

Abstract
The heme oxygenase (HO) enzymes catalyze the rate-limiting step of heme breakdown. Prior studies have demonstrated that the vulnerability of neurons and astrocytes to hemoglobin is modified in cells lacking HO-2, the constitutive isoform. The present study assessed the effect of the inducible isoform, HO-1. Wild-type astrocytes treated for 3–5 days with 3–30 μM hemoglobin sustained no loss of viability, as quantified by LDH and MTT assays. The same treatment resulted in death of 25–50% of HO-1 knockout astrocytes, and a fourfold increase in protein oxidation. Cell injury was attenuated by transfer of the HO-1 gene, but not by bilirubin, the antioxidant heme breakdown product. Conversely, neuronal protein oxidation and cell death after hemoglobin exposure were similar in wild-type and HO-1 knockout cultures. These results suggest that HO-1 induction protects astrocytes from the oxidative toxicity of Hb, but has no effect on neuronal injury.
Introduction
Hemoglobin (Hb) is the most abundant protein in blood, where it is present at a concentration of 2–2.5 mM in the healthy adult. After CNS hemorrhage, some of this Hb enters the extracellular space due to erythrocyte lysis, which may be mediated by complement activation [1]. Although small quantities of free Hb may be removed by binding to its transport protein haptoglobin [2], deposition of iron in tissue surrounding experimental hematomas is consistent with local catabolism. A growing body of evidence suggests that free heme, iron and perhaps other Hb degradation products may contribute to oxidative injury after hemorrhagic insults [3].
Breakdown of the heme moieties of Hb is catalyzed by the heme oxygenase (HO) enzymes, yielding iron, carbon monoxide, and biliverdin. Two isoforms have been characterized in the CNS. Under normal conditions, HO-1 is expressed at a very low level, but it is rapidly induced by Hb, heme, and various oxidants [4]. HO-2 is constitutively expressed, primarily by neurons [4], although it also has been detected in cultured astrocytes and endothelial cells [5; 6].
The consequence of heme breakdown by the heme oxygenases has been investigated in several CNS injury models, which have used pharmacologic or genetic approaches to alter HO activity. Somewhat conflicting results have been reported to date. A beneficial effect of HO has been observed in models of ischemia and trauma [7; 8; 9; 10], and in cultured astrocytes exposed to hemin [11]; cytoprotection is usually attributed to conversion of heme to biliverdin [12], which has an antioxidant effect per se [13], and is rapidly reduced to the antioxidant bilirubin. Conversely, a pro-oxidant effect of HO, attributed to iron release [14], has also been observed. The latter appears to predominate in neurons exposed to Hb [15; 16; 17; 18], which may reflect the relative inability of this cell type to sequester and detoxify excess iron [19; 20].
All studies addressing the effect of HO on Hb toxicity in CNS cells have used either HO inhibitors or HO-2 knockout (KO) mice [15; 16; 18; 21]. Unfortunately, both approaches are somewhat limited by nonspecific effects. Currently available inhibitors are rather reactive compounds that may enhance the toxicity of oxidants by mechanisms unrelated to HO, including inhibition of caspases, guanyl cyclase, and nitric oxide synthase [22; 23]. Models using HO-2 KO mice are putatively more specific. However, it has recently been reported that the HO-2 protein alters the vulnerability of HEK 293 cells to oxidative injury by a mechanism that is completely unrelated heme breakdown [24]. Although the molecular basis for this phenomenon remains undefined, it is noteworthy that HO-2 binds heme at two non-catalytic heme regulatory motifs, in addition to its catalytic site [25]. It is therefore possible that an alternate function of either mobilizing or sequestering heme may contribute to its effect on Hb toxicity and other oxidative injuries.
In order to further investigate the effect of HO on Hb toxicity in astrocytes and neurons, we have established a colony of mice lacking the HO-1 gene. In light of our prior observations using HO-2 KO cells, in the present study we tested the following hypotheses: 1) HO-1 KO astrocytes are more susceptible than wild-type (WT) to Hb; 2) HO-1 KO neurons are less sensitive to Hb.
