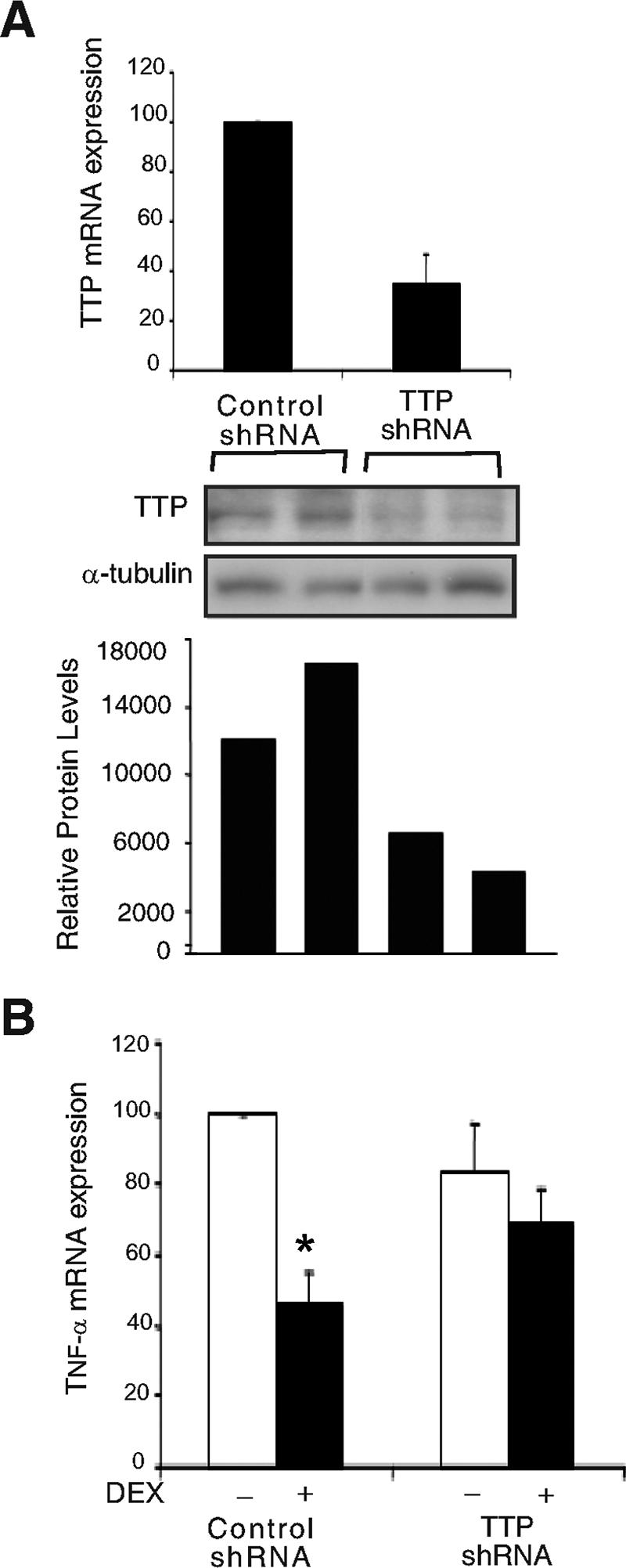
Dexamethasone-induced TTP is critical for inhibition of TNF-α mRNA expression. (A) Real-time RT-PCR analysis of RNA extracted from A549 cells stably expressing control (nontarget) shRNA or A549 cells stably expressing a shRNA to TTP. Values are the means ± standard errors of the means of four independent experiments (upper panel). Western blot analysis of A549 cells stably expressing control shRNA or shRNA to TTP is shown in the center panel. Lysates from two wells expressing control shRNA (first two lanes) and two wells expressing shRNA to TTP (second two lanes) were probed with antibody to human TTP. Densitometric analysis of immunoreactive bands is shown in the lower panel. (B) Real-time RT-PCR analysis of RNA extracted from A549 cells stably expressing either nontarget control shRNA or shRNA to TTP and treated with dexamethasone (2 h, 100 nM). Values are the means ± standard errors of the means for four independent experiments plotted as the percentage of TNF-α mRNA from hormone-free control shRNA. *, P = 0.02 (relative to hormone-free control shRNA).
