

Abstract
Ral GTPase activity is a crucial cell-autonomous factor supporting tumor initiation and progression. To decipher pathways impacted by Ral, we have generated null and hypomorph alleles of the Drosophila melanogaster Ral gene. Ral null animals were not viable. Reduced Ral expression in cells of the sensory organ lineage had no effect on cell division but led to postmitotic cell-specific apoptosis. Genetic epistasis and immunofluorescence in differentiating sensory organs suggested that Ral activity suppresses c-Jun N-terminal kinase (JNK) activation and induces p38 mitogen-activated protein (MAP) kinase activation. HPK1/GCK-like kinase (HGK), a MAP kinase kinase kinase kinase that can drive JNK activation, was found as an exocyst-associated protein in vivo. The exocyst is a Ral effector, and the epistasis between mutants of Ral and of msn, the fly ortholog of HGK, suggest the functional relevance of an exocyst/HGK interaction. Genetic analysis also showed that the exocyst is required for the execution of Ral function in apoptosis. We conclude that in Drosophila Ral counters apoptotic programs to support cell fate determination by acting as a negative regulator of JNK activity and a positive activator of p38 MAP kinase. We propose that the exocyst complex is Ral executioner in the JNK pathway and that a cascade from Ral to the exocyst to HGK would be a molecular basis of Ral action on JNK.
The Ral pathway is an essential component of physiological Ras signaling as well as Ras-driven oncogenesis. It can be instrumental in oncogenic transformation, and an activated form of a Ral exchange factor, Rlf, recapitulates the capacity of Ras to transform immortalized human cell cultures, either alone or together with other Ras effectors (26, 60). Reciprocally, the lack of RalGDS, another Ral exchange factor, reduces tumorigenesis in a multistage skin carcinogenesis model and transformation by Ras in tissue culture (24). The molecular basis of the Ral contribution to oncogenesis remains to be elucidated.
None of the Ral effectors and their attributed cellular functions are obvious actors in oncogenesis. One of the two well-documented Ral effectors, RLIP76/RalBP1, is involved in endocytosis (38, 56). The other, the exocyst complex, is involved in secretion, polarized exocytosis, and migration and can be found at the tip of filopods and at tight junctions (8, 43, 53, 57, 64, 69). The exocyst complex is composed of eight proteins, which have been initially identified via mutants of secretion in the budding yeast. Exocyst complexes are bound to vesicles and are supposed to participate in vesicle trafficking and tethering to the plasma membrane. Globally, Ral appears to be a regulator of vesicle trafficking with consequences on cell proliferation, cell fate, and cell signaling (2, 13, 17, 23, 30, 41).
In order to gain insight into Ral function, we have undertaken a genetic and cell biology approach using Drosophila melanogaster, which has a single Ral gene. We have generated null and hypomorph alleles of Ral and shown that Ral is an essential gene. Ral loss-of-function has dramatic effects on the differentiation of sensory organ precursor cells and leads to caspase-8-independent cell death by releasing ectopic tumor necrosis factor (TNF) receptor-associated factor 1-c-Jun N-terminal kinase (TRAF1-JNK) signaling. We further show that sensory organ cell survival in Ral mutants is rescued by an activation of p38 mitogen-activated protein (MAP) kinase, revealing an antiapoptotic function of this latter. We find that the influence of Ral on sensory organ cell fate is directly mediated by the exocyst complex together with a novel interaction partner, the MAP4K4 (also known as hepatocyte progenitor kinase-like/germinal center kinase-like kinase [HGK] in mammals and Misshapen [MSN] in flies). This suggests that a Ral/exocyst/JNK regulatory axis may represent a key component of developmental regulatory programs.
MATERIALS AND METHODS
Genetics and fly handling.
Fly crosses were performed at 25°C unless noted otherwise. For some of the genetic studies involving ectopic expression, we have employed the GAL4-UAS transcription system in which GAL4 produced in a particular pattern acts in trans to activate the expression of UAS transgenes in the corresponding pattern (7). Expression in the proneural cluster was obtained with the scabrous-GAL4 (sca-GAL4) driver, which initiates expression at the second larval instar and progresses to the late pupal stages (55). Deletions in the Ral gene were generated by excision of the P elements in lines PG69 and PG89. Flies were prepared for scanning electron microscopy as described previously (40). The mounted samples were ion coated and observed with a scanning electron microscope (Hitachi Instruments, Inc.). Preparation of flies for Nomarski optics was performed in Hoyer's solution following standard fixation procedures in glycerol-acetic acid (1:4) (75).
Estimation of phenotypes and their modulation.
For each genotype, at least 50 flies were examined and distributed in five classes according to their microchaeta numbers with attributed coefficient: flies with no missing microchaetae (wild-type), coefficient 0; 1 to 10 microchaetae absent, coefficient 1; 11 to 30 microchaetae absent, coefficient 2; >30 absent but >20 present microchaetae, coefficient 4; fewer than 20 microchaetae present, coefficient 5. The percentage of flies in each class was multiplied by the coefficient of the class, and the sum of these numbers gives a semiquantitative view of the phenotype of each genotype. Minimum and maximum scores are zero and 500 for wild-type flies and flies with no microchaetae, respectively. Ral35d males scored 280, and RalS25N males scored 136, on average. Suppression of the Ral35d phenotype was strong or weak if scores were less than 140 ([280 + 0]/2) or between 140 and 280, respectively. Enhancement of the Ral35d phenotype was strong or weak if the score was more than 390 ([280 + 500]/2) or between 280 and 389, respectively. The same calculation was used with RalS25N, using 136 as a medium score on the scale.
Drosophila stocks.
The w; UAS-H2B-YFP; neuP72/SM5:TM6b, Tb line (3) was used for live imaging. DIAP mutants (th) were gifts of K. White (45) and A. Muller (77); DRONC strains were provided by K. Basler (52), p38b strains were provided by T. Adachi-Yamada (1), strains carrying bsk mutations or expressing dominant-negative forms of D-Jun and D-Fos were provided by C. Yanicostas (62), UAS-p35 strains were provided by B. Limbourg-Bouchon, Dredd mutants (44) were provided by B. Lemaitre, the d-mekk1 mutant was provided by H. Inoue (34), and sca-GAL4 flies were provided by F. Schweisguth. The Sec5 mutant was a kind gift of T. Schwarz (54), and msn domain mutants were kindly provided by Y. Rao (65, 66). For experiments with puckered, pucE69 (puc-lacZ) was used (49). Other mutants were provided from Bloomington and Szeged stock centers and from the GenEXel collection. The w1118 strain was used as a wild-type stock for genetic interaction.
Immunocytochemistry.
Dissected nota from staged pupa (25 to 30 and 31 to 35 h after pupal formation [APF]) of Ral35d and control (w118 or sca-Gal4) males were processed as described previously (21) and analyzed using mouse anti-Cut (1:500; Developmental Studies Hybridoma Bank, University of Iowa), rat anti-Su(H) (1:1,000; gift from F Schweisguth), mouse anti-Senseless (1:500), guinea pig anti-Numb (1:100; gift from Y. Jan), rabbit anti-Baz (1:2,000; gift from A. Wodarz), and rabbit anti-horseradish peroxidase (HRP) (1:500; ICN Biomedicals). Cy3- and Cy5-coupled (Jackson Laboratories) and Alexa 488-coupled (Molecular Probes) secondary antibodies were used at 1:1,000. Images were acquired on Leica TCS or SP2 confocal microscopes and assembled using Adobe Photoshop.
DNA fragmentation was assayed by terminal deoxynucleotidyltransferase (TdT)-mediated dUTP nick end labeling (TUNEL kit; Roche) and visualized using Cy3-conjugated streptavidin (1:1,000; Jackson Laboratories). The nota at 20 to 25, 26 to 30, 31 to 35, and 36 to 38 h APF of non-Tb male pupa were selected from the following crosses: (i) w118 × w/Y; UAS-H2B-YFP; neuP72/SM5:TM6b, Tb and (ii) Ral35d × w/Y; UAS-H2B-YFP; neuP72/SM5:TM6b, Tb. Rabbit anti-activated caspase-3 (Cell Signaling Technology) was used at 1:20; rabbit anti-β-galactosidase (β-Gal; Cappel) was used at 1:1,000.
Images were acquired on Leica DMRA and SP2 confocal microscopes and assembled using Photoshop software.
Time-lapse microscopy.
In vivo imaging was carried out as described previously (20). Images were acquired every 4 min on a Leica TCS confocal microscope or an Olympus BX41 fluorescence microscope (20× objective) equipped with a CoolSnap camera driven by Metaview software. Time-lapse movies were assembled using NIH Image software.
Biochemistry.