Materials and Methods
Cell cultures
HO-1 KO mice with a 129/Sv X BALB/c genetic background were descendants of those generated by Yet et al.[26]. Animal care and treatments followed guidelines described in “Principles of Laboratory Animal Care” (NIH publication No. 80–23, revised 1996). Genotype was determined by PCR analysis of DNA extracted from tail clippings, using previously published primers [27]. Cortical astrocyte cultures were prepared from 1 to 3 day postnatal HO-1 WT and KO mice, using a culture method that has already been described [11]. Mixed neuron/astrocyte cultures were prepared from fetal mice at 15–17 days gestation, also using a previously detailed method [16]. Genotype of cultures was confirmed by PCR and immunoblotting.
Hemoglobin exposure
Purified human Hb A was a gift from Hemosol Corp. (Mississauga, Ontario, Canada). Cultures were washed free of growth medium, and Hb was added to cultures at defined concentrations that were diluted in minimal essential medium containing 10 mM glucose. At the end of the exposure interval, cell viability was quantified with the MTT and lactate dehydrogenase (LDH) assays, as previously detailed [11].
Detection of protein oxidation
Protein oxidation was assayed using the Oxyblot kit (Chemicon, Inc., Temecula, CA), which detects oxidatively-generated carbonyl groups. Cells were collected in ice-cold lysis buffer (210 mM mannitol, 70 mM sucrose, 5 mM HEPES, 1 mM EDTA, 0.1 % sodium dodecyl sulfate, 0.1 % Triton X-100). 2-mercaptoethanol (1%) was immediately added to lysates to prevent further protein oxidation. Carbonyl groups were derivatized to 2,4-dintrophenylhydrazone by reaction with 2,4-dinitrophenylhydrazine, following the manufacturer’s instructions. Proteins were then separated on a 12% polyacrylamide gel and were transferred to a PVDF membrane. Carbonylated proteins were detected with rabbit anti-DNP primary antibody (1:150) followed by goat anti-rabbit HRP-conjugated secondary antibody (1:300). Immunoreactive proteins were visualized using Super Signal West Femto Reagent (Pierce) and Kodak ImageStation 400; lane densities were analyzed with Kodak 1D software.
Adenoviral gene transfer
Preparation of an adenovirus containing the coding sequence of the human HO-1 gene has been previously described [28]. Viruses were propagated in HEK 293 cells; titer was quantified by cytopathic effect assay, using HEK 293 cells exposed to serial dilutions of virus for 10 days. Prior experiments have determined that optimal transfection in this culture system is accomplished by treatment with a viral dose of 100 MOI (multiplicity of infection) for 24h, in medium containing 3.3% equine serum [29]; this method was used exclusively.
Cell hemin assay
Cell hemin content was quantified using the method of Motterlini et al. [30]. Cells were washed once with 0.75 ml DMEM (Invitrogen, Grand Island, NY), which was then completely aspirated and replaced with 200 μl formic acid (Sigma, St. Louis, MO). After five minutes, the lysate was collected, and its absorbance at 398 nm was determined. Hemin content was extrapolated from a hemin in formic acid standard curve, and was expressed as nmol/mg protein.
Results
Vulnerability of astrocytes and neurons to hemoglobin
Consistent with prior observations [21], treatment of WT astrocyte cultures with 3–30 μM Hb for 75–120h resulted in no cell injury, as detected by LDH assay (Fig. 1A). Higher concentrations resulted in Hb precipitation that impeded inspection of the cells, and so were not investigated. In concomitantly treated HO-1 KO cultures, Hb produced injury within 75h, which was readily detected by inspection of cultures, and which was statistically significant when quantified by LDH assay. This progressed to about 50% cell lysis after exposure to 10 μM Hb for 120h; however, no cell death was observed at 24h. Increased injury in knockout cultures was confirmed with the MTT assay, which measures the ability of viable cells to reduce MTT to a colored formazan product (cell viability after 120h exposure to Hb 10 μM: 94.2 ± 8.2% in WT vs. 21.4 ± 8.3% in KO cultures).
Fig. 1.
