

Abstract
The homeobox gene Nanog is a key intrinsic determinant of self renewal in embryonic stem (ES) cells, and its repression leads ES cells to selectively differentiate into primitive endoderm. Although Nanog repression occurs at the outermost layer of ES cell aggregates independent of the leukemia inhibitory factor (LIF)/STAT3 pathway, it is largely undetermined what external cues and intracellular signals cause the event. Of interest, addition of the tyrosine phosphatase inhibitor, sodium vanadate, selectively repressed Nanog transcription without any detectable changes in upstream transcriptional regulators Oct3/4 and Sox2. Furthermore, sodium vanadate induced primitive endoderm differentiation, even in the inner cells of ES cell aggregates. Expression of Gata6 and Zfp42, two putative downstream Nanog effectors, was also increased and decreased by the addition of sodium vanadate, respectively, but these changes were eliminated by exogenous Nanog expression. The effects of sodium vanadate were abrogated by Grb2 deficiency or by the addition of the Mek inhibitor, PD98059. Indeed, PD98059 prevented Nanog repression induced by ES cell aggregation as well. Furthermore, transfection of a constitutive active Mek mutant into ES cells induced Nanog repression and primitive endoderm differentiation. These data indicate that the Grb2/Mek pathway primarily mediates Nanog gene repression upon ES cell differentiation into primitive endoderm.
The homeoprotein Nanog was found to play an essential role in the self renewal of embryonic stem (ES) cells (5, 23). Increased expression of Nanog can maintain mouse ES cells in an undifferentiated state regardless of the presence of leukemia inhibitory factor (LIF)/STAT3 signaling. Nanog expression during embryonic development is restricted to a pluripotent cell population including the inner cell mass (ICM) of blastocysts and primordial germ cells (5, 38). Disruption of the Nanog gene in ES cells caused a loss of pluripotency and resulted in differentiation towards the primitive endoderm lineage. In addition, a DNA motif predicted to be recognized by the Nanog protein was found in the promoter region of Gata6, a gene known to play a critical role during primitive endoderm differentiation (23). Thus, the primary function of Nanog is considered to be preventing ES cells from differentiating into primitive endoderm.
Primitive endoderm is the first differentiated cell type to arise from the inner cell mass (ICM) in blastocysts during mammalian embryogenesis. Derivation of primitive endoderm from the ICM can be recapitulated by forming ES cell aggregates in suspension culture (10). As observed in the ICM during development, primitive endoderm differentiation is restricted to the outermost layer of the ES cell aggregates, and the rest of the inner cells retain their pluripotency in the presence of LIF (25, 31). Our previous study identified that the Nanog gene was selectively repressed in the outermost layer of ES cell aggregates in the presence of LIF, and it identified that Nanog repression was essential for primitive endoderm differentiation in the outermost layer (13). However, it is largely undetermined what external cues and intracellular signals cause this repression.
Previous studies using gene targeting suggested that several signaling molecules may be involved in primitive endoderm specification. Disruption of FGFR1 or FGFR2, which are family members of receptor tyrosine kinases, inhibited primitive endoderm differentiation (2, 11). In addition, when fibroblast growth factor (FGF) signaling was blocked by the overexpression of a dominant-negative mutant of FGFR2 or by the FGF receptor (FGFR)-specific inhibitor SU5042, the formation of the primitive endoderm layer was abolished (8, 17, 18). These studies strongly suggest that the FGF-FGFR interaction is pivotal for primitive endoderm specification. Of interest, the phosphatidylinositol 3-kinase (PI3K)-Akt pathway rather than Erk signaling was predominately affected by the dominant-negative mutant of FGFR (8). Furthermore, the addition of a PI3K inhibitor to ES cell aggregates reduced primitive endoderm differentiation (8). These studies imply that the PI3K-Akt pathway may play a role in FGF-mediated primitive endoderm differentiation.
Another signaling pathway involved in primitive endoderm differentiation is the Ras-Erk pathway. Targeted disruption of Grb2, which links phosphotyrosine to intracellular signaling pathways, abrogated the formation of primitive endoderm in blastocysts. This phenotype was rescued by the expression of a Grb2-Sos1 fusion protein, which activated the downstream Ras-Erk pathway (9). Further, another study showed that introducing an active mutant of Ras into murine ES cells resulted in primitive endoderm differentiation (39). These studies suggest that the Grb2/Sos/Ras/Erk pathway plays an essential role in primitive endoderm specification. Thus, it is still controversial which intracellular signaling pathway is predominately involved in primitive endoderm specification. Moreover, none of the previous studies clearly demonstrated how these signaling pathways affected Nanog gene expression.
In the present study, we have attempted to reveal the intracellular signaling pathways controlling Nanog gene expression during ES cell differentiation into primitive endoderm. Using various pharmacologically active small molecules as well as genetic or biochemical modifications of signaling molecules, we determined that Nanog gene repression was primarily mediated by the Grb2/Mek pathway.
MATERIALS AND METHODS
ES cell culture.
The following ES cell lines were used in this study. Afp-GFP ES cells and TRE-Nanog ES cells were previously described (13). Nanog β-geo ES cells were kindly gifted from S. Yamanaka (23). Grb2 null ES cells were kindly gifted from T. Pawson (9). All ES cells were maintained in an undifferentiated state on gelatin-coated dishes in Knock-out Dulbecco's modified Eagle medium (GIBCO BRL, Grand Island, NY) containing 10% knockout serum replacement (GIBCO BRL), 1% fetal bovine serum (Atlanta Biologicals, Norcross, GA), 2 mM l-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 25 mM HEPES (GIBCO BRL), 300 μM monothioglycerol (Sigma, St. Louis, MO), and 1,000 U/ml recombinant mouse LIF (ESGRO) (Chemicon, Temecula, CA). Aggregation of ES cells was induced by making hanging drops (2,000 cells/drop) in LIF-containing medium. The following reagents were used in this study: sodium orthovanadate (S-6508), PD98059 (P-215), doxycycline, FGF1 (F-5542), and FGF2 (F0291) (Sigma); and LY294002 (440204), SP600125 (420128), and SU5402 (572630) (Calbiochem, La Jolla, CA).
Flow cytometry analysis.
A single-cell suspension was prepared from aggregates of ES cells by treating them with 0.05% trypsin (GIBCO-BRL). Dissociated cells were separated by FACS Sort (Becton Dickinson, Franklin Lakes, NJ). Data analysis was performed by using CellQuest Acquisition software (Becton Dickinson).