For preparation of protein extracts, embryos (8 to 24 h) were collected, washed with water, dechorionated in 14% sodium hypochlorite (3 min), rinsed with water, and frozen at −80°C. Frozen embryos were resuspended in lysis buffer (0.5 ml for “100 μl” of embryos; 25 × 10−3 M HEPES, pH 7.5, 0.15 M NaCl, 10−3 M dithiothreitol, 10−3 M EDTA, protease inhibitors [Complete Protease Inhibitors; Roche]) and ground to obtain a homogenous suspension; samples were centrifuged (at 4°C for 10 min at 16,000 × g), and the supernatant, after a second centrifugation, was used for Western blotting. A total of 50 μg of protein/lane was separated in 12% acrylamide sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels and transferred on nitrocellulose membranes; Ral proteins were detected using anti-human RalB antibodies (Transduction Laboratories, BD) and an ECL detection system (Amersham).
For immunoprecipitation from HeLa and NRK cells, cells were lysed in 20 mM Tris-HCl, pH 7.4, 100 mM NaCl, 1 mM MgCl2, 0.1 mM dithiothreitol, 1% Triton X-100, and 10% glycerol, and immunoprecipitation and further analysis were performed according to standard procedures.
Cell culture and transfection.
HeLa and NRK cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Life Technologies). For MAP kinase experiments, before transfection, cells were seeded into 35-mm culture dishes and grown to 60% confluence. Small interfering RNAs (siRNAs; 200 pM) were transfected using Oligofectamine (Life Technologies). For suspension cultures, cells were detached from plates by treatment with trypsin and maintained in 1% Seakem agarose (Cambrex)-coated plates for 24 h. When cells were transfected both with siRNA and expression plasmids, after 4 h in the presence of siRNA in Oligofectamine, the supernatant was removed, and cells were immediately transfected following a standard Ca precipitate method. Synthetic siRNAs targeting RalA and Sec5 were designed by standard methods using the following sense sequences: 5′-GACAGGUUUCUGUAGAAGAdTdT-3′ (RalA), 5′-CAGAGCUGAGCAGUGGAAUTdTdT-3′ (RalA), and 5′-GGUCGGAAAGACAAGGCAGAUdTdT-3′ (SEC5). Scrambled RalA sequence (5′-ACGACGGUGAACUGAAUGGdTdT-3′) or siRNA targeting luciferase was used as controls. The following antibodies were used: mouse monoclonal anti-RalA (BD Transduction Laboratories); mouse monoclonal anti-phospho-SAPK/JNK (Thr183/Tyr185) (Cell Signaling Technology); rabbit polyclonal anti-phospho-c-Jun (Ser73) (Cell Signaling Technology); rabbit polyclonal anti-JNK (Santa Cruz Biotechnology), rabbit polyclonal anti-ERK1/2 (Santa Cruz Biotechnology); monoclonal anti-EXO70 (72), polyclonal anti-SEC5 (58), and monoclonal anti-myc (Roche); and polyclonal anti-phospho-p38 (Thr180/Tyr182) (Cell Signaling).
Two-hybrid screens.
Baits were amplified by PCR using Pfu polymerase (Stratagene) and cloned in the two-hybrid bait plasmid pB27. All constructs were checked by sequencing. The construction of the library as well as screening procedures and bioinformatics analysis have been described previously (14, 19, 59).
RESULTS
Ral is an essential gene.
Mutagenesis of the X chromosome with P element insertions using the P{GawB } transposon yielded two insertions within the Rala locus, hereafter referred to as Ral (6). One was in the first exon (strain PG69), and the other was in the first intron of Ral (strain PG89) (Fig. 1A). In both strains males died during embryogenesis from stage 13 through the first-instar larval stage.
FIG. 1.
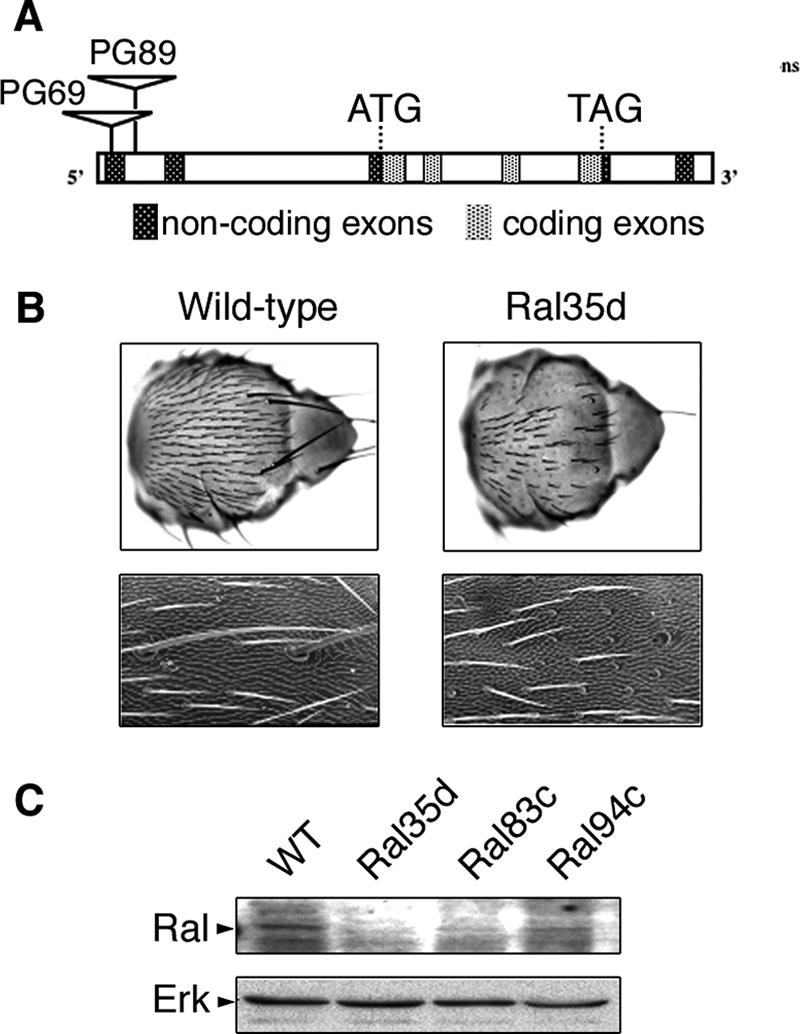
Characteristics of Ral mutants. (A) Schematic representation of the Ral locus and of the sites of insertion of transposons P{GawB} in strains PG69 and PG89, which were further excised to generate null and hypomorph mutants. (B) Scanning electron microscopy of complete nota (upper row) and fragments of nota (lower row) images documenting the loss-of-bristle phenotype of wild-type (left) and Ral35d (right) males. Higher magnification (lower row) shows that sockets are present and only shafts are missing, both for microchaetae and macrochaetae. (C) Western blot analysis of Ral protein expression in wild-type and in Ral35d, Ral83c, and Ral94c mutants. A total of 50 μg of protein from whole embryos was tested using antibodies against human RalB, which detects Drosophila Ral protein. Erk was used as a loading control.
Viability of PG89 and PG69 males was restored by precise excision of the P transposon as well as by expression of a RalWT cDNA under the ubiquitous daughterless (da-GAL4) promoter. These results indicated that lethality was due to insertions in the Ral locus and that Ral is an essential gene.
Hypomorph Ral mutants display defects in sensory organ lineage development.
Two classes of mutants, lethal or viable but displaying a loss-of-bristle phenotype in males (Fig. 1B), were obtained from PG69 and PG89 flies by P-excision mutagenesis. Expression of RalWT driven by da-GAL4 or sca-GAL4 rescued both lethal and loss-of-bristle phenotypes, verifying that the defects were due to mutation of the Ral locus. In some mutants, the loss-of-bristle phenotype was associated with female sterility. Further work was carried out using three viable Ral excision mutations generated from PG89, Ral35d, Ral83c, and Ral94c. Males carrying any of these mutations displayed a highly penetrant loss-of-bristle phenotype (Fig. 1B): the external parts of their sensory organs were made of empty sockets and no shaft. This phenotype (referred to below as the Ral phenotype) was very similar to that observed with a dominant negative allele of Ral (RalS25N) expressed in sensory organs (51, 67).
Genomic characterization of the Ral locus in the Ral35d, Ral83c, and Ral94c mutants by reverse PCR showed that most of the PG89 transposon sequences had been excised while both P terminal repeats had been retained. These flies expressed less RAL protein than wild-type flies (Fig. 1C), indicating that Ral35d, Ral83c, and Ral94c are hypomorph alleles. Females homozygous for these Ral alleles presented a wild-type phenotype, suggesting that two copies of these alleles provided enough Ral activity to fulfill Ral function.