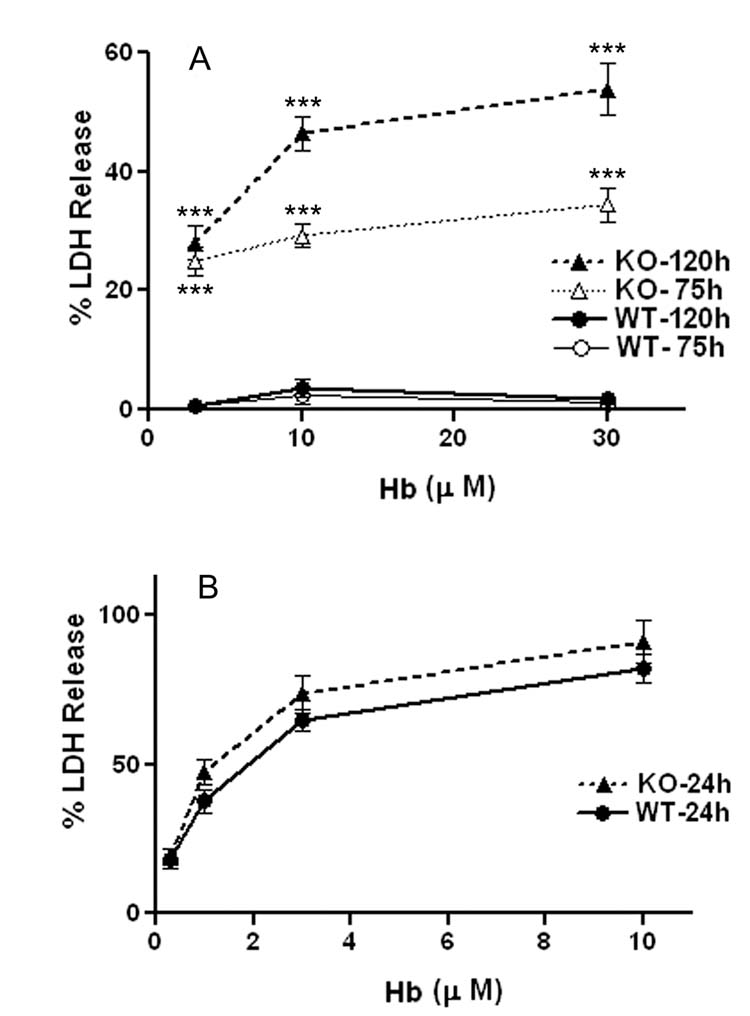
Effect of HO-1 gene deletion on vulnerability of astrocytes and neurons to Hb. A) Mean medium LDH (± S.E.M., 14–20/condition) in astrocyte cultures treated for 75–120 h with indicated concentrations of Hb. LDH values were scaled to those in sister cultures treated with 0.3% Triton X-100 (= 100), which produces 100% cell lysis. B) Mixed neuron/astrocyte cultures (27–31/condition) were treated with indicated Hb concentrations for 24h. LDH values were scaled to those in sister cultures treated with 300 μM NMDA, which releases all neuronal LDH without injuring astrocytes. *** P < 0.001 versus signal in corresponding WT culture, Bonferroni multiple comparisons test.
Also consistent with prior observations [16], treatment of WT mixed neuron/astrocyte cultures with Hb produced widespread neuronal death by 24h (Fig. 1B), without morphologic change in the astrocyte feeder layer. KO astrocytes in mixed cultures likewise sustained no morphologic evidence of injury at 24 hours; however, neuronal injury, as assessed by LDH release, was similar to that in WT mixed cultures. Due to the high background signal from the viable astrocyte monolayer, the MTT assay was not used to assess cell viability in mixed cultures.
Protein carbonylation is a sensitive marker of oxidation in this culture system [29]. In both WT and KO astrocyte cultures, the level of protein carbonyls after medium exchange only was barely detectable (Fig. 2). A fourfold increase was observed in KO astrocyte cultures after 75h exposure to 10 μM Hb, which was significantly reduced in WT cultures. In WT mixed neuron-astrocyte cultures, a 6.7-fold increase in protein oxidation was observed after 24h treatment with 10 μM Hb, which was very similar to that seen in KO cultures.
Fig. 2.

