

Abstract
Human herpesvirus 8 (HHV-8, also called KSHV) is linked to the etiopathogenesis of Kaposi’s sarcoma (KS), multicentric Castleman’s disease (MCD), and primary effusion lymphoma (PEL). The universal presence of HHV-8 in early KS has not yet been shown. We used a mAb (LN53) against latent nuclear antigen-1 (LNA-1) of HHV-8 encoded by ORF73 to study the distribution of the cell types latently infected by HHV-8 in patch, plaque, and nodular KS, MCD, and PEL. In early KS, HHV-8 is present in <10% of cells forming the walls of ectatic vessels. In nodular KS, HHV-8 is present in cells surrounding slit-like vessels and in >90% of spindle cells, but not in normal vascular endothelium. In addition, HHV-8 colocalizes with vascular endothelial growth factor receptor-3 (VEGFR-3), a marker of lymphatic and precursor endothelium. In early KS lesions, VEGFR-3 is more extensively expressed than LNA-1, indicating that HHV-8 is not inducing the proliferation of VEGFR-3-positive endothelium directly. In MCD, HHV-8 is present in mantle zone large immunoblastic B cells. No staining for LNA-1 is seen in samples from multiple myeloma, prostate cancer, and angiosarcoma, supporting the absence of any etiological link between these diseases and HHV-8.
Human herpesvirus 8 [HHV-8 or Kaposi’s sarcoma (KS)-associated herpesvirus] is causally linked to KS, multicentric Castleman’s disease (MCD), and primary effusion lymphoma (PEL) (1, 2). PCR analysis shows that HHV-8 is present in all epidemiologic types of KS (i.e., AIDS-associated KS, classic KS, endemic KS, and posttransplant KS) (1, 2). By various in situ hybridization techniques (including PCR in situ and RNA in situ hybridization), HHV-8 DNA is present in most of the tumor cells, so-called spindle cells, in KS lesions (3–8). In situ hybridization and in situ lysis Gardella gels indicate that most cells in KS are latently infected (6, 9), although lytic infection is clearly also present in a small proportion of cells, as demonstrated by in situ hybridization (6, 10), immunohistochemistry (11), and electron microscopy (12). The cell type(s) and percentage of cells infected in early lesions (i.e., patch and plaque) have not been studied systematically.
Besides KS, HHV-8 is also causally linked to MCD and the rare PEL (13, 14). HHV-8 is present in 100% of HIV-positive MCD cases and nearly 50% of HIV-negative cases (13, 15–17). The cell type(s) in MCD infected by HHV-8 is unclear (16, 17). PEL is a rare effusion-based lymphoma (18), although limited solid-tumor formation has been demonstrated (19–21). HIV-positive PEL cases appear to harbor HHV-8 and Epstein—Barr virus (EBV), whereas HIV-negative cases harbor only HHV-8 (14, 22–24). Both types lack c-myc rearrangements, and cell lines from both types have been established (25–28). In PEL cells from effusions and in cell lines established from them, HHV-8 episomes in the presence or absence of viral particles are present in every cell (25–29).
The role of HHV-8, if any, in the pathogenesis of other tumors, including multiple myeloma and prostate cancer remains controversial (6, 30–33). We generated mAbs against one of the latent nuclear antigens (LNAs) of HHV-8 encoded by viral ORF73 (34). ORF73 encodes the major immunogenic LNA identified as a nuclear stippling pattern in PEL cells when exposed to sera of patients with KS (35–37). ORF73 encodes a 1,162 amino acid protein with a 337 amino acid N-terminal domain, a hydrophilic 585 amino acid central domain consisting of multiple repeat elements, and a 240 amino acid C-terminal domain. ORF73 has no sequence similarity to any of the EBV LNAs. One of the antibodies, LN53, recognizes an EQEQE repeat epitope in ORF73 and reacts with antigens in paraffin-embedded tissue (34). We use this antibody here to study the distribution of the cell types infected latently by HHV-8 in patch, plaque, and nodular KS, MCD, in a lymph node of a patient with PEL, and in other tumors where HHV-8 might play a role.
METHODS
We studied HHV-8 ORF73 expression in 14 cutaneous paraffin-embedded biopsies of KS (11 early, i.e., patch or plaque lesions and three nodular lesions) from four patients with AIDS-associated KS and three patients with classic KS; one lymph node biopsy affected by KS in a patient with AIDS-associated KS; four lymph nodes and four spleen samples from five HIV-infected patients with MCD, and one lymph node from an HIV-negative patient with an anaplastic large cell CD30-positive lymphoma. These biopsies were all positive for HHV-8 DNA by PCR (data not shown). We also studied paraffin-embedded biopsies from five prostate carcinoma, five hemangiosarcoma, one lymph node from an HIV-negative patient with MCD, and 10 bone marrow samples with multiple myeloma. These cases did not contain HHV-8 DNA by nested PCR using primers derived from the conserved HHV-8 major capsid protein gene, ORF25 (38) (not shown).
Glutathione S-transferase and polyhistidine fusion proteins from the C terminus of ORF73 were used to immunize rats. Four mAbs (named LN20, LN53, LN69, and LN72) were generated and characterized by Western blot, immunoprecipitation, immunofluorescence assays, and FACS analysis (34). Among the four mAbs, LN53 was the most reactive and robust for LNA-1 antigen in sections from paraffin-embedded biopsies and works well for immunohistochemistry (34).