Lack of Ral leads to postmitotic cell-specific death by apoptosis.
Bristles are mechanosensory organs formed by four cells: a sheath, a shaft, a socket cell, and a neuron. These cells arise from four rounds of divisions of primary precursor cells (pIs) (20, 37). Ral hypomorph mutants have fewer bristles than wild-type flies.
We have investigated the cellular basis of the Ral phenotype in our hypomorph mutants. At 24 to 36 h APF, nota were stained for markers allowing the identification of all four cells of the mechanosensory organs (5, 21, 36). Four cells were present in microchaeta and macrochaeta organs of wild-type flies. In Ral35d flies, neuron, socket, and sheath cells were present, but shaft cells were often missing, both in macro- and microchaetae (Fig. 2). In some cases, a sheath cell was also missing in microchaetae. During the course of this work, we tested bristle sensory organs in flies overexpressing RalS25N and observed the same absence of shaft cells (data not shown). A Ral bristle phenotype obtained with a dominant negative allele of Ral has been previously attributed to abortive actin polymerization with all four cells present in microchaeta organs (67). It is possible that the discrepancy between the two results is due to the fact that Sawamoto observed microchaeta organs before 32 h APF, a period when we observed exceptional loss of shaft cells. The actin phenotype at that stage might be an early consequence of entrance in apoptosis.
FIG. 2.

Absence of shaft cells in sensory organs of Ral mutants. Wild-type and Ral35d nota were dissected at 30 h APF and 35 h APF, respectively. Both macrochaeta (A) and microchaeta (B) organs in wild-type flies were composed of four cells (visualized by Cut immunoreactivity [5]; green): one socket cell [large nuclear size and Su(H) immunoreactivity (21); red], a neuron (small nuclear size and HRP immunoreactivity [36]; blue), a shaft cell [large nuclear size and Su(H) negative], and a sheath cell (small nuclear size and HRP negative). In Ral mutants, the shaft cell was absent, and sensory organs were composed of three cells: the neuron, the socket, and the sheath cells. Each panel was obtained after merging about 10 horizontal confocal sections.
To determine if the loss of shaft cells was a consequence of cell death or a defect in cell division during bristle development, bristle lineages were filmed (20). At 18 h APF, all four cells of macrochaeta organs were present, and the first precursor cells (pI) that lead to microchaetae started to divide. Five to eight hours later, shaft cells in the macrochaeta organs began to loose contact with other cells of the cluster and became fragmented. Figure 3A shows the fate of cells of a macrochaeta organ in a Ral35d male: the nucleus of a cell, whose position and size corresponded to a shaft cell, became fragmented and eventually disappeared (see Film S1 in the supplemental material). The same phenomenon was observed in microchaeta organs around 33 to 35 h APF (Fig. 3B; see also Film S2 in the supplemental material). Because macrochaetae arise earlier than microchaetae, it is likely that the delay in shaft cell fragmentation between the two lineages was related to differences in the timing of determination of both organs (32, 71). These data show that death occurs after the appropriate cell divisions and determination had taken place.
FIG. 3.

Shaft cell die by apoptosis in Ral mutants. (A and B) All four cells appear in sensory organs, and then one dies. Cell division in sensory organs of nota of live Ral35d; neuP72 UAS-H2B::YFP pupae was recorded by time-lapse microscopy (confocal for the macrochaeta lineage in panel A or standard epifluorescent for the microchaeta lineage in panel B). Time (hours:minutes) APF is indicated on each panel. Shaft cells (small arrows) were identified by size of their nuclei and their relative positions with respect to the other cells in the cluster. Shaft nuclear fragments (arrowheads) are observed at 26:36 h APF in the macrochaeta lineage (A) and at 33:53 h in the microchaeta lineage (B). Prior to fragmentation, shaft cells lost contact with the other cells in the cluster (26:28 h APF in macrochaeta in panel A; better views for both lineages are available in the films in the supplemental material). Cell movement was frequently associated with changes in the shape of the nucleus and DNA condensation that was observed as bright spots inside the nucleus. In panel A, each image corresponds to the merging of 10 horizontal confocal optical sections. The anterior is on the left. (C) Asymmetric distributions of Numb (Nb; green) and Baz (red) in wild-type and Ral35d dividing pI (upper row) and pIIa (lower row) cells. In both wild-type (WT; n = 10) and Ral35d (n = 17) mutant pI cells (identified by Senseless [Ss]; blue) at prometaphase, Numb was localized at the anterior cortex whereas Baz was localized at the posterior pI cell cortex. In both wild-type (n = 17) and Ral35d mutant (n = 21) pIIa cells, Numb was localized at the anterior cortex whereas Baz was enriched at the posterior pIIa cortex. The dividing pIIa cells were identified by cytoplasmic Cut staining and the presence of the pIIb daughter cells. In the lower row, the insets show the maximal projections of several confocal planes to illustrate the position of the dividing pIIa cells and the anterior pIIIb cells. Nota were dissected at 17 h APF to see pI division and at 21 to 22 h APF to see pIIa and pIIIb divisions. Scale bar, 5 μm. (D and E) The shaft cells die by apoptosis. Cell death was characterized by TUNEL staining performed on macrochaeta (upper row) and microchaeta (lower row) sensory organs at 25 h APF and 35 h APF, respectively. All four cells were detected by green fluorescence due to expression of H2B::YFP, and TUNEL-positive cells were detected by red fluorescence (Cy3-conjugated streptavidin). TUNEL-positive cells were identified as shaft cells by nuclear size and position. (E) Cell apoptosis was also visualized by the presence of an activated caspase-3 in one cell per organ in macrochaetae (upper row) and microchaetae (middle row), as opposed to wild-type organs (lower row). All four cells were detected by Cut immunoreactivity.
The specification of the sensory organ cells relies on the asymmetric localization of Bazooka (Baz), which controls the asymmetric segregation of the cell fate determinant Numb during the asymmetric cell divisions of the sensory organs (4, 63). Having established that the bristle lineage is identical in wild-type and Ral35d mutant organs, we analyzed whether the loss of shaft cells in Ral35d mutant organs might be caused by defects in the asymmetric distribution of either Numb or Baz in shaft precursor cells pI and/or pIIa. We observed that the Numb and Baz localizations were identical in wild-type and Ral35d dividing pI cells, which produce the pIIa and pIIb cells (Fig. 3C). Furthermore, in the dividing pIIa cells, which produce the socket and shaft cells, the localizations of both Numb and Baz were identical in wild-type and Ral35d pupae (Fig. 3C). We therefore conclude that the loss of shaft cells in Ral35d organs is not due to defects in the asymmetric localization of either Numb or Baz during the division of the precursor cells that produce the shaft cell.
Since the exocyst seems crucial for Ral action (see below), we have also analyzed the distributions of Numb and Baz in sec5 mutant pI cells. In agreement with the results in Ral35d mutant, no defects in the asymmetric distributions of Baz and Numb were observed in sec5 mutant dividing pI cells (data not shown). This result is consistent with recently reported data where a sec15 loss-of-function did not affect the asymmetric localization of Baz, Pins, and Numb in pI cells (35). We could not analyze the distribution of Baz and Numb in sec5 mutant pIIa cells, as the loss of SEC5 results in cytokinesis defects during the pI cell division (J. Langevin, unpublished data).
Cell death in macrochaetae and microchaetae is due to apoptosis.
Ral35d mutant nota were stained for a TUNEL test around 20 to 25 h APF, and one cell per organ was labeled positive in many macrochaeta organs (Fig. 3D). The large size and position of the positive cell suggested that it was an offspring of the pIIa cell that gives rise to the shaft and socket cells. This was consistent with the observed absence of shaft cells in macrochaeta organs (Fig. 2A). TUNEL labeling between 31 to 38 h APF of microchaeta organs also revealed that one cell was often scored positive (Fig. 3D). These results were confirmed with activated caspase-3 staining in both macrochaeta and microchaeta organs in Ral mutants (Fig. 3E).
In Ral-dependent apoptosis, Ral acts as an upstream negative regulator of the JNK pathway.
In flies, a major pathway leading to apoptosis involves the Hid, Reaper, and Grim proteins that inhibit the antiapoptotic activity of DIAP1 (10, 25, 42, 61, 73, 76). DIAP1, the fly ortholog of mammalian inhibitor of apoptosis protein), inhibits a caspase-8-dependent pathway, and a cascade where TRAF1 binds Misshapen (msn), a MAP4K4 orthologous to vertebrate HGK/Nck-associated kinase (42, 46). MSN participates in the activation of the JNK (encoded by basket in flies), which is a positive regulator of apoptosis in a caspase-8-independent manner (33, 52). Caspase-9 (DRONC) in association with APAF1 (DARK) was suggested to participate in a JNK-dependent apoptosis (52), although DRONC and JNK might also work in parallel (76). We examined which if any of these pathways were engaged in mediating the cell death observed in Ral mutants.