Hb-induced protein oxidation in HO-1 knockout astrocytes and neurons. Bars represent mean carbonyl signal intensities (±S.E.M., 4/condition) from cultures treated with Hb 10 μM for 75h (astrocytes) or 24h (mixed), or subjected to media exchange only (wash). *** P < 0.001 versus signal in HO-1 knockout cultures treated with Hb.
Since HO-1 had no detectable effect on the neurotoxicity of Hb in our mixed culture system, additional experiments were conducted on astrocyte cultures only.
Hb toxicity in HO-1 knockout astrocytes is attenuated by HO-1 gene transfer
In order to further test the hypothesis that the increased vulnerability of KO astrocytes was due to HO-1 deficiency, HO-1 was expressed in KO cells by incubation with 100 MOI of an adenovirus encoding the human HO-1 gene, which produces a sixfold increase in HO-1 expression in these cultures [29]. Vulnerability to Hb was markedly diminished by this pretreatment (Fig. 3), but not by pretreatment with a control virus lacking the HO-1 gene.
Fig. 3.

HO-1 gene transfer protects HO-1 knockout astrocytes from Hb. Cultures (12/condition) were pretreated for 24h with 100 MOI of adenovirus encoding the human HO-1 gene (HO-1-Hb), control virus (Null-Hb), or culture medium only, then incubated with 10 μM Hb for 75h and 120h. LDH values were scaled to those in sister cultures treated with 0.3% Triton X-100 for 1 h (=100). ***P<0.001 versus cultures exposed to Hb with control virus pretreatment or without any virus treatment.
Increased hemin in HO-1 knockout astrocytes
The heme groups of Hb have a high affinity for globin, and the likelihood of their release or transfer to other molecules is negligible [31]. However, at 37°C, Hb autoxidizes to methemoglobin, which more readily releases oxidized heme, also known as hemin, or transfers it to other proteins and lipids [32]. Since free hemin is a potent oxidant, and is catabolized primarily by heme oxygenase, we hypothesized that its accumulation would be greater in knockout astrocytes. Consistent with this hypothesis, KO astrocytes exposed to 10 μM Hb for 24h had a hemin content of 1.64 ± 0.19 nmol/mg protein, versus 0.86 ± 0.13 nmol/mg protein in WT astrocytes (P = 0.005, unpaired t test). Hemin was not detectable by this method in control cultures subjected to medium exchange only.
Bilirubin does not protect knockout astrocytes from Hb
The antioxidant effect of heme oxygenase is often attributed to increased cell bilirubin. Nanomolar concentrations of exogenous bilirubin protect HO-2 knockout neurons from the toxicity of hydrogen peroxide [12]. Its efficacy against Hb toxicity in astrocyte cultures was therefore assessed. Concomitant treatment with 50–500 nM bilirubin had no effect (Table 1); higher concentrations were toxic.
Table 1.
HO-1 knockout astrocyte cultures were treated with Hb 10 μM alone or with indicated bilirubin concentrations for 120h. Injury was assessed by LDH release and MTT assays.
| %LDH Release | % Cell Viability | |
|---|---|---|
| Hb 10 μM | 31.9±3.2 | 57.1±1.9 |
| Hb + Bilirubin 50 nM | 32.2±3.4 | 58.3±3.9 |
| Hb + Bilirubin 500 nM | 34.9±3.0 | 64.6±3.1 |
Bilirubin Does Not Protect HO-1 Knockout Astrocytes from Hb
Discussion
The present results suggest that HO-1 is essential for astrocyte resistance to Hb. As expected from prior studies, prolonged (3–5 day) exposure of cultured wild-type astrocytes to highly neurotoxic concentrations of Hb produced no loss of cell viability. Astrocytes from mice that were genetically identical except that they lacked the HO-1 gene sustained significant oxidative injury after the same treatment. These results are very similar to those observed when wild-type astrocyte cultures were treated with Hb in the presence of HO inhibitors [21], and indicate that the exacerbation of injury by these inhibitors was due at least in part to HO inhibition rather than some nonspecific effect.