Whole-mount X-Gal staining.
Cells were fixed in solution containing 2% formaldehyde, 0.2% glutaraldehyde, 5 mM EGTA, 2 mM MgCl2, and 0.02% NP-40 for 10 min at room temperature. After washing twice with phosphate-buffered saline (PBS) containing 0.1% Tween 20 (PBST), the samples were stained in 5-bromo-4-chloro-3-indolyl-d-galactoside (X-Gal) mix [containing 5 mM K3Fe(CN)6, 5 mM K4Fe(CN)6, 2 mM MgCl2, 10% dimethyl sulfoxide, 0.01% sodium deoxycholate, 0.02% NP-40, and 1 mg/ml X-Gal] for 8 to 12 h at 37°C and then rinsed in PBST. Poststain fix was performed in 3.7% formaldehyde at 4°C overnight and followed by paraffin embedding. Sections (7 μm) were counterstained by eosin Y.
Plasmid construction and transfection.
The Grb2 cDNA was PCR amplified by LA-Taq polymerase (TAKARA Bio, Otsu, Japan) using cDNAs prepared from wild-type ES cells as a template. Primer sequences, which contain BamHI and NheI restriction sites, are the following: sense, 5′-CGGGATCATGGAAGCCATCGCCAAA; antisense, 5′-CTAGCTAGCTTAGACGTTCCGGTTCACTG. The cDNA fragment was cloned into pCAG-IRES-hygromycin vector. The resultant vector was transfected into Grb2 null ES cells using Fugene 6 (Roche, Indianapolis, IN), and colonies constitutively expressing Grb2 were selected by hygromycin B (100 mg/ml) (Invitrogen, Carlsbad, CA). Plasmids containing the Mek1 constitutively active mutant (ΔN3 S218E, S222D) and the kinase-dead mutant (K97M) were kindly provided by N. Ahn (20). The coding region was shuttled into NotI and MluI sites of pCAG-hygromycin vector. The expression vectors were transfected by Fugene 6 (Roche), and stable colonies were selected by hygromycin B (100 μg/ml) (Invitrogen, Carlsbad, CA).
Reverse transcription-PCR (RT-PCR).
Total RNA was extracted by using an RNA aqueous kit (Ambion Inc., Austin, TX). The cDNA was synthesized by using a SuperScript II first-strand synthesis system with oligo(dT) (GIBCO-BRL). PCR was performed by using Taq DNA polymerase (Eppendorf, Westbury, NY). For each gene, the DNA primers were originated from different exons to ensure that the PCR product represents the specific mRNA species and not genomic DNA (primer sequences are available on request).
Western blotting.
Cells were lysed in radioimmunoprecipitation assay buffer, and 10 μg of total proteins was separated with sodium dodecyl sulfate-10% polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. The following were used as primary antibodies: Nanog, AB5731 (Chemicon); Oct3/4, C-10; Sox2, H-65; and phosphotyrosine, PY20 (Santa Cruz, Santa Cruz, CA); phospho-Erk1/2, E10 (#9106); phospho-Akt, #9271; and phospho-SAPK/Jnk, #9251 (Cell signaling, Beverly, MA). Peroxidase-conjugated immunoglobulin G (Santa Cruz) was used as the secondary antibody, followed by enhanced chemiluminescence (ECL) detection (Amersham, Piscataway, NJ).
RESULTS
Increased protein tyrosine phosphorylation effectively induced primitive endoderm differentiation and Nanog gene repression.
When murine ES cells are aggregated, only the outermost layer will differentiate into primitive endoderm (25, 31). This cell fate specification occurs regardless of the presence of LIF or Bmp4 (13). Although FGFR activation has been shown to be essential for primitive endoderm differentiation, it is not likely sufficient for the initial cell fate specification. Simple addition of FGF1 or FGF2 to the culture medium during ES cell aggregation does not increase the efficiency of differentiation towards primitive endoderm (data not shown). Thus, we postulated that additional signaling factors were attenuating the effects of the FGF-FGFR endoderm-promoting interaction in the inner cells of ES cell aggregates. Activation of receptor-type tyrosine kinases, including FGF receptors, is negatively regulated by various tyrosine phosphatases, which act to attenuate growth factor stimulations (27, 40). In order to block the feedback loop of protein tyrosine phosphorylation, we added sodium vanadate to our cell culture. Sodium vanadate inhibits a broad range of protein tyrosine phosphatases (14).
To monitor primitive endoderm differentiation, we used transgenic ES cells carrying a green fluorescent protein (GFP) reporter under the control of the α-fetoprotein promoter (Afp-GFP ES cells) (13). As we previously reported, when these ES cells were aggregated, only the outermost layer became GFP positive. In contrast, within 48 h after sodium vanadate treatment, GFP-positive cells appeared in the inner core of the aggregates in addition to the outer layer (Fig. 1A). The percentage of total GFP-positive cells in the aggregates was evaluated by flow cytometry analysis after the cells were dissociated into a suspension of single cells. Without sodium vanadate treatment, GFP-positive cells appeared only in the outermost layer and accounted for 2.2% (±0.2% [standard deviation]) of the total cells, whereas in sodium vanadate-treated cells, the percentage of GFP-positive cells reached a high of 86.7% (±1.9% standard deviations) of the population when the maximum dose (100 μM) was added (Fig. 1B). Figure 1C demonstrates that sodium vanadate increases protein tyrosine phosphorylation in a dose-dependent manner.
FIG. 1.

Increases in tyrosine phosphorylation induced primitive endoderm differentiation. (A) Afp-GFP ES cells were treated with sodium vanadate at the indicated concentrations for 48 h in the presence of LIF. Phase-contrast images are shown in the upper panels, and corresponding live fluorescence images are shown in the middle panels. Bars, 200 μm. The ES cells were dissociated with trypsin and analyzed by flow cytometry, which is shown in the lower panels. (B) Representative dose-response curve of sodium vanadate on primitive endoderm (PrE) differentiation. Primitive endoderm cells were estimated based on GFP expression in Afp-GFP ES cells by flow cytometry analysis. (C) Western blotting against antiphosphotyrosine (pTyr) antibody and anti-Nanog antibody. Protein tyrosine phosphorylation increased upon sodium vanadate treatment in Afp-GFP ES cells, whereas Nanog protein decreased. (D) Downregulation of Nanog expression in ES cells upon sodium vanadate treatment. Nanog β-geo ES cells were treated with the indicated concentrations of sodium vanadate for 48 h, and whole-mount X-Gal staining (blue) was performed. The paraffin sections were counterstained with eosin Y (pink). Bars, 100 μm.