Paraffin-embedded sections (3- to 5-μm) were cut onto sialin-coated slides. Sections were deparaffinized with xylene and 100% ethanol and microwaved in 10 mmol/L citrate buffer, pH 6.0, at 780 W for 6 min (39). After treatment with 20% acetic acid, sections were incubated with LN53 (dilution 1 in 500 in PBS) for 1 hr at 22°C. Slides were then washed twice with 0.1% Tween in PBS. Incubation of the primary antibody was followed by a streptavidin–biotin complex alkaline phosphatase system (Vector Laboratories) and the sections were counterstained with hematoxylin.
Endothelial cells were stained with mAbs to CD31 (Dako), CD34 (NovoCastra, Newcastle, U.K.) or factor VIII (Immunostain EuroDPC, Llanberis U.K.). Lymphatic endothelium was stained with mAb 9D9, which stains the vascular endothelial growth factor receptor-3 (VEGFR-3) and is specific for lymphatic or proliferating endothelial cells (40). Lymphocyte subpopulations, follicular dendritic cells, and monocytes–macrophages were stained with mAbs to CD3 (Dako), CD45RO (Dako), CD20 (Dako), CD21 (NovoCastra), CD23 (NovoCastra), CD25 (Dako), CD43 (NovoCastra), CD10 (NovoCastra), CD38 (Serotec), CD5 (NovoCastra), CD40 (Harlan Laboratories, Haslett, MI), CD79a-cy (Dako) and CD68 (Dako), CD30 (NovoCastra) and epithelial membrane antigen (Dako). These antibodies and LN53 were stained on adjacent sections. For costaining experiments, LN53 was revealed with fast blue chromogen (Vector Laboratories) and the second primary antibody (CD23, CD25, CD43, CD10, CD38, CD5, CD79a-cy, CD30, VEGFR-3/9D9) was revealed with red chromogen (Vector Laboratories).
The expression of the EBV latent membrane protein-1 was tested on samples from patients with MCD by using a mAb from Dako. Detection of EBER 1,2 mRNAs and BHLF1 mRNAs was performed by using FITC-labeled specific oligonucleotides. The hybridization product was detected with a mouse anti-FITC mAb (Dako). As a third layer, the APAAP complex (Dako) was used with 5-bromo-4-chloro-3-indolyl phosphate nitroblue tetrazolium as a chromogen and nuclear fast red as counterstaining (41).
RESULTS
KS.
We show HHV-8 LNA-1 antigen in all stages of KS (Table 1). In patch-stage lesions, signals are found in spindle-like cells surrounding small ectatic vessels (Fig. 1B). LNA-1 is present in <10% of the cells surrounding vascular spaces (Fig. 1B). High-magnification microscopy (×160) confirms that HHV-8 is present in fusiform-shaped cells forming the walls of slit-like and angulated vessels in patch stage KS (Fig. 1 D and E). In plaque lesions, LNA-1 is present in spindle-like cells forming the walls of ectatic vessels (not shown) and in infiltrating spindle cells (Fig. 2A). In some of the plaque lesions up to 50% of spindle cells were positive (Fig. 2A). In nodular KS lesions, HHV-8 is latently present in the vast majority (>90%) of spindle cells (Fig. 2 B and C) and no signals are seen in surrounding normal dermis or overlaying epidermis (not shown). In nodular KS, LNA-1 is also found in endothelial cells surrounding angulated vessels (Fig. 2B) (neoangiogenic vessels or lymphatic endothelium) but not in well-formed (established) vascular endothelium (Fig. 2 B and C and Fig. 3B), suggesting that HHV-8 is predominantly present in endothelial cells surrounding lymphatic or neoangiogenic vessels and not in normal vascular endothelium in KS lesions.
Table 1.
Results of staining with the mAb LN53 to LNA-1
| Diagnosis (sample tested) | No. Tested | Positive by PCR | Positive for LNA-1 |
|---|---|---|---|
| Kaposi’s sarcoma (skin/patch and plaque) | 11 | 11 | 8 |
| Kaposi’s sarcoma (skin/nodular stage) | 3 | 3 | 3 |
| Kaposi’s sarcoma (lymph node) | 1 | 1 | 1 |
| Multicentric Castleman’s disease HIV+ (lymph node) | 4 | 4 | 4 |
| Multicentric Castleman’s disease HIV+ (spleen) | 4 | 4 | 3 |
| Multicentric Castleman’s disease HIV− (lymph node) | 1 | 0 | 0 |
| Primary effusion lymphoma (lymph node) | 1 | 1 | 1 |
| Multiple myeloma (bone marrow) | 10 | 0 | 0 |
| Hemangiosarcoma (tumor) | 5 | 0 | 0 |
| Prostate carcinoma (prostate) | 5 | 0 | 0 |
Figure 1.
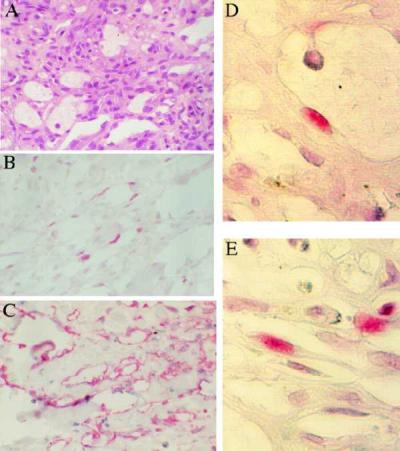
Patch stage KS (×40). (A) Hematoxylin-eosin (H/E) stain of patch KS lesion. (B) LNA-1 staining of adjacent sections shows that latent HHV-8 infection is present in fusiform cells forming the walls of ectatic spaces; staining is nuclear. (C) VEGFR-3 staining of adjacent section shows expression by all cells forming the walls of ectatic vessels; staining is cytoplasmic. (D and E) Higher magnification (×160) of patch KS stained with LNA-1 showing (D) one positive cell in the wall of an ectatic vessel and (E) positive cells forming the walls of slit-like vessels.