Expression of the caspase-8 inhibitor p35 in males carrying Ral35d or removal of one copy of DREDD, the closest fly ortholog of caspase-8 (11, 29), had no effect on the Ral bristle phenotype (28), suggesting that it is caspase-8 independent (Table 1). On the contrary, expression of a dominant negative allele of fly caspase-9 (DRONCDN) suppressed the loss-of-macrochaeta and -microchaeta phenotypes of Ral35d. The requirement of an effective caspase-9/apoptosome complex for the apoptosis of shaft cells in Ral mutants suggests that Ral acts as a negative regulator of this complex (Table 1).
TABLE 1.
Epistatic relationships between Ral mutants and mutants of the JNK pathway and of Ral effectorsa
| Gene/protein | Removal of one copy/ overexpression of dominant negative allele | Overexpression of WTb | Ral loss-of-bristle phenotype
|
|
|---|---|---|---|---|
| Microchaetae | Macrochaetae | |||
| DREDD/caspase-8 | dreddB118 **, dreddL23 ** | No effect | No effect | |
| p35 | p35 */** | No effect | No effect | |
| TRAF1 | Df(2L)M24F * | Suppressed (strong) | Suppressed | |
| TRAF1EP578 * | Enhanced (strong) | Enhanced | ||
| MSN | msn102 *, msn172 * | Suppressed (weak) | Suppressed | |
| msnWT *, msnEP549 * | Enhanced (strong) | Enhanced | ||
| msnD160N | No effect | No effect | ||
| msnΔPXXP | Enhanced (strong) | Enhanced | ||
| JNK | bsk1 */**, bsk2 */**, bskDN ** | Suppressed (weak) | Suppressed | |
| Jun | D-JunDN */** | Suppressed (strong) | Suppressed | |
| Fos | D-FosDN * | Suppressed (strong) | Suppressed | |
| Puckered | pucE69 * | Enhanced (strong) | Enhanced | |
| DRONC | DRONCDN * | Suppressed (weak) | Suppressed | |
| MEKK1 | mekk1Ur36* | Enhanced (weak) | Enhanced | |
| p38 MAP kinases | Df(3R)crb89F-4*(p38a) | Enhanced (strong) | Enhanced | |
| p38bDN ** | Enhanced (strong) | Enhanced | ||
| p38bWT ** | Suppressed (rescue) | Suppressed | ||
| RLIP1 | RLIPGE28345 * | Suppressed (weak) | Suppressed | |
| RLIP1WT */** | Enhanced (weak) | Enhanced | ||
| Exocyst complex | sec5E10 * | Enhanced (strong) | Enhanced | |
| sec15GE24290 * | Enhanced (strong) | Enhanced | ||
| exo70GE23538 * | Enhanced (weak) | Enhanced | ||
Females carrying the Ral35d mutation (*) or flies expressing dominant negative RalS25N (**) were crossed with flies carrying the listed loss-of-function mutations or transgenes. Males were analyzed for the loss-of-bristle phenotype compared to the phenotype in Ral35d/Y (*). In crosses using RalS25N (**), both males and females were analyzed and compared with flies expressing only RalS25N. Transgenes were expressed under control of the sca-Gal4 driver. None of the transgenes or heterozygote mutants used in this study produced a bristle phenotype on its own.
Wild-type (WT) alleles have been overexpressed in all these experiments, except the two indicated msn alleles: msnD160N, which is a kinase-dead mutant, and msnΔPXXP, which is an hyperactivated kinase mutant.
To assess how JNK signaling and Ral function are integrated in the hierarchy of signaling pathways regulating apoptosis, we analyzed genetic modulation of Ral-induced apoptosis by second-site mutations or coexpression of transgenes (Fig. 4A). Reducing the dose of JNK pathway activators and effectors (msn, TRAF1, bsk, D-Jun, and D-Fos) suppressed Ral-induced apoptosis, and, conversely, increasing activators (TRAF1 and msn) or reducing negative regulators (puckered) enhanced Ral-induced apoptosis (Table 1). The molecular impact of Ral on JNK pathway regulation likely occurs upstream of TRAF1/msn. That JNK pathway mutants appeared epistatic on Ral mutants suggested that Ral is an upstream negative regulator of JNK, a result consistent with a previous genetic study (67). A possible explanation for this suppression is that in Ral mutants, a JNK-based apoptosis system gets rid of cells whose fate is perturbed by a lack of Ral signaling. This possibility is unlikely since not only is apoptosis suppressed by JNK loss-of-function mutants but also normal bristle differentiation is achieved.
FIG. 4.

Ral brakes JNK, a conserved function in flies and human. (A) Ral behaves as a negative regulator of the JNK pathway in flies. Nomarsky microscopy of adult nota documents examples of genetic interactions between Ral mutants and mutants of the JNK pathway. The loss-of-bristle phenotype of Ral35d mutant flies (left image) was either suppressed (middle image) when the JNK pathways were down-regulated (as in the present example with a dominant negative allele of Jun expressed under the control the sca-Gal4 driver) or enhanced (as in the example of the right image, where the amount of MSN was increased under the control of the sca-Gal4 driver). Results of all tested interactions are presented in Table 1. (B) JNK is unduly activated in shaft cells of Ral mutants. lacZ expression under control of the Puckered promoter in the pucE69 mutant (49) is a reporter for JNK activity. lacZ-positive neurons are present in both wild-type and Ral mutant flies, but lacZ staining in shaft cells is observed only in Ral mutants. Cut staining reveals all four cells; HRP detects the neuron. (C and D) RalA inhibits basic JNK activation in human cells and impairs TNF-α activation of p38 MAP kinase. HeLa cells were transfected with control or RalA siRNAs. In panel C, at 72 h posttransfection, whole-cell lysates of attached cells (“attached”) or cells detached from plates with trypsin and maintained in suspension for 24 h (“suspension”) were prepared. Equivalent total protein was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the indicated proteins (phospho-JNK corresponds to JNK1 and 2 phosphorylated on Thr183 and Tyr185; phospho-c-Jun corresponds to c-Jun phosphorylated on Ser73). JNK1/2 and ERK1/2 are shown as a loading control. In panel D, at 72 h posttransfection cells were treated with 10 ng/ml of TNF-α for the indicated times, and cell extracts were an analyzed for p38 MAP kinase phosphorylation as well as for RalA depletion. ERK1/2 is shown as a loading control.
Epistatic relationships between Ral and the JNK pathway were studied also in the context of an activated Ral allele (RalG20V). Flies expressing constitutively active RalG20V under control of sca-Gal4 exhibited a specific bristle phenotype, which can be described as a mislocalization of shafts of dorso-central macrochaetae and some morphology modification (51), and instead of growing from inside their sockets, shafts grew beside their sockets. RalG20V flies display a 100% penetrance of this phenotype, with two to four out of the four dorso-central macrochaetae concerned. Activation of the JNK pathway by puckered mutation (pucE69) suppressed the RalG20V mislocalization phenotype, both in terms of penetrance and expressivity. Symmetrically, a TRAF mutant (TRAFEP578), which down-regulates the JNK pathway, enhanced the RalG20V phenotype, and almost all flies exhibited a mislocalization of all four dorso-central macrochaetae (data not shown).
This epistatic relationship applies also to Ral lethal mutants, which die before the second larval stage. The PG69 insertion drives expression of GAL4 along the pattern of expression of Ral (data not shown). Overexpression of dominant negative D-Jun (D-JunDN) directed by GAL4 expressed from PG69 insertion rescued PG69 males up to the pharate adult stage. Similarly, PG69 males were rescued by the removal of one copy of basket (bsk2). PG89 males were rescued by D-JunDN expressed under a da-Gal4 driver. These results suggest that during embryogenesis, Ral controls cell death via the JNK pathway.
JNK signaling in Drosophila is reflected by the expression levels of puckered, a gene encoding a dual-specificity phosphatase (49, 50). Ral regulation of JNK signaling was confirmed at the cellular level in sensory organs of Ral mutants using a puc enhancer trap line (pucE69), in which the puc-lacZ reporter gene is induced in cells undergoing JNK pathway activation (22, 50). β-Gal-positive neurons were observed in all organs independently of Ral35d mutation, whereas β-Gal-positive shaft cells were observed only in Ral mutants, at a time when all mitotic divisions were completed (Fig. 4B).
p38 MAP kinase displays antiapoptotic properties.