However, these results fail to support the hypothesis that HO-1 knockout neurons are less vulnerable to Hb. The basis for this hypothesis was our previous observation that HO-2 knockout neurons were less sensitive to Hb than their wild-type counterparts, both in cell culture and in vivo [16; 18]. The disparate effect of HO-1 and HO-2 may reflect a predominance of HO-2 in central neurons after Hb exposure [4; 10]. Cultured astrocytes, but not neurons, rapidly induce HO-1 in response to Hb or hemin, although baseline expression is similar in both cell types [33]. HO-1 may therefore have little effect on neuronal vulnerability to Hb because its contribution to overall HO activity is negligible in this HO-2-expressing cell population. Alternatively, the deleterious effect of HO-2 on Hb neurotoxicity may be due to another function of this protein that is not provided by HO-1, specifically heme mobilization. McCoubrey and Maines [25] observed that HO-2 is a hemoprotein that may bind up to two heme moieties at sites not involved in heme breakdown, and hypothesized that it may thereby be involved in physiologic free radical generation and redox signaling. Under conditions of gross heme excess after hemorrhage, this binding may contribute to oxidative stress, and its absence may account for the cytoprotection provided by HO-2 gene knockout. Further investigation of this putative function of HO-2 seems warranted. It is noteworthy, however, that Hb neurotoxicity is iron-dependent, since it is attenuated in cell culture and in vivo by iron chelators [34; 35]. Heme breakdown by HO-2 is therefore likely to be at least somewhat relevant to heme-mediated neuronal injury.
Since Hb is a rather poor substrate for HO compared with hemin [36], the effect of HO-1 on astrocytes suggests that Hb toxicity may be mediated by hemin transfer to cells from methemoglobin, rather than by the action of the intact protein. Evidence for this mechanism is provided by the observation that hemin accumulates in astrocytes treated with Hb, and that its level is significantly higher in HO-1 knockouts. Since hemin, but not free iron, is highly toxic to cultured astrocytes [11], the benefit of HO-1 may be to prevent cell hemin concentrations from reaching lethal levels. Other protective effects of HO, such as increased production of carbon monoxide [37], cannot be excluded. However, the present data do not support a significant contribution from bilirubin in this model.
Although HO-1 knockout astrocytes were injured by 3–5 day Hb treatment, they were considerably more resistant than neurons, most of which are killed by a 24h exposure to similar Hb concentrations. This is not surprising, since cultured neurons are fastidious cells that are injured more rapidly than astrocytes when subjected to a variety of oxidative insults [38; 39]. The greater vulnerability of neurons to Hb, compared with wild-type or mutant astrocytes, is likely due to both their lower level of endogenous antioxidants and their greater dependence on the function of membrane cation pumps, which are readily inactivated by oxidative stress [40; 41].
These results suggest that nonselective HO inhibitors, which are active against both HO-1 and HO-2, may increase oxidative injury to astrocytes after CNS hemorrhage, particularly with prolonged use. They also suggest that selective inhibition of HO-2, if feasible, would provide maximal neuroprotection, while mitigating the pro-oxidant consequences of nonselective inhibitors on astrocytes. Although several studies have demonstrated the efficacy of nonselective inhibitors against the toxicity of intracerebral blood or Hb in vivo [15; 17; 42], an adverse effect may be difficult to detect in rodent or other small animal models, since the ratio of astrocytes to neurons in these species is a fraction of that of the human brain [43]. Further investigation in large animal models, with administration for therapeutically relevant durations, seems advisable prior to any clinical trials of HO inhibitors after CNS hemorrhage.
Acknowledgments
This study was supported by grants from the National Institutes of Health (NS042273 and NS050662). We thank Dr. Shaw-Fang Yet for providing HO-1 knockout breeders, and Dr. Lee-Young Chau for providing the adenoviral HO-1 vector and controls.