Figure 2.

(A) In a plaque KS lesion, up to 50% of spindle-shaped cells surrounding the typical lacs vasculaires are positive for HHV-8 by LN53 (×40). (B and C) Nodular KS stained for HHV-8 with LN53 (×40). >90% of spindle cells are positive. The typical nuclear stippling pattern is seen. Endothelial cells surrounding an angulated vessel are positive (black arrowheads). However, in both figures the endothelial cells surrounding mature vascular spaces are negative for HHV-8 (white arrowheads).
Figure 3.

Expression of VEGFR-3 in nodular KS (×40). (A) Most spindle cells express VEGFR-3, and endothelial cells surrounding ectatic vessels are positive for VEGFR-3 (black arrowheads), whereas endothelial cells surrounding a normal blood vessel are negative (white arrowhead). (B) Adjacent section of A stained with LN53 shows that LNA-1 positive cells have a similar distribution to VEGFR-3, although fewer cells are positive for LNA-1. The same vessel negative for VEGFR-3 is also negative for LNA-1 (white arrowhead). (C) Costaining of VEGFR-3 (cytoplasmic, pink) and HHV-8 (nuclear, dark blue stippling) confirms that cells containing HHV-8 express VEGFR-3.
We therefore studied the distribution of lymphatic endothelium in KS lesions and compared this distribution with that of HHV-8. A mAb (9D9) against VEGFR-3 was used (40). In patch and plaque KS, VEGFR-3 was expressed in all fusiform cells surrounding ectatic vessels (Fig. 1C). A smaller number of these cells expressed LNA-1 antigen (Fig. 1B). In nodular KS lesions, VEGFR-3 antibody does not stain well formed vascular endothelium (Fig. 3A), similar to that observed for LNA-1 (Figs. 2 B and C and 3B). In nodular KS, more than 90% of spindle cells stain for VEGFR-3 (Fig. 3A). Costaining experiments confirm that LNA-1 and VEGFR-3 colocalize in nodular KS (Fig. 3C).
MCD.
By using the LN53 antibody, the HHV-8-positive cells localized in the mantle zone of the follicles (Fig. 4B). Ten to 30% of the cells in the mantle zone were positive with the characteristic stippling patterns associated with LNA-1 (Fig. 5 A and B) (34). HHV-8-positive cells are larger than surrounding CD79a-cy positive lymphocytes and have large central or marginal nuclei with one or two nucleoli. Morphologically, these cells resemble immunoblasts. The results of staining of adjacent sections and of costaining experiments are summarized in Table 2. These cells do not stain with common T-cell markers but stain with the B-cell marker CD20 (Fig. 5C). They do not stain with plasma cell or immunoblast markers such as CD79a-cy (which stains B cells) (Fig. 4D) and CD38 (not shown). We did not observe any staining for LNA-1 in follicular dendritic cells using either CD21 and CD23 (Fig. 4C). Immunocytochemistry failed to detect EBV latent membrane protein expression in any of the cases. With in situ hybridization for EBER, only rare small lymphocytes were positive in one lymph node and one spleen sample from two different patients with MCD.
Figure 4.

Expression of CD23, CD79a-cy, and HHV-8 LNA-1 in a lymph node biopsy from MCD. (A) H/E staining of a follicle (×10). (B) Adjacent section stained with LN53 showing many positive cells in the mantle zone (nuclear red) (×10). (C) Costaining with CD23 (cytoplasmic, dark purple) and LN53 (nuclear, light blue) confirms that the positive cells for HHV-8 do not express CD23 (×20). (D) Costaining with CD79a-cy (plasma membrane, pink) and LN53 (nuclear, blue) shows that HHV-8 positive cells do not express CD79 (×40).
Figure 5.

High magnification (×140) of MCD mantle zone. (A and B) The HHV-8-positive cells (black arrowheads) are large, have prominent nuclei with one or two nucleoli, and resemble immuno- or plasmablastic cells. Staining for LNA-1 shows the typical nuclear stippling (black arrowhead). Lymphocytes surrounding the HHV-8-positive cells are negative (white arrowheads). (C) CD20 staining shows that the large immunoblastic cells are positive (black arrowhead).
Table 2.
Immunophenotype of HHV-8-positive cells in MCD, compared to HHV-8-positive PEL cells
| mAbs | PEL cells (27, 28) | Parraviccini et al. (17) | Present study |
|---|---|---|---|
| T-cell markers | |||
| CD3 | neg | neg | neg |
| CD5 | neg | ND | neg |
| CD45RO | neg | neg | neg |
| B-cell markers | |||
| CD20 | neg | neg | + |
| CD79a | neg | ND | neg |
| CD10 | neg | ND | neg |
| Activation markers | |||
| CD23* | + | ND | neg |
| CD25 | + | ND | neg |
| CD30 | +/neg | neg | neg |
| CD38 | + | neg | neg |
| CD40 | neg | ND | neg |
| Follicular dendritic cells | |||
| CD21 | neg | ND | neg |
| Monocyte/macrophages | |||
| CD68 | ND | neg | neg |
| Others | |||
| CD43 | + | ND | neg |
| EMA | + | neg | neg |
| CD34 | ND | ND | neg |
| VEGFR-3 | ND | ND | neg |
CD23 also stains follicular dendritic cells; CD21 also stains B-cells. ND, not done; neg, negative; +; positive.
PEL.
A 68-yr-old HIV-negative Israeli patient presented with a pleural effusion. Diagnostic tap revealed a CD30-positive anaplastic large cell lymphoma. We stained a lymph node biopsy from this patient and showed that the lymphoma cells were positive for CD30 (Fig. 6B) and for epithelial membrane antigen (not shown). The neoplastic cells are positive for LNA-1 (Fig. 6C). Viral antigen and CD30 colocalized to the same cells (Fig. 6D).