DISCUSSION
We present evidence here for a new anti-inflammatory pathway for glucocorticoids via induction of TTP mRNA and protein synthesis. This inductive mechanism differs from classical anti-inflammatory pathways, which involve transrepressive mechanisms, including NF-κB or AP-1. While glucocorticoids repress TNF-α at the promoter level or via other transcriptional mechanisms in A549 cells, our experiments with the TNF-UTRLuc reporter indicate that dexamethasone also decreases TNF-α through its 3′ UTR. Glucocorticoid regulation of the COX-2 3′ UTR has previously been reported to be mediated by MKP-1, which blocks activation of the p38 pathway (24, 25). MKP-1 is induced by glucocorticoids in A549 cells (16); however, treatment with three different MKP-1 inhibitors had no effect on dexamethasone-mediated inhibition of TNF-UTR luciferase activity (data not shown), ruling out the possibility that dexamethasone mediates its destabilization effects on TNF-α through induction of MKP-1. Conversely, siRNAs to TTP significantly rescued the dexamethasone-mediated reduction in TNF-UTRLuc activity, suggesting that glucocorticoid induction of TTP has a significant effect on destabilization of TNF-α mRNA and/or translation via this mechanism. Analyses of luciferase mRNA mirrored those results observed with luciferase protein, indicating that dexamethasone induction of TTP likely destabilizes TNF-α mRNA. Our experiments with TTP-depleted A549 cells demonstrate diminished glucocorticoid-mediated suppressive effects on TNF-α. The finding that glucocorticoids possess posttranscriptional control of TNF-α through TTP provides new insight into the anti-inflammatory signaling of these drugs.
TTP mRNA expression has previously been reported to be induced in various cell types by a number of different stimuli (10, 14, 23, 27, 30, 33, 39), each reflecting a transient, immediate-early response after which TTP mRNA returns to basal or near-basal levels. In contrast, our work shows that glucocorticoid treatment resulted in a sustained level of mRNA induction in A549 cells. The labile nature of TTP mRNA (8) makes this finding unprecedented. Induction of TTP mRNA was observed as early as 30 min after treatment with dexamethasone. This observation, along with our transcription run-on assays and experiments with actinomycin D, cycloheximide, and RU486, indicates that glucocorticoid induction of TTP mRNA is a transcriptional event, likely mediated directly by the glucocorticoid receptor. Although a putative GRE has been proposed to reside in the murine TTP promoter region (14), our analysis of the entire human TTP gene did not reveal such a sequence. However, a half-GRE site is present in the 3′ flanking region of this gene, and our ChIP assay results indicate that GR is recruited to this site as well as to another region of the TTP promoter that contains putative binding sites for STATs and Smads, which are important for TTP transcriptional regulation by other stimuli (30, 36) and have been shown to function in concert with GR to regulate other genes (2, 34). Together, these data suggest a transcriptional mechanism by which the glucocorticoid receptor binds to other transcription factors via promoter interactions and perhaps the 3′ UTR to induce hTTP gene transcription. In this regard, TTP promoter reporters (30), generously provided by Yan Chen (Indiana University School of Medicine and the Walther Cancer Institute, Indianapolis, Ind.), were unresponsive to dexamethasone treatment (data not shown), suggesting either that the native conformation of the gene within chromatin is critical for this regulation or that the 3′ UTR binding site is also necessary. Supporting this hypothesis, a recent report investigating dexamethasone inhibition of LPS-induced murine TTP suggested that histone deacetylation is critical for negative regulation of TTP through transcriptional silencing (18).