References
- 1.Hua Y, Xi G, Keep RF, Hoff JT. Complement activation in the brain after experimental intracerebral hemorrhage. J Neurosurg. 2000;92:1016–1022. doi: 10.3171/jns.2000.92.6.1016. [DOI] [PubMed] [Google Scholar]
- 2.Panter SS, Sadrzadeh SM, Hallaway PE, Haines JL, Anderson VE, Eaton JW. Hypohaptoglobinemia associated with familial epilepsy. J Exp Med. 1985;161:748–54. doi: 10.1084/jem.161.4.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoff JT, Xi G. Brain edema from intracerebral hemorrhage. Acta Neurochir Suppl. 2003;86:11–5. doi: 10.1007/978-3-7091-0651-8_3. [DOI] [PubMed] [Google Scholar]
- 4.Matz P, Turner C, Weinstein PR, Massa SM, Panter SS, Sharp FR. Heme-oxygenase-1 induction in glia throughout rat brain following experimental subarachnoid hemorrhage. Brain Res. 1996;713:211–222. doi: 10.1016/0006-8993(95)01511-6. [DOI] [PubMed] [Google Scholar]
- 5.Scapagnini G, D’Agata V, Calabrese V, Pascale A, Colombrita C, Alkon D, Cavallaro S. Gene expression profiles of heme oxygenase isoforms in the rat brain. Brain Res. 2002;954:51–59. doi: 10.1016/s0006-8993(02)03338-3. [DOI] [PubMed] [Google Scholar]
- 6.Parfenova H, Basuroy S, Bhattacharya S, Tcheranova D, Qu Y, Regan RF, Leffler CW. Glutamate induces oxidative stress and apoptosis in cerebral vascular endothelial cells: contributions of HO-1 and HO-2 to cytoprotection. Am J Physiol Cell Physiol. 2006;290:C1399–410. doi: 10.1152/ajpcell.00386.2005. [DOI] [PubMed] [Google Scholar]
- 7.Takizawa S, Hirabayashi H, Matsushima K, Tokuoka K, Shinohara Y. Induction of heme oxygenase protein protects neurons in cortex and striatum, but not hippocampus, against transient forebrain ischemia. J Cereb Blood Flow Metab. 1998;18:559–569. doi: 10.1097/00004647-199805000-00011. [DOI] [PubMed] [Google Scholar]
- 8.Doré S, Sampei K, Goto S, Alkayed NJ, Guastella D, Blackshaw S, Gallagher M, Traystman RJ, Hurn PD, Koehler RC, Snyder SH. Heme oxygenase-2 is neuroprotective in cerebral ischemia. Mol Med. 1999;5:656–663. [PMC free article] [PubMed] [Google Scholar]
- 9.Panahian N, Yoshiura M, Maines MD. Overexpression of heme oxygenase-1 is neuroprotective in a model of permanent middle cerebral artery occlusion in transgenic mice. J Neurochem. 1999;72:1187–1203. doi: 10.1111/j.1471-4159.1999.721187.x. [DOI] [PubMed] [Google Scholar]
- 10.Chang EF, Wong RJ, Vreman HJ, Igarashi T, Galo E, Sharp FR, Stevenson DK, Noble-Haeusslein LJ. Heme oxygenase-2 protects against lipid peroxidation-mediated cell loss and impaired motor recovery after traumatic brain injury. J Neurosci. 2003;23:3689–96. doi: 10.1523/JNEUROSCI.23-09-03689.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Regan RF. Heme oxygenase-2 gene deletion increases astrocyte vulnerability to hemin. Biochem Biophys Res Commun. 2004;318:88–94. doi: 10.1016/j.bbrc.2004.03.187. [DOI] [PubMed] [Google Scholar]
- 12.Doré S, Takahashi M, Ferris CD, Hester LD, Guastella D, Snyder SH. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc Natl Acad Sci USA. 1999;96:2445–2450. doi: 10.1073/pnas.96.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–6. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 14.Lamb NJ, Quinlan GJ, Mumby S, Evans TW, Gutteridge JMC. Haem oxygenase shows pro-oxidant activity in microsomal and cellular systems: implications for the release of low-molecular-mass iron. Biochem J. 1999;344:153–158. [PMC free article] [PubMed] [Google Scholar]