Figure 6.

(A) The lymph node of a patient with PEL stained with H/E showing large anaplastic cells (×20). (B and C) These neoplastic cells express CD30 (B) and HHV-8 LNA-1 (C) (×40). (D) Costaining confirms that CD30 (plasma membrane, red) and LNA-1 (nuclear stippling, purple) colocalize in the neoplastic cells (×40).
Others.
By using LN53, we did not observe any staining in tumors that were negative by PCR for HHV-8: ten multiple myeloma bone marrow biopsies, the lymph node of the HIV-negative patient with MCD, five hemangiosarcoma, or five prostate carcinoma samples (Table 1). The hemangiosarcomas were also stained for VEGFR-3; one of five showed staining in a few endothelial cells, but no staining was seen in the sarcoma cells (not shown).
DISCUSSION
We demonstrate latent HHV-8 infection in all stages of KS, in MCD, and in a lymph node of a patient with PEL. HHV-8 is present in cells belonging to the endothelial lineage in KS, in immunoblastic cells belonging to the B-cell lineage in MCD, and in CD30-positive epithelial membrane antigen-positive lymphoma cells in PEL.
KS is a vascular tumor with a complex histology and an uncertain histogenesis (2, 42, 43). Most of the spindle cells in KS lesions express endothelial markers, including CD31 and CD34 (44, 45). However, it was also shown that KS spindle cells express markers for smooth muscle cells (46), macrophages, and dendritic cells (47, 48), suggesting that spindle cells are either derived from pluripotent mesenchymal precursors or represent a heterogeneous population of cells. Importantly, KS spindle cells do not stain with PAL-E, a marker expressed on vascular endothelium but not on lymphatic endothelium (49).
HHV-8 is present in all the different epidemiologic types of nodular KS (2); however, the universal presence of HHV-8 in early KS has not yet been shown (50). It has been argued that HHV-8 could be reactivated in early KS lesions by an inflammatory cytokine presence (43, 51), or that HHV-8 is brought into early lesions by infiltrating macrophages (10, 52). By PCR in situ hybridization (3, 5) and in situ hybridization (4, 7, 8), HHV-8 DNA or RNA has been localized to spindle, endothelial, and monocytes in nodular KS. However, these techniques have proved inadequate to detect infected cell type/s in early KS lesions (plaque or patch). Moreover, in situ hybridization techniques have produced controversial results including the detection of HHV-8 in prostatic carcinoma samples (6) and bone marrow dendritic cells in MM (30), although these results have not been confirmed (31–33, 38, 53, 54).
Using an antibody against a LNA of HHV-8 (34), we show latent HHV-8 infection in patch, plaque, and nodular KS. In all three stages, staining is seen in fusiform- or spindle-shaped cells forming the walls of ectatic vessels (Fig. 1B, Fig. 2 A–C). In plaque and nodular KS, most spindle cells between these vessels are also positive (Fig. 2 A–C). However, very notably, mature endothelial cells surrounding normal vascular blood vessels are negative in all cases studied (Fig. 2 B and C and Fig. 3B). This staining for HHV-8 is reminiscent of that for VEGFR-3: a marker of lymphatic vessels (40, 55–57). VEGFR-3 is the receptor for VEGF-C (55). During early embryogenesis, VEGFR-3 mRNA is detected in most endothelial cells, but later it becomes restricted to lymphatic endothelium of adult tissues. VEGF-C stimulates the proliferation of lymphatic endothelium, and VEGF-C transgenic mice exhibit lymphatic endothelium proliferation (56). VEGFR-3 expression in human skin correlates with the known distribution of the lymphatic vasculature (57). Like HHV-8, VEGFR-3 stains KS spindle cells but not normal vascular endothelium (Fig. 3A) (40, 57). We therefore tested the expression of VEGFR-3 in adjacent HHV-8 LNA-1-stained early and nodular KS. VEGFR-3 is highly expressed in patch and plaque KS, whereas fewer cells, but with a similar distribution and morphology, stain for HHV-8 (Fig. 1 B and C). In nodular KS, the same cells costain for VEGFR-3 and HHV-8 (Fig. 3C).
The extensive expression of VEGFR-3 in early KS suggests that KS is either a proliferation of lymphatic endothelium or of immature endothelial cells (precursors). The similar distribution of VEGFR-3 and HHV-8 in early and late KS lesions implies that HHV-8 either infects lymphatic endothelium and that spindle cells are derived from lymphatic endothelium or that HHV-8 infection induces expression of this lymphatic marker in spindle cells. However, VEGFR-3 is expressed in many more cells than HHV-8 in early KS (Fig. 1 B and C) suggesting that HHV-8 does not induce the expression of VEGFR-3 directly and that HHV-8 does not directly promote the proliferation of VEGFR-3 positive cells in very early KS.
Other observations also support the hypothesis that KS is a proliferation of lymphatic or precursor endothelium: (i) our failure to demonstrate HHV-8 in normal vascular endothelium; (ii) the observation that KS does not occur in organs that lack a lymphatic system (i.e., brain and eye) (58, 59); (iii) the fact that spindle cells do not stain with PAL-E (49, 60); (iv) the association of primary HHV-8 infection with lymph nodal KS in HIV-infected patients (61) and possibly in African children (2).