Our measurements of the TTP mRNA half-life also exclude the possibility that dexamethasone may be functioning to stabilize TTP transcripts in A549 cells. These experiments were imperative, since reports have indicated that TTP is regulated posttranscriptionally through its own AU-rich 3′ UTR, with destabilization occurring through an autoregulatory mechanism and stabilization occurring through either the ERK (THP-1 cells) or p38 (RAW264.7 cells) pathway following LPS activation (7, 38). TTP has been extensively investigated in mouse macrophage (RAW) cell lines due to its abundant expression level in this cell type. However, we observed that RAW cells have very low levels of GR and fail to show dexamethasone induction of TTP mRNA or protein. Despite the likelihood of cell-type-specific regulation in vitro, our analysis of rat tissues indicates that glucocorticoid induction of TTP mRNA and protein is widespread, occurring in the lung, thymus, and liver. Enhancement of TTP protein was also found in the spleen in the presence of a low level of mRNA induction, demonstrating the characteristic stability of this protein (8). The systemic effect of glucocorticoids on TTP in multiple tissues conveys a strong argument for the physiological relevance of this regulation. Currently we cannot distinguish the relative importance of transcriptional transrepression versus TTP-mediated mRNA degradation for explaining the anti-inflammatory activity of glucocorticoids. However, glucocorticoid inhibition of TNF-α and other proinflammatory cytokines through this novel signaling pathway may have wide-ranging clinical implications, particularly as the design of new glucocorticoids is focused on transrepression-selective compounds.
Acknowledgments
We thank Grace Kissling, Paul Housley, and members of the Cidlowski and Blackshear labs for technical assistance.
This research was supported by the Intramural Research Program of the NIH, NIEHS.
Footnotes
Published ahead of print on 18 September 2006.
REFERENCES
- 1.Almawi, W. Y., and O. K. Melemedjian. 2002. Molecular mechanisms of glucocorticoid antiproliferative effects: antagonism of transcription factor activity by glucocorticoid receptor. J. Leukoc. Biol. 71:9-15. [PubMed] [Google Scholar]
- 2.Aurrekoetxea-Hernandez, K., and E. Buetti. 2004. Transforming growth factor β enhances the glucocorticoid response of the mouse mammary tumor virus promoter through Smad and GA-binding proteins. J. Virol. 78:2201-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bakheet, T., B. R. Williams, and K. S. Khabar. 2003. ARED 2.0: an update of AU-rich element mRNA database. Nucleic Acids Res. 31:421-423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barnes, P. J. 1998. Anti-inflammatory actions of glucocorticoids: molecular mechanisms. Clin. Sci. (London) 94:557-572. [DOI] [PubMed] [Google Scholar]
- 5.Boutaud, O., D. A. Dixon, J. A. Oates, and H. Sawaoka. 2003. Tristetraprolin binds to the COX-2 mRNA 3′ untranslated region in cancer cells. Adv. Exp. Med. Biol. 525:157-160. [DOI] [PubMed] [Google Scholar]
- 6.Brooks, S. A., J. E. Connolly, R. J. Diegel, R. A. Fava, and W. F. Rigby. 2002. Analysis of the function, expression, and subcellular distribution of human tristetraprolin. Arthritis Rheum. 46:1362-1370. [DOI] [PubMed] [Google Scholar]
- 7.Brooks, S. A., J. E. Connolly, and W. F. Rigby. 2004. The role of mRNA turnover in the regulation of tristetraprolin expression: evidence for an extracellular signal-regulated kinase-specific, AU-rich element-dependent, autoregulatory pathway. J. Immunol. 172:7263-7271. [DOI] [PubMed] [Google Scholar]
- 8.Cao, H., J. S. Tuttle, and P. J. Blackshear. 2004. Immunological characterization of tristetraprolin as a low abundance, inducible, stable cytosolic protein. J. Biol. Chem. 279:21489-21499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carballo, E., W. S. Lai, and P. J. Blackshear. 2000. Evidence that tristetraprolin is a physiological regulator of granulocyte-macrophage colony-stimulating factor messenger RNA deadenylation and stability. Blood 95:1891-1899. [PubMed] [Google Scholar]