In mammals, JNK and p38 MAP kinase are often coactivated in response to various stresses including TNF (47, 48). In flies, p38 MAP kinase is also activated in response to various stresses (15, 78), but TNF-dependent apoptosis requires only the JNK pathway (33, 42, 52). The promiscuity of the JNK and p38 MAP kinase activation prompted us to test the contribution of p38 MAP kinase to Ral apoptotic signaling.
Drosophila harbors two p38-encoding genes, p38a and p38b (1, 27). Removal of one copy of p38a (Df(3R)crb89F-4) as well as expression of a dominant negative p38bDN enhanced dramatically the Ral bristle phenotype. Conversely, overexpression of a wild-type p38b rescued the Ral bristle phenotype (Table 1). These data raised the possibility that, in Ral-dependent apoptosis in the bristle lineage, Ral behaves as a positive regulator of p38 MAP kinases, which behave as antiapoptotic molecules. Consistently, a loss-of-function mutant of MEKK1, an upstream activator of p38 MAP kinase (34), enhances the Ral bristle phenotype (Table 1).
The role of Ral as a negative and positive regulator of JNK and p38 MAP kinase, respectively, is conserved in human cells. siRNA-mediated depletion of RalA resulted in dramatic induction of JNK and c-Jun phosphorylation (Fig. 4C). Similar results were obtained in cells maintained in suspension and in attached cells (Fig. 4C), despite the fact that RalA is required for anchorage-independent proliferation in the former but not in the latter (13). RalB was not tested because its depletion causes premature cell death (13). Also, consistent with the genetic observations in flies, p38 MAP kinase activation in response to TNF-α was blunted by RalA depletion (Fig. 4D).
The exocyst likely mediates Ral-dependent sensory cell survival.
We tested mutants of two Ral effectors, RLIP1 (the fly ortholog of RalBP1/RLIP76) and the exocyst, for their contribution to Ral-dependent cell survival (9, 18). Overexpression of the RLIP1 protein from a UAS-RLIP1 transgene in Ral35d flies enhanced the Ral phenotype. The most likely explanation is that overexpression of RLIP1 blocks access of Ral to its effectors required for its antiapoptotic function. Conversely, an insertional mutation in the RLIP1 gene suppressed the loss-of-bristle phenotype of Ral35d males (Table 1).
Loss-of-function mutants of three subunits of the exocyst (Sec5, Sec15, and Exo70) were tested in combination with Ral35d. All mutations enhanced the Ral loss-of-bristle phenotype. Thus, the lack of a functional exocyst enhances Ral-dependent cell death.
Molecular basis for an exocyst-JNK functional connection.
That the exocyst, primarily understood as a secretory vesicle trafficking machine, might carry activities regulating MAP kinase was surprising and prompted us to search the molecular avatar(s) of this activity. In a two-hybrid screen performed with rat SEC5 as the bait and a human placenta random-primed two-hybrid cDNA library, we have identified HGK as a partner of SEC5. Reciprocally, a screen performed with HGK as a bait identified Sec5 as a partner (data not shown). HGK, also known as MAP4K4, is the ortholog of Drosophila MSN. It belongs to the STE20 kinase family, and displays a kinase domain (amino acids 25 to 289) and a citron homology (CNH) domain (amino acids 1002 to 1320) separated by a region containing a PXXP motif that binds to the SH3 domain of DOCK (66, 68). The smallest identified region of interaction between HGK and Sec5 comprises amino acids 745 to the C terminus and contains the CNH domain (Fig. 5A). Reciprocally, a two-hybrid screen with a C-terminal fragment of HGK starting at amino acid 735 identified SEC5 as a partner. The smallest identified region of interaction was comprised of amino acids 224 to 693 of SEC5 (data not shown).
FIG. 5.

The exocyst complex interacts with HGK/MAP4K4, an activator of the JNK pathway. (A) Schematic representation of human and Drosophila MAP4K4 HGK and MSN. Domains are indicated as well as the percent identity/similarity between the different regions of the human and fly proteins. The global identity and similarity for the whole proteins are 43 and 53%, respectively. For the sake of simplicity, we show only isoform 1 of HGK (NP_004825). The difference between the three reported isoforms affects the regions flanking the kinase domain and the CNH domain. (B) Sec5 interacts with HGK. HeLa cells were transfected with the indicated plasmids and the indicated siRNAs. Whole-cell extracts were immunoprecipitated with the indicated antibodies. Myc-HGK was present in immunoprecipitates obtained with anti-Sec5 antibodies (58) in cells transfected with a plasmid expressing myc-HGK. This result was specific: when Sec5 protein was depleted by specific siRNA, less Sec5 and less myc-HGK were detected in the anti-Sec5 immunoprecipitate, showing that myc-HGK was not precipitated directly by the anti-Sec5 antibodies. (C) siRNAs against Sec5 are specific. The cells extracts used in panel B for immunoprecipitation were probed for the presence of the indicated proteins. The siRNA against Sec5 (siSec5) reduced strongly the amount of Sec5, while myc-HGK looks only marginally affected. (D) HGK is immunoprecipitated with the Exo70 subunit of the exocyst complex. NRK cells transfected with a plasmid expressing myc-HGK were lysed, and cell extracts were immunoprecipitated with the indicated antibodies. Immunoprecipitates were tested for the presence of other subunits of the exocyst and of HGK. HGK was detected in the immunoprecipitate with anti-Exo70. IgG, immunoglobulin G; siLuc, siRNA against luciferase.
We tested domains of MAP4K4 for their contribution to the genetic interaction between Ral and the JNK pathway, using overexpression of msn mutants in Drosophila. While overexpression of wild-type msn enhanced the Ral bristle phenotype (Table 1), a kinase-dead mutant (msnD160N) (65, 68) had no effect (Table 1). Mutant msnΔPXXP codes for a protein deleted of amino acids 332 to 667, still contains the kinase domain and the CNH domain, and is considered a constitutively active mutant (31, 68). Overexpression of this mutant under control of sca-Gal4 displayed no bristle phenotype on its own but enhanced the Ral loss-of-bristle phenotype of Ral35d and RalS25N mutants. This enhancement was even stronger than the one observed with overexpression of wild-type msn (Table 1). The requirement of the kinase activity of the MAP4K4 Msn for the genetic interaction of msn with Ral35d strengthens the connection of Ral to JNK activation.
The interaction between SEC5 and HGK was confirmed in vivo. Since endogenous HGK could not be detected in HeLa cells, experiments were performed using cells transfected with a plasmid expressing myc-HGK. SEC5 was immunoprecipitated, and HGK was present in the immunoprecipitate (Fig. 5B and C). This coimmunoprecipitation was specific: it was not detected either with immunoglobulin G or when the experiment was performed in cells cotransfected with an siRNA against SEC5 (Fig. 5B and C). Endogenous HGK was found immunoprecipitated with anti-EXO70 antibodies (72) in NRK cells, suggesting that HGK interacts with the exocyst complex, not only with SEC5 (Fig. 5D).
DISCUSSION
We have generated mutants of the unique Ral locus of Drosophila, which have enabled us to show that Ral is an essential gene. Hypomorph mutations of Ral displayed a loss-of-bristle phenotype with sockets without shafts, as do flies expressing dominant negative alleles of Ral (51, 67). Whereas Ral is expressed in many if not all tissues (58), the only situation where a decreased level of Ral appears compatible with adult viability leads to a developmental phenotype in the bristle sensory organs. In Ral mutants, the pI precursor cells undergo the right number of divisions with a correct timing, but afterward shaft cells die by apoptosis, showing that death hits after cell division and determination has taken place, during the subsequent differentiation stage (Fig. 2 and 3).
We have explored the various pathways that lead to apoptosis for their interactions with Ral. The caspase-8-mediated pathway did not contribute to the Ral phenotype, as opposed to a caspase-9-mediated pathway. We tested the JNK pathway, a cascade of four kinases starting with MSN (MAP4K4 or HGK in human), which requires formation of a complex with TRAF1 for its full activity, and ending at the Jun N-terminal kinase (46, 47). Puckered is a phosphatase that dephosphorylates and deactivates JNK.
Loss-of bristle and apoptosis phenotypes due to decrease of Ral signaling were suppressed by down-regulation of the JNK pathway and enhanced by its up-regulation. Symmetrically, a phenotype due to a hyperactivation of the Ral pathway by the overexpression of RalG20V was suppressed and enhanced by enhancing or decreasing JNK signaling, respectively.
The fact that the enhancement and suppression described here can be induced by genetic alterations of TRAF and MSN as well as of JNK proteins (Table 1) suggests that Ral is a general negative regulator of this cascade. Dominant negative alleles of transcriptional effectors of the JNK, Jun itself but also Fos, suppressed the Ral phenotype, suggesting that Ral regulates transcriptional events involved positively or negatively in apoptosis.