We also analyzed the effects of sodium vanadate on Nanog transcription. Here we used ES cells in which the β-galactosidase gene was knocked in under the control of the endogenous Nanog gene promoter locus (Nanog β-geo ES cells) (23). As we previously reported, β-galactosidase activity was downregulated upon ES cell aggregation in the outermost layer of the aggregates but remained largely active at the inner core in the presence of LIF (13). When we enhanced protein tyrosine phosphorylation by adding sodium vanadate to the culture, β-galactosidase activity was downregulated, even in the inner core of the aggregates, within 48 h (Fig. 1D). Upon sodium vanadate treatment, the inside cells in the aggregates were also enlarged, resembling the primitive endoderm cells that are usually seen in the outermost layer of the ES cell aggregates. The decrease in Nanog expression resulting from the addition of sodium vanadate was also confirmed by immunoblotting (Fig. 1C). These data suggest that protein tyrosine phosphorylation in ES cells promotes primitive endoderm differentiation accompanied by Nanog gene repression.
Effects of sodium vanadate on gene expression of pluripotency and differentiation markers: time course and dose responses.
To further explore the mechanisms by which sodium vanadate represses Nanog, we examined mRNA expression levels of Nanog and other pluripotency-related genes as well as primitive endoderm marker genes within 32 h after sodium vanadate treatment. As shown in Fig. 2A, Nanog mRNA levels significantly decreased within 24 h after sodium vanadate treatment (25 μM). Previously, putative downstream target genes of Nanog were reported based on a predicted Nanog binding consensus sequence (23). Zfp42/Rex1, which is suggested to be positively regulated by Nanog, was also downregulated upon sodium vanadate treatment. In contrast, Gata6, which is suggested to be negatively regulated by Nanog, was upregulated at the same time point that Nanog mRNA decreased. Other primitive endoderm marker genes such as Gata4 and Bmp2 mRNA increased following Gata6 expression (Fig. 2A).
FIG. 2.

(A) Changes of pluripotency and differentiation marker gene expression in ES cells during sodium vanadate treatment. Total RNA was isolated from Nanog β-geo ES cells after being treated with 25 μM sodium vanadate for the indicated periods of time (hours) and was subjected to RT-PCR analysis. (B) Sodium vanadate treatment of ES cells with or without exogenous Nanog expression. TRE-Nanog ES cells contain an inducible Nanog transgene under a tet-off gene induction system (13). The cells were treated with 25 μM sodium vanadate in the presence or absence of doxycycline (1 μg/ml) (DOX) for 24 h and subjected to RT-PCR analysis. The expression of endogenous Nanog mRNA decreased upon sodium vanadate treatment regardless of Nanog transgene expression, whereas the expression of Zfp42 and Gata6 mRNA was not changed by the addition of sodium vanadate when the Nanog transgene was expressed. (C) Protein levels of Nanog, Oct3/4, and Sox2 after sodium vanadate treatment. Nanog β-geo ES cells were treated with sodium vanadate for 48 h at the indicated concentrations and subjected to Western blotting. Only the Nanog protein decreased within 48 h after treatment.
To distinguish whether the changes in Zfp42 and Gata6 expression were the consequence of a loss of Nanog expression or a result of sodium vanadate addition independent of Nanog expression, we overexpressed Nanog exogenously during sodium vanadate treatment. Previously, we generated transgenic ES cells, TRE-Nanog ES cells, through which Nanog could be overexpressed by a tetracycline-inducible vector (13). TRE-Nanog ES cells can be maintained in an undifferentiated state in the absence of LIF. Using these ES cells, we examined mRNA levels by RT-PCR after 24 h of sodium vanadate treatment (25 μM). As shown in Fig. 2B, Gata6 and Zfp42 expression did not change upon sodium vanadate treatment when exogenous Nanog was present. In contrast, endogenous Nanog gene expression decreased upon sodium vanadate treatment even in the presence of exogenous Nanog expression. To exclude the possibility of a clonal artifact, the same TRE-Nanog ES cells were also maintained in a LIF-containing medium in the presence of doxycycline (1 μg/ml). The addition of doxycycline reduced expression of the Nanog transgene to an undetectable level. In this condition, the expression of Zfp42 as well as endogenous Nanog was downregulated by sodium vanadate treatment, whereas that of Gata6 was upregulated.
Oct3/4 and Sox2 are critical transcription factors that maintain the pluripotency of ES cells (3). Increasing evidence suggests that both factors regulate Nanog gene transcription by binding to the proximal promoter region of the Nanog gene (3, 16, 30). In our study, the mRNA levels of the Oct3/4 gene were not affected by sodium vanadate treatment throughout the time course (Fig. 2A). Sox2 mRNA started decreasing at a later time point (32 h) after Nanog was already repressed (Fig. 2A). Furthermore, the protein expression levels of Oct3/4 and Sox2 did not decrease within 48 h after treatment, whereas Nanog protein significantly decreased at this time point (Fig. 2C). These data indicate that sodium vanadate primarily suppresses Nanog expression among pluripotency-related genes and that Nanog repression is not likely a consequence of a loss of Oct3/4 and Sox2 expression in ES cells.
Grb2 deficiency abrogated the effects of sodium vanadate on Nanog repression.
Grb2 has an SH2 domain that specifically binds to a peptide motif containing a phosphotyrosine. Thus, it works as an adaptor protein, linking the signals received by receptor tyrosine kinases, such as FGF receptors, to their downstream targets. Grb2 is also known to be essential to the formation of the primitive endoderm layer both in vivo in the ICM of a blastocyst stage embryo and in vitro during ES cell aggregation (9). We therefore examined whether Grb2 was involved in sodium vanadate-induced Nanog repression. We treated both wild-type and Grb2 null ES cells with sodium vanadate (25 to 100 μM) and compared their morphological changes and gene expression patterns by RT-PCR. In wild-type ES cells, the cells were enlarged and dissociated in suspension culture. In contrast to wild-type ES cells, there were no obvious morphological changes in Grb2 null ES cells treated with sodium vanadate (Fig. 3A). In addition, Nanog expression was not affected by sodium vanadate in Grb2 null ES cells, and there was no sign of differentiation towards the primitive endoderm lineage. In wild-type ES cells, Nanog and Zfp42 genes were repressed, while Gata6, along with other primitive endoderm marker genes, was upregulated upon sodium vanadate treatment (Fig. 3B). To further confirm the role of Grb2 in sodium vanadate-mediated Nanog repression, we added back a Grb2 expression vector into the Grb2 null ES cells and established stable clones. In the Grb2 add-back ES cells, Nanog and Zfp42 genes were now repressed and Gata6 was upregulated upon sodium vanadate treatment (Fig. 3C). These data indicate that sodium vanadate-induced Nanog repression and primitive endoderm differentiation are mediated through Grb2.