In early lesions, only a small proportion (<10%) of spindle- or fusiform-shaped cells are positive for HHV-8 (Fig. 1B). In nodular lesions, most (>90%) spindle cells express LNA-1 (Fig. 2 B and C). These data indicate that early KS is not a monoclonal expansion of HHV-8 infected cells and are consistent with studies showing that nodular KS can be oligo- or polyclonal (62, 63) and may not necessarily be monoclonal (64, 65). However, the fact that most spindle cells in nodular KS are positive for HHV-8 suggests that HHV-8 latent proteins provide a growth advantage to infected cells.
LNA-1 and VEGFR-3 appear to be robust markers for the immunohistological diagnosis of early KS. LNA-1 positive cells in all stages of KS show the typical nuclear stippling pattern also described in latently infected PEL cells exposed to sera of KS patients (26, 66). The stippling pattern is also seen in HHV-8-positive cells in MCD (Fig. 5 A and B), indicating that LNA-1 associates with the same nuclear structures in all three cell types.
MCD is a lymphoproliferative disorder that has been reported in close association with KS in both HIV-seropositive and -seronegative patients (67, 68) Exacerbations of symptoms related to MCD correlate with an increased HHV-8 viral load in peripheral blood mononuclear cells, supporting a role for HHV-8 in MCD pathogenesis (69). IL-6 enhances B-cell survival and proliferation, and IL-6 overexpression has been implicated in the pathogenesis of MCD (70, 71). HHV-8 encodes an IL-6 homolog (vIL-6) (11, 72, 73). By using a polyclonal antibody against vIL-6, it was shown that vIL-6 is indeed expressed in a few cells (2–13%) surrounding the germinal centers of the follicles in Castleman’s disease (17). Although we here show a similar distribution of HHV-8-infected cells, we found more cells (10–30%) positive for LNA-1 than shown for vIL-6 (17). This could either be because our MCD cases are from HIV-positive patients or because vIL-6 is not expressed in all latently infected cells, unlike LNA-1 (11, 72–74).
The cells harboring HHV-8 in MCD are larger than the surrounding lymphocytes (Fig. 5 A–C) and have prominent central or marginal nuclei. Morphologically they resemble immunoblastic cells (Fig. 5 A–C) (17). These HHV-8-positive cells do not stain for T-cell (i.e., CD 3, CD45RO) or dendritic cell markers (CD21, CD23), but stain with the B-lymphocyte marker CD20 (Fig. 5C) (Table 2). However, the cells lack expression of B-activation markers such as CD23, CD38, and CD30. In none of the MCD cases tested here did we demonstrate EBV infection by immunohistochemistry or by in situ hybridization in any significant number of cells, supporting the lack of a pathogenic link between EBV and MCD (68, 75). HHV-8-positive MCD cases appear to have a poorer prognosis than HHV-8-negative cases (16, 17). Indeed, all cases described here died within a median of 10 mo after the diagnosis of MCD. It remains to be seen whether HHV-8-induced proliferation of infected lymphocytes leads to the outgrowth of monoclonal aggressive MCD or associated lymphoma (67, 68, 76, 77).
Although VEGFR-3 is expressed by lymphatic cells in lymph nodes affected by MCD, these cells do not stain for LNA-1. HHV-8 is present only in hematopoietic cells in MCD, compared with the viral presence in mesenchymal cells in KS.
PEL is a rare lymphoma that is characterized by effusions in body cavities, the lack of B- or T-lymphocyte antigens, and the absence of c-myc rearrangements (22). HHV-8 has been detected in all PEL cases, in association or not with EBV. The patient described here was diagnosed with an anaplastic large cell lymphoma after lymph node biopsy. However, in retrospect, the predominant effusion phenotype of his lymphoma, the lack of common B- or T-cell markers, and the expression of epithelial membrane antigen, CD30, and HHV-8 by the tumor cells indicate that he indeed had PEL. HHV-8, but not EBV, is present in the tumor cells correlating with other HIV-negative cases of PEL. Lymph nodal and noneffusion involvement in PEL have previously been described (20, 21).
We did not observe any staining for HHV-8 LNA-1 in bone marrow biopsies from patients with multiple myeloma. This is in accordance with other studies that could not confirm any association between HHV-8 and multiple myeloma using immunologic and molecular techniques (31–33, 53, 54).
In conclusion, using a mAb against a LNA of HHV-8, we show HHV-8 latently infected cells in three distinct cell types in the three major neoplasms associated with this virus. Both KS and MCD are complex lesions and HHV-8 links reactive inflammatory processes with neoplastic formation in both diseases. In PEL HHV-8 is present in every cell (Fig. 6 C and D), and this is clearly a monoclonal tumor of CD30-positive plasmacytoid cells (22). LNA-1 is strongly expressed in large immunoblastic cells belonging to the B-cell lineage in MCD. In KS, the presence of HHV-8-infected cells in early lesions supports a role for this virus in the etiopathogenesis of KS. Spindle cells belong to the endothelium lineage that expresses VEGFR-3, i. e., precursor endothelium (including neoangiogenic endothelium) and lymphatic endothelium. Here we have shown that HHV-8 infects these cells. The fact that VEGFR-3 is expressed by more cells in early KS compared with HHV-8 indicates that paracrine mechanisms are important in the initiation and progression of KS.
Acknowledgments
We thank Martin Burgin for technical assistance, Hugh Paterson for confocal microscopy, and Christine Shotton for purifying mAb LN53. This work was supported by the United Kingdom Cancer Research Campaign, La Ligue Nationale contre le Cancer, la Fondation Cancer et Solidarité and la Fondation René Touraine. Chris Boshoff is a Glaxo Wellcome Prize Fellow.