Down-regulating the JNK pathway is not only suppresses apoptosis in Ral-defective cells but also rescues normal bristle development. Together with data in S2 cells, where Ral behaves also as a negative regulator of JNK in the absence of any cell death (67), our results suggest a functional relationship between Ral and the JNK pathway wherein Ral activation keeps JNK down. Data using activated and dominant negative alleles of Ral in mammalian cell culture support a positive effect of Ral on JNK activation (16, 17). We do not understand the source of this discrepancy, which might be due to cell- and/or context-specific interactions of Ral with the JNK pathway. However, our data obtained by RNA interference in HeLa cells are consistent with our fly model.
Epistatic relationships between Ral and p38 MAP kinase mutants revealed another actor in Ral-dependent apoptosis: the p38 MAP kinase behaves as an antiapoptotic kinase, which could be positively regulated by Ral.
A control of the basic JNK activity might serve two purposes. First, it minimizes JNK activity and avoids undesirable cell death in normal conditions. Second, a low level of basal JNK activity allows better differential in activation of JNK when this activation happens in response to stresses that lead eventually to apoptosis.
The molecular basis of Ral action on the JNK pathway were addressed genetically and biochemically. The model that comes out is that the exocyst complex is the matchmaker between Ral and the JNK pathway, and the simplest interpretation of genetic data is that the exocyst works like a negative regulator of HGK activity. Finally, the exocyst complex was found to bind in vivo to HGK, providing a biochemical basis for the functional effect of Ral on JNK.
Decreasing the JNK pathway seems to favor the oncogenic capacity of Ras in mouse primary fibroblasts (39). Our results can explain one of the contributions of the Ral pathway to oncogenesis (24, 70, 74): cancer cells have to sustain proliferative signals and relieve proapoptotic signals, and Ral via the exocyst complex might be in charge, at least, of this latter task in oncogenesis. Finally, it has been recently shown that the exocyst complex carries enzymatic activities working in the NF-κB pathway (12). These data together with the present report widen the role of the exocyst to functions other than directing vesicle traffic and contributing to exocytosis.
Supplementary Material
Acknowledgments
We are very grateful to our colleagues from the Bloomington Stock Centre for kindly providing several of the lines used in this study, as well as to K. White, A. Muller, F. Schweisguth, C. Yanicostas, and Y. Rao. We are especially indebted to Thomas Schwarz who provided the Sec5 mutant before publication, to Shu-Chan Hsu for anti-EXO70 monoclonal antibodies, and to Sean Munro for a superb polyclonal anti-Sec5 affinity-purified antibody. Sophie Malinsky and Nathalie Remillieux-Leschelle provided expert technical help. None of the electronic images would have been possible without the dedicated and skillful help of Michèle Grasset at the Centre Interuniversitaire de Microscopie Electronique (CIME; IFR 2062 CNRS).
This work was supported by grant 4397 from Association de Recherche sur la Cancer (ARC) and by Association Christelle Bouillot to J.C., by a grant from Fondation Simone et Cino del Duca, and grant OMS034 “Centre de Ressources Biologiques” from Ministère de la Recherche to J.A.L., and by Welch grant I-1414 and NIH grant CA71443 to M.W. M.B. and S.V.L. were supported by grant QLK3-CT-1999-00875 from the European Community. M.B. was further supported by a postdoctoral fellowship from Institut Curie, and C.R. was supported by a fellowship from ARC.
Footnotes
Published ahead of print on 25 September 2006.
Supplemental material for this article may be found at http://mcb.asm.org/.
REFERENCES
- 1.Adachi-Yamada, T., M. Nakamura, K. Irie, Y. Tomoyasu, Y. Sano, E. Mori, S. Goto, N. Ueno, Y. Nishida, and K. Matsumoto. 1999. p38 mitogen-activated protein kinase can be involved in transforming growth factor β superfamily signal transduction in Drosophila wing morphogenesis. Mol. Cell. Biol. 19:2322-2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agapova, L. S., J. L. Volodina, P. M. Chumakov, and B. P. Kopnin. 2004. Activation of Ras-Ral pathway attenuates p53-independent DNA damage G2 checkpoint. J. Biol. Chem. 279:36382-36389. [DOI] [PubMed] [Google Scholar]
- 3.Bellaiche, Y., M. Gho, J. A. Kaltschmidt, A. H. Brand, and F. Schweisguth. 2001. Frizzled regulates localization of cell-fate determinants and mitotic spindle rotation during asymmetric cell division. Nat. Cell Biol. 3:50-57. [DOI] [PubMed] [Google Scholar]
- 4.Bellaiche, Y., A. Radovic, D. F. Woods, C. D. Hough, M. L. Parmentier, C. J. O'Kane, P. J. Bryant, and F. Schweisguth. 2001. The partner of inscuteable/discs-large complex is required to establish planar polarity during asymmetric cell division in Drosophila. Cell 106:355-366. [DOI] [PubMed] [Google Scholar]
- 5.Blochlinger, K., R. Bodmer, L. Y. Jan, and Y. N. Jan. 1990. Patterns of expression of cut, a protein required for external sensory organ development in wild-type and cut mutant Drosophila embryos. Genes Dev. 4:1322-1331. [DOI] [PubMed] [Google Scholar]
- 6.Bourbon, H. M., G. Gonzy-Treboul, F. Peronnet, M. F. Alin, C. Ardourel, C. Benassayag, D. Cribbs, J. Deutsch, P. Ferrer, M. Haenlin, J. A. Lepesant, S. Noselli, and A. Vincent. 2002. A P-insertion screen identifying novel X-linked essential genes in Drosophila. Mech. Dev. 110:71-82. [DOI] [PubMed] [Google Scholar]
- 7.Brand, A. H., and N. Perrimon. 1993. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118:401-415. [DOI] [PubMed] [Google Scholar]
- 8.Brymora, A., V. A. Valova, M. R. Larsen, B. D. Roufogalis, and P. J. Robinson. 2001. The brain exocyst complex interacts with RalA in a GTP-dependent manner: identification of a novel mammalian Sec3 gene and a second Sec15 gene. J. Biol. Chem. 276:29792-29797. [DOI] [PubMed] [Google Scholar]
- 9.Camonis, J. H., and M. A. White. 2005. Ral GTPases: corrupting the exocyst in cancer cells. Trends Cell Biol. 15:327-332. [DOI] [PubMed] [Google Scholar]
- 10.Chen, P., W. Nordstrom, B. Gish, and J. M. Abrams. 1996. grim, a novel cell death gene in Drosophila. Genes Dev. 10:1773-1782. [DOI] [PubMed] [Google Scholar]
- 11.Chen, P., A. Rodriguez, R. Erskine, T. Thach, and J. M. Abrams. 1998. Dredd, a novel effector of the apoptosis activators reaper, grim, and hid in Drosophila. Dev. Biol. 201:202-216. [DOI] [PubMed] [Google Scholar]
- 12.Chien, Y., S. Kim, R. Bumeister, Y.-M. Loo, S. Kwon, C. Johnson, M. Balakireva, Y. Romeo, L. Kopelovich, M. Gale Jr., C. Eaman, J. Camonis, Y. Zhao, and M. White. 2006. A RalB/Exocyst/TBK1 activation complex couples innate immune signaling to tumor cell survival. Cell 127:157-170. [DOI] [PubMed]
- 13.Chien, Y., and M. A. White. 2003. RAL GTPases are linchpin modulators of human tumour-cell proliferation and survival. EMBO Rep. 4:800-806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Colland, F., X. Jacq, V. Trouplin, C. Mougin, C. Groizeleau, A. Hamburger, A. Meil, J. Wojcik, P. Legrain, and J. M. Gauthier. 2004. Functional proteomics mapping of a human signaling pathway. Genome Res. 14:1324-1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Craig, C. R., J. L. Fink, Y. Yagi, Y. T. Ip, and R. L. Cagan. 2004. A Drosophila p38 orthologue is required for environmental stress responses. EMBO Rep. 5:1058-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Ruiter, N. D., R. M. Wolthuis, H. van Dam, B. M. Burgering, and J. L. Bos. 2000. Ras-dependent regulation of c-Jun phosphorylation is mediated by the Ral guanine nucleotide exchange factor-Ral pathway. Mol. Cell. Biol. 20:8480-8488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Essers, M. A., S. Weijzen, A. M. de Vries-Smits, I. Saarloos, N. D. de Ruiter, J. L. Bos, and B. M. Burgering. 2004. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 23:4802-4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feig, L. A. 2003. Ral-GTPases: approaching their 15 minutes of fame. Trends Cell Biol. 13:419-425. [DOI] [PubMed] [Google Scholar]