FIG. 3.

Grb2 deficiency abrogated the effects of sodium vanadate on Nanog repression. (A) Wild-type and Grb2 null ES cells were in suspension culture with or without sodium vanadate (50 μM) for 48 h. Live phase-contrast images were shown. Bars, 200 μm. (B) RT-PCR analysis of gene expression in wild-type and Grb2 null ES cells. Total RNA was prepared after 48 h of sodium vanadate treatment at the indicated concentrations. (C) RT-PCR analysis of gene expression in Grb2 null ES cells and Grb2 addback-Grb2 null ES cells treated with or without sodium vanadate (50 μM) for 48 h.
Inhibition of Mek blocked the effects of sodium vanadate on Nanog repression.
Increased tyrosine phosphorylation by sodium vanadate could potentially trigger the activation of multiple downstream signaling pathways. To investigate other downstream signaling pathways for their potential role in primitive endoderm lineage specification, we examined the phosphorylation status of Erk, Akt, and Jnk. Using various models, it has previously been suggested that these three kinases are involved in primitive endoderm differentiation (8, 15, 33, 34). As shown in Fig. 4A, all of these signaling molecules (Erk1/2, Akt, and Jnk) were phosphorylated upon sodium vanadate treatment.
FIG. 4.
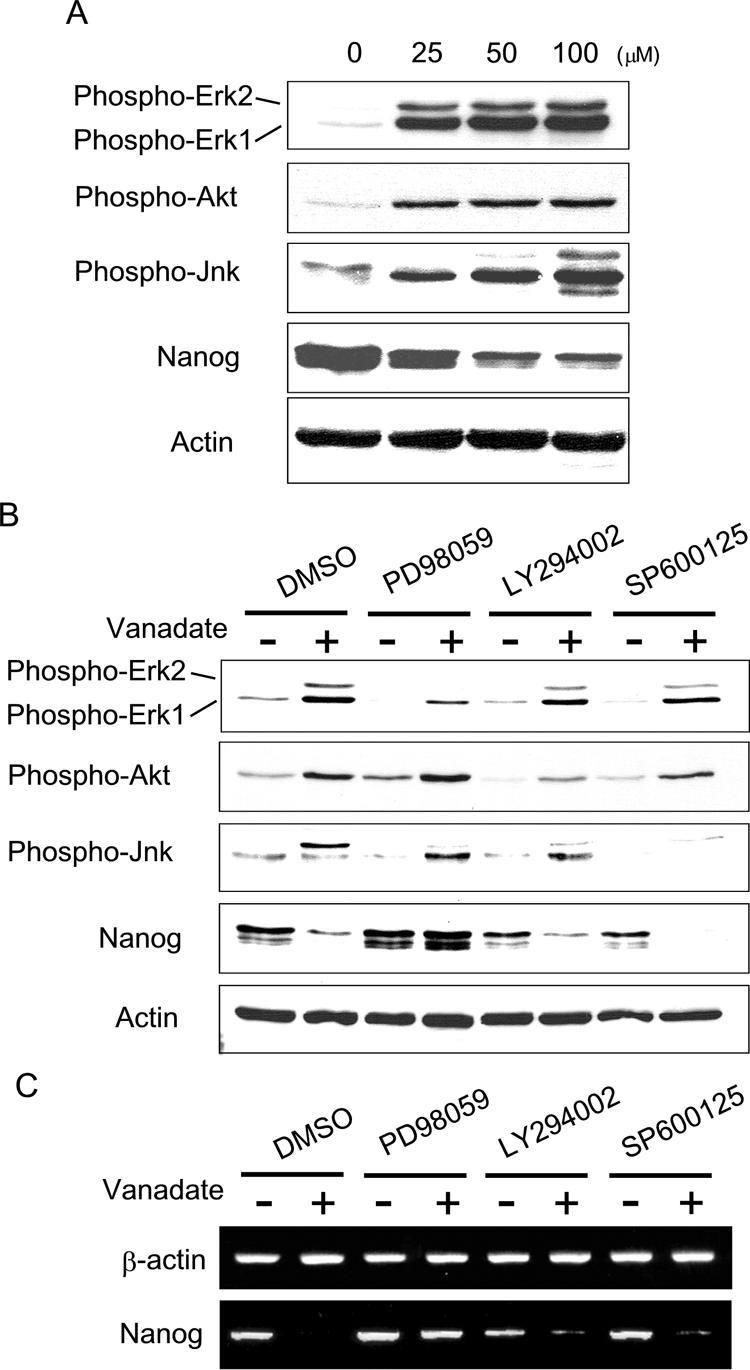
Inhibition of Mek blocked the effects of sodium vanadate on Nanog repression. (A) Western blotting using phosphospecific antibodies against Erk, Akt, or Jnk. Nanog β-geo ES cells were treated with sodium vanadate for 48 h at the indicated concentrations. (B) The effect of specific kinase inhibitors on sodium vanadate-induced Nanog repression. Western blotting was performed after 48 h of treatment with or without sodium vanadate (25 μM) and/or kinase inhibitors, as indicated. (C) RT-PCR analysis was performed after 48 h of treatment with or without sodium vanadate (25 μM) and/or kinase inhibitors, as indicated. DMSO, dimethyl sulfoxide.
To determine which signaling pathway is responsible for Nanog repression, we examined the effects of kinase-specific inhibitors on sodium vanadate-induced Nanog repression. Nanog β-geo ES cells were treated with sodium vanadate in the presence of the Mek inhibitor (PD98059), the PI3K inhibitor (LY294002), or the Jnk inhibitor (SP600125) for 48 h. As shown in Fig. 4B, PD98059 specifically inhibited phosphorylation of Erk1/2, whereas LY294002 and SP600125 inhibited phosphorylation of Akt and Jnk, respectively. We found that only PD98059 blocked sodium vanadate-induced Nanog repression (Fig. 4B and C). We also confirmed that PD98059 blocked the effects of sodium vanadate on Nanog by whole-mount X-Gal staining of Nanog β-geo ES cell aggregates (data not shown). These data suggest that Mek activation is responsible for sodium vanadate-induced Nanog repression.