- 15.Huang FP, Xi G, Keep RF, Hua Y, Nemoianu A, Hoff JT. Brain edema after experimental intracerebral hemorrhage: role of hemoglobin degradation products. J Neurosurg. 2002;96:287–293. doi: 10.3171/jns.2002.96.2.0287. [DOI] [PubMed] [Google Scholar]
- 16.Rogers B, Yakopson V, Teng ZP, Guo Y, Regan RF. Heme oxygenase-2 knockout neurons are less vulnerable to hemoglobin toxicity. Free Rad Biol Med. 2003;35:872–881. doi: 10.1016/s0891-5849(03)00431-3. [DOI] [PubMed] [Google Scholar]
- 17.Koeppen AH, Dickson AC, Smith J. Heme oxygenase in experimental intracerebral hemorrhage: the benefit of tin-mesoporphyrin. J Neuropathol Exp Neurol. 2004;63:587–597. doi: 10.1093/jnen/63.6.587. [DOI] [PubMed] [Google Scholar]
- 18.Qu Y, Chen J, Benvenisti-Zarom L, Ma X, Regan RF. Effect of targeted deletion of the heme oxygenase-2 gene on hemoglobin toxicity in the striatum. J Cereb Blood Flow Metab. 2005;25:1466–1475. doi: 10.1038/sj.jcbfm.9600143. [DOI] [PubMed] [Google Scholar]
- 19.Kress GJ, Dineley KE, Reynolds IJ. The relationship between intracellular free iron and cell injury in cultured neurons, astrocytes, and oligodendrocytes. J Neurosci. 2002;22:5848–55. doi: 10.1523/JNEUROSCI.22-14-05848.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moos T, Morgan EH. The metabolism of neuronal iron and its pathogenic role in neurological disease: review. Ann N Y Acad Sci. 2004;1012:14–26. doi: 10.1196/annals.1306.002. [DOI] [PubMed] [Google Scholar]
- 21.Regan RF, Guo YP, Kumar N. Heme oxygenase-1 induction protects murine cortical astrocytes from hemoglobin toxicity. Neurosci Lett. 2000;282:1–4. doi: 10.1016/s0304-3940(00)00817-x. [DOI] [PubMed] [Google Scholar]
- 22.Grundemar L, Ny L. Pitfalls using metalloporphyrins in carbon monoxide research. Trends Pharmacol Sci. 1997;18 doi: 10.1016/s0165-6147(97)01065-1. [DOI] [PubMed] [Google Scholar]
- 23.Blumenthal SB, Kiemer AK, Tiegs G, Seyfried S, Holtje M, Brandt B, Holtje HD, Zahler S, Vollmar AM. Metalloporphyrins inactivate caspase-3 and -8. FASEB J. 2005;19:1272–1279. doi: 10.1096/fj.04-3259com. [DOI] [PubMed] [Google Scholar]
- 24.Kim YS, Dore S. Catalytically inactive heme oxygenase-2 mutant is cytoprotective. Free Radic Biol Med. 2005;39:558–64. doi: 10.1016/j.freeradbiomed.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 25.McCoubrey WK, Huang TJ, Maines MD. Heme oxygenase-2 is a hemoprotein and binds heme through heme regulatory motifs that are not involved in heme catalysis. J Biol Chem. 1997;272:12568–12574. doi: 10.1074/jbc.272.19.12568. [DOI] [PubMed] [Google Scholar]
- 26.Yet SF, Perrella MA, Layne MD, Hsieh CM, Maemura K, Kobzik L, Wiesel P, Christou H, Kourembanas S, Lee ME. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J Clin Invest. 1999;103:R23–9. doi: 10.1172/JCI6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujita T, Toda K, Karimova A, Yan SF, Naka Y, Yet SF, Pinsky DJ. Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med. 2001;7:598–604. doi: 10.1038/87929. [DOI] [PubMed] [Google Scholar]
- 28.Juan SH, Lee TS, Tseng KW, Liou JY, Shyue SK, Wu KK, Chau LY. Adenovirus-mediated heme oxygenase-1 gene transfer inhibits the development of atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2001;104:1519–1525. doi: 10.1161/hc3801.095663. [DOI] [PubMed] [Google Scholar]