ABBREVIATIONS
- HHV-8
human herpesvirus 8
- KS
Kaposi’s sarcoma
- MCD
multicentric Castleman’s disease
- PEL
primary effusion lymphoma
- LNA
latent nuclear antigen
- EBV
Epstein—Barr virus
- VEGFR-3
vascular endothelial growth factor receptor-3
References
- 1.Moore P S, Chang Y. Am J Epidemiol. 1998;147:217–221. doi: 10.1093/oxfordjournals.aje.a009440. [DOI] [PubMed] [Google Scholar]
- 2.Boshoff C, Weiss R A. In: Advances in Cancer Research. Van de Woude G, Klein G, editors. Vol. 75. San Diego: Academic; 1998. pp. 57–86. [DOI] [PubMed] [Google Scholar]
- 3.Boshoff C, Schulz T F, Kennedy M M, Graham A K, Fisher C, Thomas A, McGee J O, Weiss R A, O’Leary J J. Nat Med. 1995;1:1274–1278. doi: 10.1038/nm1295-1274. [DOI] [PubMed] [Google Scholar]
- 4.Li J J, Huang Y Q, Cockerell C J, Friedman Kien A E. Am J Pathol. 1996;148:1741–1748. [PMC free article] [PubMed] [Google Scholar]
- 5.Foreman K E, Bacon P E, Hsi E D, Nickoloff B J. J Clin Invest. 1997;99:2971–2978. doi: 10.1172/JCI119492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Staskus K A, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson D J, Ganem D, et al. J Virol. 1997;71:715–719. doi: 10.1128/jvi.71.1.715-719.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Davis M A, Sturzl M, Blasig C, Schreier A, Guo H, Reitz M, Opalenik S R, Browning P J. J Natl Cancer Inst. 1998;89:1868–1874. doi: 10.1093/jnci/89.24.1868. [DOI] [PubMed] [Google Scholar]
- 8.Dittmer D, Lagunoff M, Renne R, Staskus K, Haase A, Ganem D. J Virol. 1998;72:8309–8315. doi: 10.1128/jvi.72.10.8309-8315.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Decker L L, Shankar P, Khan G, Freeman R B, Dezube B J, Lieberman J, Thorley Lawson D A. J Exp Med. 1996;184:283–288. doi: 10.1084/jem.184.1.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blasig C, Zietz C, Haar B, Neipel F, Esser S, Brockmeyer N H, Tschachler E, Colombini S, Ensoli B, Sturzl M. J Virol. 1997;71:7963–7968. doi: 10.1128/jvi.71.10.7963-7968.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moore P S, Boshoff C, Weiss R A, Chang Y. Science. 1996;274:1739–1744. doi: 10.1126/science.274.5293.1739. [DOI] [PubMed] [Google Scholar]
- 12.Orenstein J M, Alkan S, Blauve H A, Jeang K-T, Weinstein M D, Ganem D, Herndier B. AIDS. 1997;11:735–745. doi: 10.1097/00002030-199705000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals Hatem D, Babinet P, d’Agay M F, Clauvel J P, Raphael M, Degos L. Blood. 1995;86:1276–1280. [PubMed] [Google Scholar]
- 14.Cesarman E, Chang Y, Moore P S, Said J W, Knowles D M. N Engl J Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 15.Gessain A, Sudaka A, Briere J, Fouchard N, Nicola M A, Rio B, Arborio M, Troussard X, Audouin J, Diebold J, et al. Blood. 1996;87:414–416. [PubMed] [Google Scholar]
- 16.Chadburn A, Cesarman E, Nador R G, Liu Y F, Knowles D M. Cancer. 1997;80:788–797. [PubMed] [Google Scholar]
- 17.Parravicini C, Corbellino M, Paulli M, Magrini U, Lazzarino M, Moore P S, Chang Y. Am J Pathol. 1997;6:1517–1522. [PMC free article] [PubMed] [Google Scholar]
- 18.Knowles D M, Inghirami G, Ubriaco A, Dalla-Favera R. Blood. 1989;73:792–799. [PubMed] [Google Scholar]
- 19.Nador R G, Cesarman E, Chadburn A, Dawson D B, Ansari M Q, Said J, Knowles D M. Blood. 1996;88:645–656. [PubMed] [Google Scholar]
- 20.Morassut S, Vaccher E, Balestreri L, Gloghini A, Gaidano G, Volpe R, Tirelli U, Carbone A. Radiology. 1997;205:459–463. doi: 10.1148/radiology.205.2.9356629. [DOI] [PubMed] [Google Scholar]
- 21.Gessain A, Briere J, Angelin Duclos C, Valensi F, Beral H M, Davi F, Nicola M A, Sudaka A, Fouchard N, Gabarre J, et al. Leukemia. 1997;11:266–272. doi: 10.1038/sj.leu.2400549. [DOI] [PubMed] [Google Scholar]
- 22.Nador R G, Cesarman E, Knowles D M, Said J W. N Engl J Med. 1995;333:943. doi: 10.1056/NEJM199510053331417. [DOI] [PubMed] [Google Scholar]
- 23.Said W, Chien K, Takeuchi S, Tasaka T, Asou H, Cho S K, de Vos S, Cesarman E, Knowles D M, Koeffler H P. Blood. 1996;87:4937–4943. [PubMed] [Google Scholar]
- 24.Ansari M Q, Dawson D B, Nador R, Rutherford C, Schneider N R, Latimer M J, Picker L, Knowles D M, McKenna R W. Am J Clin Pathol. 1996;105:221–229. doi: 10.1093/ajcp/105.2.221. [DOI] [PubMed] [Google Scholar]