Cell aggregation-induced Nanog repression was also blocked by Mek inhibition.
We next examined whether Mek activation was required to repress Nanog during ES cell aggregation. Nanog β-geo ES cells were cultured in suspension and were supplemented with a Mek inhibitor (PD98059). While the ES cells in the vehicle control (Fig. 5A) repressed Nanog in the outermost layer of the aggregates, the ES cell aggregates cultured in the presence of a Mek inhibitor formed no such layer (Fig. 5B). These data indicate that Mek activity is required to repress Nanog during ES cell aggregation.
FIG. 5.

FGFR-dependent Mek activation repressed the Nanog gene during ES cell aggregation. Whole-mount X-Gal staining (blue) of Nanog β-geo ES cell aggregates treated with (A) dimethyl sulfoxide, (B) Mek inhibitor (PD98059; 25 μM), (C) PI3K inhibitor (LY294002; 20 μM), (D) Jnk inhibitor (SP600125; 20 μM), or (E) FGFR-specific inhibitor (SU5402; 20 μM). The paraffin sections were counterstained with eosin Y (pink). Bars, 100 μm.
The PI3K inhibitor LY294002 has been shown to block primitive endoderm differentiation in ES cells (8). We tested the involvement of PI3K in Nanog repression during ES cell aggregation and found that Nanog gene repression in the outermost layer was not restored when PI3K was inhibited (Fig. 5C). Rather, Nanog repression was enhanced within the aggregates after the addition of LY294002. This indicates that PI3K activity is probably not essential to Nanog repression during ES cell aggregation. Nanog repression was also enhanced within the aggregates after the addition of a Jnk inhibitor (SP600125) (Fig. 5D).
The phenotypes resulting from either a targeted disruption of FGFR or from introducing a dominant-negative FGFR into ES cells have revealed the role of FGF signaling in primitive endoderm formation (2, 8, 11). We blocked the kinase activity of FGFR using SU5402, which binds to the ATP binding site of FGFR (24), and found that our results were similar to those obtained when we used a Mek inhibitor, in that Nanog was not repressed in the outermost layer (Fig. 5E). These data imply the activation of Mek during ES cell aggregation is mediated through the FGF receptor.
Of interest, addition of SU5402 increased Nanog expression even when ES cells were maintained on gelatin-coated dishes. When undifferentiated Nanog β-geo ES cells were maintained on gelatin-coated culture dishes in the presence of LIF, ES cells with reduced Nanog expression were detected sparsely using X-Gal staining (Fig. 6A, control). In order to further examine the roles of FGF and FGFR in Nanog repression, we added SU5402 to the maintenance culture of Nanog β-geo ES cells for 48 h. The SU5402-treated ES cells were more homogeneously stained with X-Gal. In addition, SU5402 treatment increased Nanog mRNA expression by approximately 1.6-fold (Fig. 6). Addition of FGF1 or FGF2 into the culture medium did not significantly alter Nanog expression in ES cells.
FIG. 6.

Inhibition of the FGFR kinase activity increased Nanog expression in ES cell maintenance culture. (A) X-Gal staining of Nanog β-geo ES cells cultured on gelatin-coated dishes in the presence of LIF. Cells were cultured with recombinant FGF1 (100 ng/ml), FGF2 (100 ng/ml), or SU5402 (20 μM) for 48 h and stained with X-Gal (blue). Phase-contrast images were captured. Bars, 100 μm. (B) RT-PCR analysis for Nanog mRNA. The ethidium bromide-stained bands were quantitated by computer-assisted densitometry (Image J).
Constitutive activation of Mek induced primitive endoderm differentiation and repressed Nanog transcription.
To confirm that Mek activation represses Nanog, we transfected ES cells with a constitutively active Mek1 mutant (ΔN3 S218E, S222D) (20). As shown in Fig. 7A, constitutive activation of Mek1 resulted in primitive endoderm differentiation of ES cells, and this was visualized by monitoring GFP expression in Afp-GFP ES cells. The enzymatic activity of this constitutive active mutant of Mek is known to be inhibited by PD98059 (1). The endoderm promoting effects of the active Mek mutant were then abrogated in the presence of PD98059. Moreover, the Mek 1 kinase-dead mutant, K97M (20), did not promote primitive endoderm differentiation. Using the same Mek mutants, we also monitored Nanog gene expression by X-Gal staining of Nanog β-geo ES cells (Fig. 7B). We confirmed that constitutive activation of Mek 1 leads to Nanog repression in ES cells.
FIG. 7.

Constitutive activation of Mek induced primitive endoderm differentiation and repressed Nanog transcription. (A) Afp-GFP ES cells were transfected with Mek mutants with or without PD98059 (25 μM) and selected with hygromycin (100 μg/ml) for 10 days. Images of representative colonies were shown using phase contrast (upper panel) and GFP filters (lower panel). Bars, 100 μm. (B) Nanog β-geo ES cells transfected with Mek mutants with or without PD98059 (25 μM) and selected with hygromycin (100 μg/ml) for 10 days. Phase-contrast images were captured after X-Gal staining (blue). Bars, 100 μm.
DISCUSSION
Although various homeoproteins, such as Nanog, have been found to play a role in both maintaining ES cell pluripotency and directing lineage-specific differentiation events, little is known about the external cues and signal transduction networks that govern them. Protein phosphorylation is a key event in the transmission of a signal from a cell surface receptor on down to its genetic targets. Such a process involves a complex network of feedback loops and a cross talk between multiple signaling pathways. In the present study, our use of pharmacological and molecular tools to modify the activities of protein kinases, phosphatases, and adaptor molecules has led us to the conclusion that the Grb2/Mek pathway mediates the repression of Nanog and the specification of primitive endoderm cell fate (Fig. 8).
FIG. 8.

Schematic summary of the signaling pathway leading to Nanog repression. Activation of the Grb2/Mek pathway induced by sodium vanadate or cell aggregation specifically represses the Nanog gene, resulting in primitive endoderm differentiation of ES cells. PTP, protein tyrosine phosphatase; Na3VO4, sodium vanadate.