- 19.Formstecher, E., S. Aresta, V. Collura, A. Hamburger, A. Meil, A. Trehin, C. Reverdy, V. Betin, S. Maire, C. Brun, B. Jacq, M. Arpin, Y. Bellaiche, S. Bellusci, P. Benaroch, M. Bornens, R. Chanet, P. Chavrier, O. Delattre, V. Doye, R. Fehon, G. Faye, T. Galli, J. A. Girault, B. Goud, J. de Gunzburg, L. Johannes, M. P. Junier, V. Mirouse, A. Mukherjee, D. Papadopoulo, F. Perez, A. Plessis, C. Rosse, S. Saule, D. Stoppa-Lyonnet, A. Vincent, M. White, P. Legrain, J. Wojcik, J. Camonis, and L. Daviet. 2005. Protein interaction mapping: a Drosophila case study. Genome Res. 15:376-384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gho, M., Y. Bellaiche, and F. Schweisguth. 1999. Revisiting the Drosophila microchaeta lineage: a novel intrinsically asymmetric cell division generates a glial cell. Development 126:3573-3584. [DOI] [PubMed] [Google Scholar]
- 21.Gho, M., M. Lecourtois, G. Geraud, J. W. Posakony, and F. Schweisguth. 1996. Subcellular localization of suppressor of hairless in Drosophila sense organ cells during notch signalling. Development 122:1673-1682. [DOI] [PubMed] [Google Scholar]
- 22.Giraldez, A. J., and S. M. Cohen. 2003. Wingless and notch signaling provide cell survival cues and control cell proliferation during wing development. Development 130:6533-6543. [DOI] [PubMed] [Google Scholar]
- 23.Goi, T., M. Shipitsin, Z. Lu, D. A. Foster, S. G. Klinz, and L. A. Feig. 2000. An EGF receptor/Ral-GTPase signaling cascade regulates c-Src activity and substrate specificity. EMBO J. 19:623-630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gonzalez-Garcia, A., C. A. Pritchard, H. F. Paterson, G. Mavria, G. Stamp, and C. J. Marshall. 2005. RalGDS is required for tumor formation in a model of skin carcinogenesis. Cancer Cell 7:219-226. [DOI] [PubMed] [Google Scholar]
- 25.Grether, M. E., J. M. Abrams, J. Agapite, K. White, and H. Steller. 1995. The head involution defective gene of Drosophila melanogaster functions in programmed cell death. Genes Dev. 9:1694-1708. [DOI] [PubMed] [Google Scholar]
- 26.Hamad, N. M., J. H. Elconin, A. E. Karnoub, W. Bai, J. N. Rich, R. T. Abraham, C. J. Der, and C. M. Counter. 2002. Distinct requirements for Ras oncogenesis in human versus mouse cells. Genes Dev. 16:2045-2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han, Z. S., H. Enslen, X. Hu, X. Meng, I. H. Wu, T. Barrett, R. J. Davis, and Y. T. Ip. 1998. A conserved p38 mitogen-activated protein kinase pathway regulates Drosophila immunity gene expression. Mol. Cell. Biol. 18:3527-3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hay, B. A., D. A. Wassarman, and G. M. Rubin. 1995. Drosophila homologs of baculovirus inhibitor of apoptosis proteins function to block cell death. Cell 83:1253-1262. [DOI] [PubMed] [Google Scholar]
- 29.Hengartner, M. O. 2000. The biochemistry of apoptosis. Nature 407:770-776. [DOI] [PubMed] [Google Scholar]
- 30.Henry, D. O., S. A. Moskalenko, K. J. Kaur, M. Fu, R. G. Pestell, J. H. Camonis, and M. A. White. 2000. Ral GTPases contribute to regulation of cyclin D1 through activation of NF-κB. Mol. Cell. Biol. 20:8084-8092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Houalla, T., D. Hien Vuong, W. Ruan, B. Suter, and Y. Rao. 2005. The Ste20-like kinase misshapen functions together with Bicaudal-D and dynein in driving nuclear migration in the developing drosophila eye. Mech. Dev. 122:97-108. [DOI] [PubMed] [Google Scholar]
- 32.Huang, F., C. Dambly-Chaudiere, and A. Ghysen. 1991. The emergence of sense organs in the wing disc of Drosophila. Development 111:1087-1095. [DOI] [PubMed] [Google Scholar]
- 33.Igaki, T., H. Kanda, Y. Yamamoto-Goto, H. Kanuka, E. Kuranaga, T. Aigaki, and M. Miura. 2002. Eiger, a TNF superfamily ligand that triggers the Drosophila JNK pathway. EMBO J. 21:3009-3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inoue, H., M. Tateno, K. Fujimura-Kamada, G. Takaesu, T. Adachi-Yamada, J. Ninomiya-Tsuji, K. Irie, Y. Nishida, and K. Matsumoto. 2001. A Drosophila MAPKKK, D-MEKK1, mediates stress responses through activation of p38 MAPK. EMBO J. 20:5421-5430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jafar-Nejad, H., H. K. Andrews, M. Acar, V. Bayat, F. Wirtz-Peitz, S. Q. Mehta, J. A. Knoblich, and H. J. Bellen. 2005. Sec15, a component of the exocyst, promotes notch signaling during the asymmetric division of Drosophila sensory organ precursors. Dev. Cell. 9:351-363. [DOI] [PubMed] [Google Scholar]
- 36.Jan, L. Y., and Y. N. Jan. 1982. Antibodies to horseradish peroxidase as specific neuronal markers in Drosophila and in grasshopper embryos. Proc. Natl. Acad. Sci. USA 79:2700-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jan, Y. N., and L. Y. Jan. 1998. Asymmetric cell division. Nature 392:775-778. [DOI] [PubMed] [Google Scholar]
- 38.Jullien-Flores, V., Y. Mahe, G. Mirey, C. Leprince, B. Meunier-Bisceuil, A. Sorkin, and J. H. Camonis. 2000. RLIP76, an effector of the GTPase Ral, interacts with the AP2 complex: involvement of the Ral pathway in receptor endocytosis. J. Cell Sci. 113:2837-2844. [DOI] [PubMed] [Google Scholar]
- 39.Kennedy, N. J., H. K. Sluss, S. N. Jones, D. Bar-Sagi, R. A. Flavell, and R. J. Davis. 2003. Suppression of Ras-stimulated transformation by the JNK signal transduction pathway. Genes Dev. 17:629-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kimmel, B. E., U. Heberlein, and G. M. Rubin. 1990. The homeodomain protein rough is expressed in a subset of cells in the developing Drosophila eye where it can specify photoreceptor cell subtype. Genes Dev. 4:712-727. [DOI] [PubMed] [Google Scholar]
- 41.Kops, G. J., N. D. de Ruiter, A. M. De Vries-Smits, D. R. Powell, J. L. Bos, and B. M. Burgering. 1999. Direct control of the forkhead transcription factor AFX by protein kinase B. Nature 398:630-634. [DOI] [PubMed] [Google Scholar]
- 42.Kuranaga, E., H. Kanuka, T. Igaki, K. Sawamoto, H. Ichijo, H. Okano, and M. Miura. 2002. Reaper-mediated inhibition of DIAP1-induced DTRAF1 degradation results in activation of JNK in Drosophila. Nat. Cell Biol. 4:705-710. [DOI] [PubMed] [Google Scholar]
- 43.Langevin, J., M. J. Morgan, C. Rossé, V. Racine, J.-B. Sibarita, S. Aresta, M. Murthy, T. Schwarz, J. Camonis, and Y. Bellaïche. 2005. Drosophila exocyst components Sec5, Sec6 and Sec15 regulate DE-Cadherin trafficking from recycling endosomes to the plasma membrane. Dev. Cell. 9:312-313. [DOI] [PubMed] [Google Scholar]
- 44.Leulier, F., A. Rodriguez, R. S. Khush, J. M. Abrams, and B. Lemaitre. 2000. The Drosophila caspase Dredd is required to resist gram-negative bacterial infection. EMBO Rep. 1:353-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lisi, S., I. Mazzon, and K. White. 2000. Diverse domains of THREAD/DIAP1 are required to inhibit apoptosis induced by REAPER and HID in Drosophila. Genetics 154:669-678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu, H., Y. C. Su, E. Becker, J. Treisman, and E. Y. Skolnik. 1999. A Drosophila TNF-receptor-associated factor (TRAF) binds the ste20 kinase Misshapen and activates Jun kinase. Curr. Biol. 9:101-104. [DOI] [PubMed] [Google Scholar]