- 25.Cesarman E, Moore P S, Rao P H, Inghirami G, Knowles D M, Chang Y. Blood. 1995;86:2708–2714. [PubMed] [Google Scholar]
- 26.Gao S J, Kingsley L, Li M, Zheng W, Parravicini C, Ziegler J, Newton R, Rinaldo C R, Saah A, Phair J, et al. Nat Med. 1996;2:925–928. doi: 10.1038/nm0896-925. [DOI] [PubMed] [Google Scholar]
- 27.Boshoff C, Gao S-J, Healy L E, Matthews S, Thomas A J, Coignet L, Warnke R A, Strauchen J A, Matutes E, Kamel, et al. Blood. 1998;91:1671–1679. [PubMed] [Google Scholar]
- 28.Drexler H G, Uphoff C C, Gaidano G, Carbone A. Leukemia. 1998;12:1507–1517. doi: 10.1038/sj.leu.2401160. [DOI] [PubMed] [Google Scholar]
- 29.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. Nat Med. 1996;2:342–346. doi: 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- 30.Rettig M B, Ma H J, Vescio R A, Pold M, Schiller G, Belson D, Savage A, Nishikubo C, Wu C, Fraser J, et al. Science. 1997;276:1851–1854. doi: 10.1126/science.276.5320.1851. [DOI] [PubMed] [Google Scholar]
- 31.Marcelin A-G, Dupin N, Bouscary D, Bossi P, Cacoub P, Ravaud P, Calvez V. Lancet. 1997;350:1144. doi: 10.1016/S0140-6736(05)63791-9. [DOI] [PubMed] [Google Scholar]
- 32.Masood R, Zheng T, Tulpule A, Arora N, Chatlynne L, Handy M, Whitman J, Jr, Kaplan M, Dosik M, Ablashi D V, et al. Science. 1997;278:1969–1970. [PubMed] [Google Scholar]
- 33.Whitby D, Boshoff C, Luppi M, Torelli G. Science. 1997;278:1971–1972. [PubMed] [Google Scholar]
- 34.Kellam, P., Bourboulia, D., Dupin, N., Shotton, C., Fisher, C., Talbot, S., Boshoff, C. & Weiss, R. A. (1999) J. Virol., in press. [DOI] [PMC free article] [PubMed]
- 35.Kellam P, Boshoff C, Whitby D, Matthews S, Weiss R A, Talbot S J. J Hum Virol. 1997;1:19–29. [PubMed] [Google Scholar]
- 36.Rainbow L, Platt G M, Simpson G R, Sarid R, Gao S-J, Stoiber H, Herrington C S, Moore P S, Schulz T F. J Virol. 1997;71:5915–5921. doi: 10.1128/jvi.71.8.5915-5921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kedes D H, Lagunoff M, Renne R, Ganem D. J Clin Invest. 1997;100:2606–2610. doi: 10.1172/JCI119804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boshoff C, Talbot S, Kennedy M, O’Leary J, Schulz T, Chang Y. Lancet. 1996;348:138. [PubMed] [Google Scholar]
- 39.Shi S R, Key M E, Kalra K L. J Histochem Cytochem. 1991;39:741–748. doi: 10.1177/39.6.1709656. [DOI] [PubMed] [Google Scholar]
- 40.Jussila L, Valtola R, Partanen T A, Salven P, Heikkila P, Matikainen M-T, Renkonen R, Kaipainen A, Detmar M, Tschachler E, et al. Cancer Research. 1998;58:1599–1604. [PubMed] [Google Scholar]
- 41.Brousset P, Butet V, Chittal S, Selves J, Delsol G. Lab Invest. 1992;67:457–464. [PubMed] [Google Scholar]
- 42.Weiss R A. Eur J Cancer. 1990;26:657–659. doi: 10.1016/0277-5379(90)90109-7. [DOI] [PubMed] [Google Scholar]
- 43.Gallo R C. Science. 1998;282:1837–1839. doi: 10.1126/science.282.5395.1837. [DOI] [PubMed] [Google Scholar]
- 44.Sturzl M, Brandstetter H, Roth W K. AIDS Res Hum Retroviruses. 1992;8:1753–1764. doi: 10.1089/aid.1992.8.1753. [DOI] [PubMed] [Google Scholar]
- 45.Nickoloff B J. Arch Dermatol. 1993;129:249–250. [PubMed] [Google Scholar]
- 46.Weich H A, Salahuddin S Z, Gill P, Nakamura S, Gallo R C, Folkmann J. Am J Pathol. 1991;139:1251–1258. [PMC free article] [PubMed] [Google Scholar]
- 47.Nickoloff B J, Griffiths C E. Science. 1989;243:1736–1737. doi: 10.1126/science.2564703. [DOI] [PubMed] [Google Scholar]
- 48.Huang Y Q, Friedman-Kien A E, Li J J, Nickoloff B J. Arch Dermatol. 1993;129:1291–1296. [PubMed] [Google Scholar]
- 49.Kaaya E E, Parravicini C, Ordonez C, Gendelman R, Berti E, Gallo R C, Biberfeld P. J Acquir Immune Defic Syndr Hum Retrovirol. 1995;10:295–305. [PubMed] [Google Scholar]
- 50.Noel J C. Lancet. 1995;346:1359. [PubMed] [Google Scholar]
- 51.Fiorelli V, Gendelman R, Sirianni M C, Chang H K, Colombini S, Markham P D, Monini P, Sonnabend J, Pintus A, Gallo R C, et al. Blood. 1998;91:956–967. [PubMed] [Google Scholar]
- 52.Siriani M C, Vincenzi L, Fiorelli V, Topino S, Scala E, Uccini S, Angeloni A, Faggioni A, Cerimele D, Cottoni F, et al. Blood. 1998;91:968–976. [PubMed] [Google Scholar]