Out of all of the genes examined in this study, the tyrosine phosphatase inhibitor, sodium vanadate, primarily repressed Nanog gene expression. Although the expressions of Zfp42 and Gata6 were also altered by the addition of sodium vanadate, these changes were eliminated by the expression of exogenous Nanog (Fig. 2B). This suggests that Nanog repression is required to alter the expression of these genes, thereby supporting the idea that these two genes truly are downstream targets of Nanog. Of interest, in the presence of exogenously overexpressed Nanog, sodium vanadate still repressed endogenous Nanog despite the fact that Gata6 expression was blocked. Taken together, these data strongly suggest that Nanog repression is the primary mechanism by which sodium vanadate induces primitive endoderm formation. The data also indicate that, although Nanog is speculated to positively autoregulate its own transcription (3), exogenous Nanog expression by itself is not sufficient to prevent sodium vanadate-induced repression of endogenous Nanog (Fig. 2B). Increasing evidence suggests that Oct3/4 and Sox2 regulate Nanog gene transcription by binding to the proximal promoter region of the Nanog gene (3, 16, 30). When we examined the kinetics of gene expression upon sodium vanadate treatment, it was found that mRNA and protein levels of Nanog decreased before there were any detectable changes in expression levels of Oct3/4 and Sox2 genes (Fig. 2A and C). These data indicate that the Nanog repression induced by sodium vanadate was not likely due to the loss of Oct3/4 and Sox2 expression. It should be noted that sodium vanadate did not alter the phosphorylation status of Oct3/4 and Sox2 (data not shown). Other transactivators or repressors, such as germ cell nuclear factor, putatively controlling Nanog gene transcription may instead be responsible. However, germ cell nuclear factor may not explain this selective Nanog repression, because it is known to repress both Nanog and Oct3/4 genes during retinoic acid-induced ES cell differentiation (12). Alternatively, Sp1, a direct Erk1/2 target, could be responsible for this Nanog-specific regulation, given that Sp1 binding sites were recently mapped on the Nanog gene promoter (37).
The effects of sodium vanadate on Nanog repression and primitive endoderm differentiation were completely abrogated by Grb2 deficiency (Fig. 3). This clearly demonstrates that the sodium vanadate-induced tyrosine phosphorylation signal to repress Nanog is transmitted via Grb2. Grb2 is an adaptor molecule with an SH2 domain that specifically binds to a peptide motif containing a phosphotyrosine. This motif links Grb2 to downstream signaling cascades, in particular to the Sos/Ras/Raf/Mek/Erk pathway (6, 29). Among the various kinase inhibitors tested, only the Mek inhibitor selectively blocked the effects of sodium vanadate on Nanog repression (Fig. 4B and C). Moreover, transfection of a constitutively active form of Mek mutant repressed Nanog and led to primitive endoderm differentiation (Fig. 7). These data indicate that tyrosine phosphorylation induces Nanog repression through the Grb2/Sos/Ras/Raf/Mek/Erk pathway. During preparation of this report, Chazaud et al. demonstrated that Grb2 deficiency abrogated Nanog repression in the ICM of blastocysts in vivo (7).
In the present study, we demonstrated that the addition of a PI3K inhibitor was not able to block sodium vanadate-induced Nanog repression (Fig. 4B and C). Further, when ES cells were aggregated in the presence of the PI3K inhibitor, Nanog repression still occurred in the outermost layer of the aggregates (Fig. 5C). These data indicate that Nanog repression is not mediated by the PI3K-Akt pathway. However, the data did not exclude the role of the PI3K-Akt pathway in primitive endoderm differentiation. Indeed, when the PI3K inhibitor was added to the aggregates of Afp-GFP ES cells in conjunction with sodium vanadate, there was a partial inhibition of GFP-positive primitive endoderm cells compared to that of cells treated only with sodium vanadate (data not shown). A previous study showed that a constitutively active Akt enhanced the synthesis of laminin and collagen IV isotypes and promoted basement membrane formation (19). Collectively, Mek/Erk activation may specifically repress Nanog gene expression and initiate primitive endoderm differentiation, whereas PI3K/Akt activation may induce the production of extracellular matrix proteins to promote further primitive endoderm differentiation.
The previous studies indicate that the FGF-FGFR interaction is pivotal for primitive endoderm specification (2, 11, 17, 18). Indeed, addition of the FGFR inhibitor (SU5402) to ES cell aggregates almost completely blocked Nanog repression (Fig. 5). Further, addition of SU5402 increased Nanog expression even when ES cells were maintained on gelatin-coated dishes (Fig. 6). The SU5402-treated ES cells were more homogeneously positive for Nanog expression. Our data imply that the signals mediated through FGFR impose a continuous pressure on ES cells to repress Nanog, even when ES cells are maintained on gelatin-coated dishes in the presence of LIF. Although FGFR1 is expressed in undifferentiated ES cells (data not shown) (22), addition of FGF1 or FGF2 into the ES cell culture did not induce Nanog repression (Fig. 6). Moreover, mouse ES cells are also known to express FGF4 (36). This implies that a molecular mechanism exists in ES cells that attenuates the FGFR/Grb2/Mek signaling pathway while maintaining Nanog expression. Alternatively, a differential expression of other FGFRs in a subpopulation of ES cells may trigger a more distinct activation of Grb2/Mek, thereby resulting in Nanog repression. Elucidating such signals will be a necessary task in order to reveal the entire signaling cascade controlling Nanog repression.
The pluripotency of murine ES cells is dependent upon LIF-induced STAT3 activity when they are cultured on gelatin-coated dishes (21, 26, 32). Although LIF also activates the Mek/Erk pathway, this Mek/Erk activation is not essential for ES cell propagation. Moreover, inhibition of Mek by PD98059 rather enhanced the growth of undifferentiated ES cells (4). The present study demonstrated that PD98059 prevented Nanog repression, which explains, at least in part, how the drug supported ES cell self renewal in the previous study. In addition, another study has shown that the PI3K-Akt pathway played a pivotal role in suppressing Erk1/2 activation during ES cell maintenance and that the addition of LY294002 in ES cells enhanced Erk1/2 activation and ES cell differentiation (28). Indeed, we observed that the addition of LY294002 repressed Nanog in the inner cells of ES cell aggregates in the presence of LIF (Fig. 5C). We did not, however, detect any elevation in Erk1/2 phosphorylation after the addition of LY294002 in our cell aggregation culture (Fig. 4B). It should be noted that introducing a myristoylated active form of Akt into ES cells was sufficient to maintain the culture of undifferentiated ES cells in the absence of LIF, and this event was not through the inhibition of Erk activity (35). Collectively, it is still unclear how the PI3K-Akt pathway facilitates the self renewal of ES cells.