- 10.Carballo, E., W. S. Lai, and P. J. Blackshear. 1998. Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 281:1001-1005. [DOI] [PubMed] [Google Scholar]
- 11.Chen, C. Y., and A. B. Shyu. 1995. AU-rich elements: characterization and importance in mRNA degradation. Trends Biochem. Sci. 20:465-470. [DOI] [PubMed] [Google Scholar]
- 12.Chrousos, G. P. 1995. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N. Engl. J. Med. 332:1351-1362. [DOI] [PubMed] [Google Scholar]
- 13.De Bosscher, K., W. Vanden Berghe, and G. Haegeman. 2003. The interplay between the glucocorticoid receptor and nuclear factor-kappa b or activator protein-1: molecular mechanisms for gene repression. Endocr. Rev. 24:488-522. [DOI] [PubMed] [Google Scholar]
- 14.DuBois, R. N., M. W. McLane, K. Ryder, L. F. Lau, and D. Nathans. 1990. A growth factor-inducible nuclear protein with a novel cysteine/histidine repetitive sequence. J. Biol. Chem. 265:19185-19191. [PubMed] [Google Scholar]
- 15.Han, J., G. Huez, and B. Beutler. 1991. Interactive effects of the tumor necrosis factor promoter and 3′-untranslated regions. J. Immunol. 146:1843-1848. [PubMed] [Google Scholar]
- 16.Hermoso, M. A., T. Matsuguchi, K. Smoak, and J. A. Cidlowski. 2004. Glucocorticoids and tumor necrosis factor alpha cooperatively regulate Toll-like receptor 2 gene expression. Mol. Cell. Biol. 24:4743-4756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hoover, R. R., and J. Floros. 1999. SP-A 3′-UTR is involved in the glucocorticoid inhibition of human SP-A gene expression. Am. J. Physiol. 276:L917-L924. [DOI] [PubMed] [Google Scholar]
- 18.Jalonen, U., A. Lahti, R. Korhonen, H. Kankaanranta, and E. Moilanen. 2005. Inhibition of tristetraprolin expression by dexamethasone in activated macrophages. Biochem. Pharmacol. 69:733-740. [DOI] [PubMed] [Google Scholar]
- 19.Joiakim, A., P. A. Mathieu, A. A. Elliott, and J. J. Reiners, Jr. 2004. Superinduction of CYP1A1 in MCF10A cultures by cycloheximide, anisomycin, and puromycin: a process independent of effects on protein translation and unrelated to suppression of aryl hydrocarbon receptor proteolysis by the proteasome. Mol. Pharmacol. 66:936-947. [DOI] [PubMed] [Google Scholar]
- 20.Kontoyiannis, D., M. Pasparakis, T. T. Pizarro, F. Cominelli, and G. Kollias. 1999. Impaired on/off regulation of TNF biosynthesis in mice lacking TNF AU-rich elements: implications for joint and gut-associated immunopathologies. Immunity 10:387-398. [DOI] [PubMed] [Google Scholar]
- 21.Lai, W. S., E. Carballo, J. R. Strum, E. A. Kennington, R. S. Phillips, and P. J. Blackshear. 1999. Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor alpha mRNA. Mol. Cell. Biol. 19:4311-4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lai, W. S., E. A. Kennington, and P. J. Blackshear. 2003. Tristetraprolin and its family members can promote the cell-free deadenylation of AU-rich element-containing mRNAs by poly(A) ribonuclease. Mol. Cell. Biol. 23:3798-3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lai, W. S., D. J. Stumpo, and P. J. Blackshear. 1990. Rapid insulin-stimulated accumulation of an mRNA encoding a proline-rich protein. J. Biol. Chem. 265:16556-16563. [PubMed] [Google Scholar]
- 24.Lasa, M., S. M. Abraham, C. Boucheron, J. Saklatvala, and A. R. Clark. 2002. Dexamethasone causes sustained expression of mitogen-activated protein kinase (MAPK) phosphatase 1 and phosphatase-mediated inhibition of MAPK p38. Mol. Cell. Biol. 22:7802-7811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lasa, M., M. Brook, J. Saklatvala, and A. R. Clark. 2001. Dexamethasone destabilizes cyclooxygenase 2 mRNA by inhibiting mitogen-activated protein kinase p38. Mol. Cell. Biol. 21:771-780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu, J. Y., and R. J. Schneider. 2004. Tissue distribution of AU-rich mRNA-binding proteins involved in regulation of mRNA decay. J. Biol. Chem. 279:12974-12979. [DOI] [PubMed] [Google Scholar]