- 53.Tarte K, Olsen S J, Yang Lu Z, Legouffe E, Rossi J F, Chang Y, Klein B. Blood. 1998;91:1852–1857. [PubMed] [Google Scholar]
- 54.Yi Q, Ekam M, Anton D, Bergenbrant S, Osterborg A, Georgii-Hemming P, Holm G, Nilsson K, Biberfeld P. Blood. 1998;92:402–404. [PubMed] [Google Scholar]
- 55.Kukk E, Lymboussaki A, Taira S, Kaipainen A, Jeltsch M, Joukov V, Alitalo K. Development (Cambridge, UK) 1996;122:3829–3837. doi: 10.1242/dev.122.12.3829. [DOI] [PubMed] [Google Scholar]
- 56.Jeltsch M, Kaipainen A, Joukov V, Meng X, Lakso M, Rauvala H, Awartz M, Fukumura D, Jain R K, Alitalo K. Science. 1997;276:1423–1425. doi: 10.1126/science.276.5317.1423. [DOI] [PubMed] [Google Scholar]
- 57.Lymboussaki A, Partanen T A, Olofsson B, Thomas-Crussells J, Fletcher C D, de Waal R M, Kaipainen A, Alitalo K. Am J Pathol. 1998;153:395–403. doi: 10.1016/S0002-9440(10)65583-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beckstead J H, Wood G S, Fletcher V. Am J Pathol. 1985;119:294–300. [PMC free article] [PubMed] [Google Scholar]
- 59.Dorfman R F. Lymphology. 1988;21:45–52. [PubMed] [Google Scholar]
- 60.Nadimi H, Saatee S, Armin A, Toto P D. J Oral Pathol. 1988;17:416–420. doi: 10.1111/j.1600-0714.1988.tb01307.x. [DOI] [PubMed] [Google Scholar]
- 61.Oksenhendler E, Cazals-Hatem D, Schultz T F, Barateau V, Grollet L, Sheldon J, Clauvel J-P, Sigaux F, Agbalika F. N Engl J Med. 1998;338:1585–1591. doi: 10.1056/NEJM199805283382204. [DOI] [PubMed] [Google Scholar]
- 62.Delabesse E, Oksenhendler E, Lebbe C, Verola O, Varet B, G. T A. J Clin Pathol. 1997;50:664–668. doi: 10.1136/jcp.50.8.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gill P, Tsai Y C, Rao A P, Spruck C H, Zheng T, Harrington W A, Cheung T, Nathwani B, Jones P. Proc Natl Acad Sci USA. 1998;95:8257–8261. doi: 10.1073/pnas.95.14.8257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rabkin C S, Bedi G, Musaba E, Biggar R J. N Engl J Med. 1995;1:257–260. [PubMed] [Google Scholar]
- 65.Rabkin C S, Janz S, Lash A, Coleman A E, Musaba E, Liotta L, Biggar R J, Zhuang Z. N Engl J Med. 1997;336:988–993. doi: 10.1056/NEJM199704033361403. [DOI] [PubMed] [Google Scholar]
- 66.Kedes D H, Operskalski E, Busch M, Kohn R, Flood J, Ganem D. Nat Med. 1996;2:918–924. doi: 10.1038/nm0896-918. [DOI] [PubMed] [Google Scholar]
- 67.Frizzera G. Hum Pathol. 1985;16:202–205. doi: 10.1016/s0046-8177(85)80002-2. [DOI] [PubMed] [Google Scholar]
- 68.Oksenhendler E, Duarte M, Soulier J, Cacoub P, Welker Y, Cadranel J, Cazals-Hatem D, Autran B, Clauvel J P, Raphael M. AIDS. 1996;10:61–67. [PubMed] [Google Scholar]
- 69.Grandadam M, Dupin N, Calvez V, Gorin I, Blum L, Kernbaum S, Sicard D, Buisson Y, Agut H, Escande J P, et al. J Infect Dis. 1997;175:1198–1201. doi: 10.1086/593567. [DOI] [PubMed] [Google Scholar]
- 70.Brandt S J, Bodine D M, Dunbar C E, Nienhuis A W. J Clin Invest. 1990;86:592–599. doi: 10.1172/JCI114749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Leger-Ravet M B, Peuchmaur M, Devergne O, Audouin J, Raphael M, Van Damme J, Galanaud P, Diebold J, Eminie D. Blood. 1991;78:2923–2930. [PubMed] [Google Scholar]
- 72.Neipel F, Albrecht J C, Ensser A, Huang Y Q, Li J J, Friedman Kien A E, Fleckenstein B. J Virol. 1997;71:839–842. doi: 10.1128/jvi.71.1.839-842.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nicholas J, Ruvolo V R, Burns W H, Sandford G, Wan X, Ciufo D, Hendrickson S B, Guo H G, Hayward G S, Reitz M S. Nat Med. 1997;3:287–292. doi: 10.1038/nm0397-287. [DOI] [PubMed] [Google Scholar]
- 74.Sarid R, Flore O, Bohenzky R A, Chang Y, Moore P S. J Virol. 1998;72:1005–1012. doi: 10.1128/jvi.72.2.1005-1012.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Murray P G, Deacon E, Young L S, Barletta J M, Mann R B, Ambinder R F, Rowlands D C, Jones E L, Ramsay A D, Crocker J. Hematol Pathol. 1995;9:17–26. [PubMed] [Google Scholar]
- 76.Xerri L, Guigou V, Lepidi H, Horschowski N, Lejeune C, Hassoun J. Arch Pathol Lab Med. 1991;115:1162–1166. [PubMed] [Google Scholar]
- 77.Wynia M K, Shapiro B, Kuvin J T, Skolnik P R. AIDS. 1995;9:814–816. doi: 10.1097/00002030-199507000-00024. [DOI] [PubMed] [Google Scholar]