The present study elucidated a signaling pathway that specifically led to Nanog gene repression in ES cells. Modifying this signaling pathway by sodium vanadate or an active Mek mutant led ES cells to differentiate into primitive endoderm with high efficiency. By further dissecting the intracellular signaling pathways governing ES cell self renewal and differentiation, we will be able to develop novel methods to direct ES cell fate, making ES cells a promising source for future cell-based therapies.
Acknowledgments
We thank T. Pawson, N. Ahn, L. Heasley, and S. Yamanaka for providing valuable materials.
This work was supported in part by National Institutes of Health grant DK59699 (to N.T.).
Footnotes
Published ahead of print on 14 August 2006.
REFERENCES
- 1.Ahn, N. G., T. S. Nahreini, N. S. Tolwinski, and K. A. Resing. 2001. Pharmacologic inhibitors of MKK1 and MKK2. Methods Enzymol. 332:417-431. [DOI] [PubMed] [Google Scholar]
- 2.Arman, E., R. Haffner-Krausz, Y. Chen, J. K. Heath, and P. Lonai. 1998. Targeted disruption of fibroblast growth factor (FGF) receptor 2 suggests a role for FGF signaling in pregastrulation mammalian development. Proc. Natl. Acad. Sci. USA 95:5082-5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyer, L. A., T. I. Lee, M. F. Cole, S. E. Johnstone, S. S. Levine, J. P. Zucker, M. G. Guenther, R. M. Kumar, H. L. Murray, R. G. Jenner, D. K. Gifford, D. A. Melton, R. Jaenisch, and R. A. Young. 2005. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122:947-956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burdon, T., C. Stracey, I. Chambers, J. Nichols, and A. Smith. 1999. Suppression of SHP-2 and ERK signalling promotes self-renewal of mouse embryonic stem cells. Dev. Biol. 210:30-43. [DOI] [PubMed] [Google Scholar]
- 5.Chambers, I., D. Colby, M. Robertson, J. Nichols, S. Lee, S. Tweedie, and A. Smith. 2003. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell 113:643-655. [DOI] [PubMed] [Google Scholar]
- 6.Chardin, P., J. H. Camonis, N. W. Gale, L. van Aelst, J. Schlessinger, M. H. Wigler, and D. Bar-Sagi. 1993. Human Sos1: a guanine nucleotide exchange factor for Ras that binds to GRB2. Science 260:1338-1343. [DOI] [PubMed] [Google Scholar]
- 7.Chazaud, C., Y. Yamanaka, T. Pawson, and J. Rossant. 2006. Early lineage segregation between epiblast and primitive endoderm in mouse blastocysts through the Grb2-MAPK pathway. Dev. Cell 10:615-624. [DOI] [PubMed] [Google Scholar]
- 8.Chen, Y., X. Li, V. P. Eswarakumar, R. Seger, and P. Lonai. 2000. Fibroblast growth factor (FGF) signaling through PI 3-kinase and Akt/PKB is required for embryoid body differentiation. Oncogene 19:3750-3756. [DOI] [PubMed] [Google Scholar]
- 9.Cheng, A. M., T. M. Saxton, R. Sakai, S. Kulkarni, G. Mbamalu, W. Vogel, C. G. Tortorice, R. D. Cardiff, J. C. Cross, W. J. Muller, and T. Pawson. 1998. Mammalian Grb2 regulates multiple steps in embryonic development and malignant transformation. Cell 95:793-803. [DOI] [PubMed] [Google Scholar]
- 10.Coucouvanis, E., and G. R. Martin. 1995. Signals for death and survival: a two-step mechanism for cavitation in the vertebrate embryo. Cell 83:279-287. [DOI] [PubMed] [Google Scholar]
- 11.Esner, M., J. Pachernik, A. Hampl, and P. Dvorak. 2002. Targeted disruption of fibroblast growth factor receptor-1 blocks maturation of visceral endoderm and cavitation in mouse embryoid bodies. Int. J. Dev. Biol. 46:817-825. [PubMed] [Google Scholar]
- 12.Gu, P., D. LeMenuet, A. C. Chung, M. Mancini, D. A. Wheeler, and A. J. Cooney. 2005. Orphan nuclear receptor GCNF is required for the repression of pluripotency genes during retinoic acid-induced embryonic stem cell differentiation. Mol. Cell. Biol. 25:8507-8519. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 13.Hamazaki, T., M. Oka, S. Yamanaka, and N. Terada. 2004. Aggregation of embryonic stem cells induces Nanog repression and primitive endoderm differentiation. J. Cell Sci. 117:5681-5686. [DOI] [PubMed] [Google Scholar]
- 14.Huyer, G., S. Liu, J. Kelly, J. Moffat, P. Payette, B. Kennedy, G. Tsaprailis, M. J. Gresser, and C. Ramachandran. 1997. Mechanism of inhibition of protein-tyrosine phosphatases by vanadate and pervanadate. J. Biol. Chem. 272:843-851. [DOI] [PubMed] [Google Scholar]
- 15.Kanungo, J., I. Potapova, C. C. Malbon, and H. Wang. 2000. MEKK4 mediates differentiation in response to retinoic acid via activation of c-Jun N-terminal kinase in rat embryonal carcinoma P19 cells. J. Biol. Chem. 275:24032-24039. [DOI] [PubMed] [Google Scholar]
- 16.Kuroda, T., M. Tada, H. Kubota, H. Kimura, S. Y. Hatano, H. Suemori, N. Nakatsuji, and T. Tada. 2005. Octamer and Sox elements are required for transcriptional cis regulation of Nanog gene expression. Mol. Cell. Biol. 25:2475-2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li, L., E. Arman, P. Ekblom, D. Edgar, P. Murray, and P. Lonai. 2004. Distinct GATA6- and laminin-dependent mechanisms regulate endodermal and ectodermal embryonic stem cell fates. Development 131:5277-5286. [DOI] [PubMed] [Google Scholar]