- 47.Liu, Z. G., and J. Han. 2001. Cellular responses to tumor necrosis factor. Curr. Issues Mol. Biol. 3:79-90. [PubMed] [Google Scholar]
- 48.Locksley, R. M., N. Killeen, and M. J. Lenardo. 2001. The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell 104:487-501. [DOI] [PubMed] [Google Scholar]
- 49.Martin-Blanco, E., A. Gampel, J. Ring, K. Virdee, N. Kirov, A. M. Tolkovsky, and A. Martinez-Arias. 1998. puckered encodes a phosphatase that mediates a feedback loop regulating JNK activity during dorsal closure in Drosophila. Genes Dev. 12:557-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McEwen, D. G., and M. Peifer. 2005. Puckered, a Drosophila MAPK phosphatase, ensures cell viability by antagonizing JNK-induced apoptosis. Development 132:3935-3946. [DOI] [PubMed] [Google Scholar]
- 51.Mirey, G., M. Balakireva, S. L'Hoste, C. Rosse, S. Voegeling, and J. H. Camonis. 2003. A RalGEF-Ral pathway is conserved in Drosophila melanogaster and sheds new light on the connectivity of the Ral, Ras, and Rap pathways. Mol. Cell. Biol. 23:1112-1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moreno, E., M. Yan, and K. Basler. 2002. Evolution of TNF signaling mechanisms: JNK-dependent apoptosis triggered by Eiger, the Drosophila homolog of the TNF superfamily. Curr. Biol. 12:1263-1268. [DOI] [PubMed] [Google Scholar]
- 53.Moskalenko, S., D. O. Henry, C. Rosse, G. Mirey, J. H. Camonis, and M. A. White. 2002. The exocyst is a Ral effector complex. Nat. Cell Biol. 4:66-72. [DOI] [PubMed] [Google Scholar]
- 54.Murthy, M., D. Garza, R. H. Scheller, and T. L. Schwarz. 2003. Mutations in the exocyst component Sec5 disrupt neuronal membrane traffic, but neurotransmitter release persists. Neuron 37:433-447. [DOI] [PubMed] [Google Scholar]
- 55.Nakao, K., and J. A. Campos-Ortega. 1996. Persistent expression of genes of the Enhancer of Split complex suppresses neural development in Drosophila. Neuron 16:275-286. [DOI] [PubMed] [Google Scholar]
- 56.Nakashima, S., K. Morinaka, S. Koyama, M. Ikeda, M. Kishida, K. Okawa, A. Iwamatsu, S. Kishida, and A. Kikuchi. 1999. Small G protein Ral and its downstream molecules regulate endocytosis of EGF and insulin receptors. EMBO J. 18:3629-3642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Polzin, A., M. Shipitsin, T. Goi, L. A. Feig, and T. J. Turner. 2002. Ral-GTPase influences the regulation of the readily releasable pool of synaptic vesicles. Mol. Cell. Biol. 22:1714-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Prigent, M., T. Dubois, G. Raposo, V. Derrien, D. Tenza, C. Rosse, J. Camonis, and P. Chavrier. 2003. ARF6 controls post-endocytic recycling through its downstream exocyst complex effector. J. Cell Biol. 163:1111-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rain, J. C., L. Selig, H. De Reuse, V. Battaglia, C. Reverdy, S. Simon, G. Lenzen, F. Petel, J. Wojcik, V. Schachter, Y. Chemama, A. Labigne, and P. Legrain. 2001. The protein-protein interaction map of Helicobacter pylori. Nature 409:211-215. [DOI] [PubMed] [Google Scholar]
- 60.Rangarajan, A., S. J. Hong, A. Gifford, and R. A. Weinberg. 2004. Species- and cell-type-specific requirements for cellular transformation. Cancer Cell 6:171-183. [DOI] [PubMed] [Google Scholar]
- 61.Richardson, H., and S. Kumar. 2002. Death to flies: Drosophila as a model system to study programmed cell death. J. Immunol. Methods 265:21-38. [DOI] [PubMed] [Google Scholar]
- 62.Riesgo-Escovar, J. R., M. Jenni, A. Fritz, and E. Hafen. 1996. The Drosophila Jun-N-terminal kinase is required for cell morphogenesis but not for DJun-dependent cell fate specification in the eye. Genes Dev. 10:2759-2768. [DOI] [PubMed] [Google Scholar]
- 63.Roegiers, F., S. Younger-Shepherd, L. Y. Jan, and Y. N. Jan. 2001. Bazooka is required for localization of determinants and controlling proliferation in the sensory organ precursor cell lineage in Drosophila. Proc. Natl. Acad. Sci. USA 98:14469-14474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosse, C., A. Hatzoglou, M. C. Parrini, M. A. White, P. Chavrier, and J. Camonis. 2006. RalB mobilizes the exocyst to drive cell migration. Mol. Cell. Biol. 26:727-734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ruan, W., H. Long, D. H. Vuong, and Y. Rao. 2002. Bifocal is a downstream target of the Ste20-like serine/threonine kinase misshapen in regulating photoreceptor growth cone targeting in Drosophila. Neuron 36:831-842. [DOI] [PubMed] [Google Scholar]
- 66.Ruan, W., P. Pang, and Y. Rao. 1999. The SH2/SH3 adaptor protein dock interacts with the Ste20-like kinase misshapen in controlling growth cone motility. Neuron 24:595-605. [DOI] [PubMed] [Google Scholar]
- 67.Sawamoto, K., P. Winge, S. Koyama, Y. Hirota, C. Yamada, S. Miyao, S. Yoshikawa, M. H. Jin, A. Kikuchi, and H. Okano. 1999. The Drosophila Ral GTPase regulates developmental cell shape changes through the Jun NH(2)-terminal kinase pathway. J. Cell Biol. 146:361-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Su, Y. C., C. Maurel-Zaffran, J. E. Treisman, and E. Y. Skolnik. 2000. The Ste20 kinase misshapen regulates both photoreceptor axon targeting and dorsal closure, acting downstream of distinct signals. Mol. Cell. Biol. 20:4736-4744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sugihara, K., S. Asano, K. Tanaka, A. Iwamatsu, K. Okawa, and Y. Ohta. 2001. The exocyst complex binds the small GTPase RalA to mediate filopodia formation. Nat. Cell Biol. 17:17. [DOI] [PubMed] [Google Scholar]
- 70.Urano, T., R. Emkey, and L. A. Feig. 1996. Ral-GTPases mediate a distinct downstream signaling pathway from Ras that facilitates cellular transformation. EMBO J. 15:810-816. [PMC free article] [PubMed] [Google Scholar]
- 71.Usui, K., and K.-I. Kimura. 1993. Sequential emergence of the evenly spaced microchaetae on the notum of Drosophila. Roux's Arch. Dev. Biol. 203:151-158. [DOI] [PubMed] [Google Scholar]
- 72.Wang, S., Y. Liu, C. L. Adamson, G. Valdez, W. Guo, and S. C. Hsu. 2004. The mammalian exocyst, a complex required for exocytosis, inhibits tubulin polymerization. J. Biol. Chem. 279:35958-35966. [DOI] [PubMed] [Google Scholar]
- 73.White, K., E. Tahaoglu, and H. Steller. 1996. Cell killing by the Drosophila gene reaper. Science 271:805-807. [DOI] [PubMed] [Google Scholar]
- 74.White, M. A., T. Vale, J. H. Camonis, E. Schaefer, and M. H. Wigler. 1996. A role for Ral guanine nucleotide dissociation stimulator in mediating Ras-induced transformation. J. Biol. Chem. 271:16439-16442. [DOI] [PubMed] [Google Scholar]
- 75.Wieschaus, E., and C. Nüsslein-Volhard. 1998. Looking at embryos, p. 179-214. In D. B. Roberts (ed.), Drosophila, a practical approach, 2nd ed. Practical Approach Series, vol. 191. IRL Press, Oxford, United Kingdom. [Google Scholar]
- 76.Wilson, R., L. Goyal, M. Ditzel, A. Zachariou, D. A. Baker, J. Agapite, H. Steller, and P. Meier. 2002. The DIAP1 RING finger mediates ubiquitination of Dronc and is indispensable for regulating apoptosis. Nat. Cell Biol. 4:445-450. [DOI] [PubMed] [Google Scholar]
- 77.Yoo, S. J., J. R. Huh, I. Muro, H. Yu, L. Wang, S. L. Wang, R. M. Feldman, R. J. Clem, H. A. Muller, and B. A. Hay. 2002. Hid, Rpr and Grim negatively regulate DIAP1 levels through distinct mechanisms. Nat. Cell Biol. 4:416-424. [DOI] [PubMed] [Google Scholar]
- 78.Zhuang, Z. H., Y. Zhou, M. C. Yu, N. Silverman, and B. X. Ge. 2006. Regulation of Drosophila p38 activation by specific MAP2 kinase and MAP3 kinase in response to different stimuli. Cell Signal. 18:441-448. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.