- 27.Mittelstadt, P. R., and A. L. DeFranco. 1993. Induction of early response genes by cross-linking membrane Ig on B lymphocytes. J. Immunol. 150:4822-4832. [PubMed] [Google Scholar]
- 28.Moffat, J., D. A. Grueneberg, X. Yang, S. Y. Kim, A. M. Kloepfer, G. Hinkle, B. Piqani, T. M. Eisenhaure, B. Luo, J. K. Grenier, A. E. Carpenter, S. Y. Foo, S. A. Stewart, B. R. Stockwell, N. Hacohen, W. C. Hahn, E. S. Lander, D. M. Sabatini, and D. E. Root. 2006. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124:1283-1298. [DOI] [PubMed] [Google Scholar]
- 29.Nishimori, T., H. Inoue, and Y. Hirata. 2004. Involvement of the 3′-untranslated region of cyclooxygenase-2 gene in its posttranscriptional regulation through the glucocorticoid receptor. Life Sci. 74:2505-2513. [DOI] [PubMed] [Google Scholar]
- 30.Ogawa, K., F. Chen, Y. J. Kim, and Y. Chen. 2003. Transcriptional regulation of tristetraprolin by transforming growth factor-beta in human T cells. J. Biol. Chem. 278:30373-30381. [DOI] [PubMed] [Google Scholar]
- 31.Ogilvie, R. L., M. Abelson, H. H. Hau, I. Vlasova, P. J. Blackshear, and P. R. Bohjanen. 2005. Tristetraprolin down-regulates IL-2 gene expression through AU-rich element-mediated mRNA decay. J. Immunol. 174:953-961. [DOI] [PubMed] [Google Scholar]
- 32.Phillips, K., N. Kedersha, L. Shen, P. J. Blackshear, and P. Anderson. 2004. Arthritis suppressor genes TIA-1 and TTP dampen the expression of tumor necrosis factor alpha, cyclooxygenase 2, and inflammatory arthritis. Proc. Natl. Acad. Sci. USA 101:2011-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Raghavan, A., R. L. Robison, J. McNabb, C. R. Miller, D. A. Williams, and P. R. Bohjanen. 2001. HuA and tristetraprolin are induced following T cell activation and display distinct but overlapping RNA binding specificities. J. Biol. Chem. 276:47958-47965. [DOI] [PubMed] [Google Scholar]
- 34.Stoecklin, E., M. Wissler, R. Moriggl, and B. Groner. 1997. Specific DNA binding of Stat5, but not of glucocorticoid receptor, is required for their functional cooperation in the regulation of gene transcription. Mol. Cell. Biol. 17:6708-6716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stoecklin, G., X. F. Ming, R. Looser, and C. Moroni. 2000. Somatic mRNA turnover mutants implicate tristetraprolin in the interleukin-3 mRNA degradation pathway. Mol. Cell. Biol. 20:3753-3763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suzuki, K., H. Nakajima, K. Ikeda, Y. Maezawa, A. Suto, H. Takatori, Y. Saito, and I. Iwamoto. 2003. IL-4-Stat6 signaling induces tristetraprolin expression and inhibits TNF-alpha production in mast cells. J. Exp. Med. 198:1717-1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taylor, G. A., E. Carballo, D. M. Lee, W. S. Lai, M. J. Thompson, D. D. Patel, D. I. Schenkman, G. S. Gilkeson, H. E. Broxmeyer, B. F. Haynes, and P. J. Blackshear. 1996. A pathogenetic role for TNF alpha in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity 4:445-454. [DOI] [PubMed] [Google Scholar]
- 38.Tchen, C. R., M. Brook, J. Saklatvala, and A. R. Clark. 2004. The stability of tristetraprolin mRNA is regulated by mitogen-activated protein kinase p38 and by tristetraprolin itself. J. Biol. Chem. 279:32393-32400. [DOI] [PubMed] [Google Scholar]
- 39.Varnum, B. C., R. W. Lim, V. P. Sukhatme, and H. R. Herschman. 1989. Nucleotide sequence of a cDNA encoding TIS11, a message induced in Swiss 3T3 cells by the tumor promoter tetradecanoyl phorbol acetate. Oncogene 4:119-120. [PubMed] [Google Scholar]
- 40.Woodworth, C. D., V. Notario, and J. A. DiPaolo. 1990. Transforming growth factors beta 1 and 2 transcriptionally regulate human papillomavirus (HPV) type 16 early gene expression in HPV-immortalized human genital epithelial cells. J. Virol. 64:4767-4775. [DOI] [PMC free article] [PubMed] [Google Scholar]