- 29.Teng ZP, Chen J, Chau LY, Galunic N, Regan RF. Adenoviral transfer of the heme oxygenase-1 gene protects cortical astrocytes from heme-mediated oxidative injury. Neurobiol Dis. 2004;17:179–187. doi: 10.1016/j.nbd.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 30.Motterlini R, Foresti R, Vandegriff K, Intaglietta M, Winslow RM. Oxidative-stress response in vascular endothelial cells exposed to acellular hemoglobin solutions. Am J Physiol. 1995;269:H648–H655. doi: 10.1152/ajpheart.1995.269.2.H648. [DOI] [PubMed] [Google Scholar]
- 31.Hebbel RP, Eaton JW. Pathobiology of heme interaction with the erythrocyte membrane. Sem Hematol. 1989;26:136–149. [PubMed] [Google Scholar]
- 32.Bunn HF, Jandl JH. Exchange of heme along hemoglobins and between hemoglobin and albumin. J Biol Chem. 1967;243:465–475. [PubMed] [Google Scholar]
- 33.Benvenisti-Zarom L, Chen-Roetling J, Regan RF. Inhibition of the ERK/MAP kinase pathway attenuates heme oxygenase-1 expression and heme-mediated neuronal injury. Neurosci Lett. 2006;398:230–4. doi: 10.1016/j.neulet.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 34.Regan RF, Rogers B. Delayed treatment of hemoglobin neurotoxicity. J Neurotrauma. 2003;20:111–120. doi: 10.1089/08977150360517236. [DOI] [PubMed] [Google Scholar]
- 35.Nakamura T, Keep RF, Hua Y, Schallert T, Hoff JT, Xi G. Deferoxamine-induced attenuation of brain edema and neurological deficits in a rat model of intracerebral hemorrhage. J Neurosurg. 2004;100:672–8. doi: 10.3171/jns.2004.100.4.0672. [DOI] [PubMed] [Google Scholar]
- 36.Abraham NG, Drummond GS, Lutton JD, Kappas A. The biological significance and physiological role of heme oxygenase. Cell Physiol Biochem. 1996;6:129–168. [Google Scholar]
- 37.Ryter SW, Otterbein LE. Carbon monoxide in biology and medicine. Bioessays. 2004;26:270–80. doi: 10.1002/bies.20005. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka S, Takehashi M, Matoh N, Iida S, Suzuki T, Futaki S, Hamada H, Masliah E, Sugiura Y, Ueda K. Generation of reactive oxygen species and activation of NF-kappaB by non-Abeta component of Alzheimer’s disease amyloid. J Neurochem. 2002;82:305–15. doi: 10.1046/j.1471-4159.2002.00958.x. [DOI] [PubMed] [Google Scholar]
- 39.Watts LT, Rathinam ML, Schenker S, Henderson GI. Astrocytes protect neurons from ethanol-induced oxidative stress and apoptotic death. J Neurosci Res. 2005;80:655–66. doi: 10.1002/jnr.20502. [DOI] [PubMed] [Google Scholar]
- 40.Leclerc L, Vasseur C, Bursaux E, Marden M, Poyart C. Inhibition of membrane erythrocyte (Ca2+ + Mg2+ )-ATPase by hemin. Biochim Biophys Acta. 1988;946:49–56. doi: 10.1016/0005-2736(88)90456-7. [DOI] [PubMed] [Google Scholar]
- 41.Makar TK, Nedergaard M, Preuss A, Gelbard AS, Perumal AS, Cooper AJ. Vitamin E, ascorbate, glutathione, glutathione disulfide, and enzymes of glutathione metabolism in cultures of chick astrocytes and neurons: evidence that astrocytes play an important role in antioxidative processes in the brain. J Neurochem. 1994;62:45–53. doi: 10.1046/j.1471-4159.1994.62010045.x. [DOI] [PubMed] [Google Scholar]
- 42.Gong Y, Tian H, Xi G, Keep RF, Hoff JT, Hua Y. Systemic zinc protoporphyrin administration reduces intracerebral hemorrhage-induced brain injury. Acta Neurochir Suppl. 2006;96:232–6. doi: 10.1007/3-211-30714-1_50. [DOI] [PubMed] [Google Scholar]
- 43.Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003;26:523–530. doi: 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]