- 18.Li, X., Y. Chen, S. Scheele, E. Arman, R. Haffner-Krausz, P. Ekblom, and P. Lonai. 2001. Fibroblast growth factor signaling and basement membrane assembly are connected during epithelial morphogenesis of the embryoid body. J. Cell Biol. 153:811-822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li, X., U. Talts, J. F. Talts, E. Arman, P. Ekblom, and P. Lonai. 2001. Akt/PKB regulates laminin and collagen IV isotypes of the basement membrane. Proc. Natl. Acad. Sci. USA 98:14416-14421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mansour, S. J., W. T. Matten, A. S. Hermann, J. M. Candia, S. Rong, K. Fukasawa, G. F. Vande Woude, and N. G. Ahn. 1994. Transformation of mammalian cells by constitutively active MAP kinase kinase. Science 265:966-970. [DOI] [PubMed] [Google Scholar]
- 21.Matsuda, T., T. Nakamura, K. Nakao, T. Arai, M. Katsuki, T. Heike, and T. Yokota. 1999. STAT3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. EMBO J. 18:4261-4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDonald, F. J., and J. K. Heath. 1994. Developmentally regulated expression of fibroblast growth factor receptor genes and splice variants by murine embryonic stem and embryonal carcinoma cells. Dev. Genet. 15:148-154. [DOI] [PubMed] [Google Scholar]
- 23.Mitsui, K., Y. Tokuzawa, H. Itoh, K. Segawa, M. Murakami, K. Takahashi, M. Maruyama, M. Maeda, and S. Yamanaka. 2003. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell 113:631-642. [DOI] [PubMed] [Google Scholar]
- 24.Mohammadi, M., G. McMahon, L. Sun, C. Tang, P. Hirth, B. K. Yeh, S. R. Hubbard, and J. Schlessinger. 1997. Structures of the tyrosine kinase domain of fibroblast growth factor receptor in complex with inhibitors. Science 276:955-960. [DOI] [PubMed] [Google Scholar]
- 25.Murray, P., and D. Edgar. 2001. The regulation of embryonic stem cell differentiation by leukaemia inhibitory factor (LIF). Differentiation 68:227-234. [DOI] [PubMed] [Google Scholar]
- 26.Niwa, H., T. Burdon, I. Chambers, and A. Smith. 1998. Self-renewal of pluripotent embryonic stem cells is mediated via activation of STAT3. Genes Dev. 12:2048-2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ostman, A., and F. D. Bohmer. 2001. Regulation of receptor tyrosine kinase signaling by protein tyrosine phosphatases. Trends Cell Biol. 11:258-266. [DOI] [PubMed] [Google Scholar]
- 28.Paling, N. R., H. Wheadon, H. K. Bone, and M. J. Welham. 2004. Regulation of embryonic stem cell self-renewal by phosphoinositide 3-kinase-dependent signaling. J. Biol. Chem. 279:48063-48070. [DOI] [PubMed] [Google Scholar]
- 29.Pouyssegur, J., V. Volmat, and P. Lenormand. 2002. Fidelity and spatio-temporal control in MAP kinase (ERKs) signalling. Biochem. Pharmacol. 64:755-763. [DOI] [PubMed] [Google Scholar]
- 30.Rodda, D. J., J. L. Chew, L. H. Lim, Y. H. Loh, B. Wang, H. H. Ng, and P. Robson. 2005. Transcriptional regulation of nanog by OCT4 and SOX2. J. Biol. Chem. 280:24731-24737. [DOI] [PubMed] [Google Scholar]
- 31.Shen, M. M., and P. Leder. 1992. Leukemia inhibitory factor is expressed by the preimplantation uterus and selectively blocks primitive ectoderm formation in vitro. Proc. Natl. Acad. Sci. USA 89:8240-8244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smith, A. G., J. K. Heath, D. D. Donaldson, G. G. Wong, J. Moreau, M. Stahl, and D. Rogers. 1988. Inhibition of pluripotential embryonic stem cell differentiation by purified polypeptides. Nature 336:688-690. [DOI] [PubMed] [Google Scholar]
- 33.Verheijen, M. H., R. M. Wolthuis, J. L. Bos, and L. H. Defize. 1999. The Ras/Erk pathway induces primitive endoderm but prevents parietal endoderm differentiation of F9 embryonal carcinoma cells. J. Biol. Chem. 274:1487-1494. [DOI] [PubMed] [Google Scholar]
- 34.Wang, H. Y., J. Kanungo, and C. C. Malbon. 2002. Expression of Galpha 13 (Q226L) induces P19 stem cells to primitive endoderm via MEKK1, 2, or 4. J. Biol. Chem. 277:3530-3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Watanabe, S., H. Umehara, K. Murayama, M. Okabe, T. Kimura, and T. Nakano. 2006. Activation of Akt signaling is sufficient to maintain pluripotency in mouse and primate embryonic stem cells. Oncogene 25:2697-2707. [DOI] [PubMed]
- 36.Wilder, P. J., D. Kelly, K. Brigman, C. L. Peterson, T. Nowling, Q. S. Gao, R. D. McComb, M. R. Capecchi, and A. Rizzino. 1997. Inactivation of the FGF-4 gene in embryonic stem cells alters the growth and/or the survival of their early differentiated progeny. Dev. Biol. 192:614-629. [DOI] [PubMed] [Google Scholar]
- 37.Wu da, Y., and Z. Yao. 2006. Functional analysis of two Sp1/Sp3 binding sites in murine Nanog gene promoter. Cell Res. 16:319-322. [DOI] [PubMed] [Google Scholar]
- 38.Yamaguchi, S., H. Kimura, M. Tada, N. Nakatsuji, and T. Tada. 2005. Nanog expression in mouse germ cell development. Gene Expr. Patterns 5:639-646. [DOI] [PubMed] [Google Scholar]
- 39.Yoshida-Koide, U., T. Matsuda, K. Saikawa, Y. Nakanuma, T. Yokota, M. Asashima, and H. Koide. 2004. Involvement of Ras in extraembryonic endoderm differentiation of embryonic stem cells. Biochem. Biophys. Res. Commun. 313:475-481. [DOI] [PubMed] [Google Scholar]
- 40.Zhang, Z. Y., B. Zhou, and L. Xie. 2002. Modulation of protein kinase signaling by protein phosphatases and inhibitors. Pharmacol. Ther. 93:307-317. [DOI] [PubMed] [Google Scholar]