

Abstract
Artemis gene mutations are responsible for the development of a severe combined immunodeficiency [radiation-sensitive (RS) SCID] characterized by a severe B and T cell deficiency and a normal natural killer cell population. To establish the feasibility of a gene therapy approach to the treatment of RS-SCID, we generated a series of lentiviral vectors expressing human Artemis from different promoters and used them to transduce highly purified hematopoietic stem cells (HSCs) from Artemis knockout mice. HSCs transduced by the different viruses were transplanted into either lethally irradiated Rag-1-deficient animals or Artemis knockout mice treated with a nonmyeloablative dose of Busulfan. In both models, transplantation of HSCs transduced by a vector that used a murine phosphoglycerate kinase (PGK) promoter led to a complete functional correction of the immunodeficiency. Corrected animals displayed rescue of mature B cells with normal levels of serum immunoglobulins, together with complete rescue of the T cell compartment as evidenced by the presence of mature T lymphocytes in peripheral blood as well as normal values of thymocytes in thymus. Those B and T cells were capable of activation, as shown both by in vitro stimulation responses and in vivo after immune challenge. Overall, the results indicate that a gene therapy approach for RS-SCID involving the transplantation of genetically modified HSCs is indeed feasible. Furthermore, our studies suggest the possibility that nonmyeloablative conditioning regimens might be effectively used to promote engraftment of genetically modified cells in the case of diseases where standard irradiation-based myeloablative bone marrow transplantation protocols may prove problematic.
Keywords: gene therapy, hematopoietic stem cells, lentivirus
Severe combined immunodeficiencies (SCID) consist of a group of rare human conditions characterized by a complete lack of T cell development. In ≈20% of SCID patients, there is a complete absence of both circulating B and T lymphocytes associated with a defect in the V(D)J recombination process (1). Mutations in the recently described Artemis gene (2) are responsible for the development of a subgroup of these SCID patients, known as radiation-sensitive (RS) SCID and SCIDA (3), in which the B-T deficiency is accompanied by an increased sensitivity to ionizing radiation (IR) of both bone marrow cells and primary skin fibroblasts (4). The function of the Artemis protein has been more precisely defined after the development of a murine knockout (KO) model of Artemis SCID (5). Artemis-deficient mice display a phenotype that closely resembles the clinical manifestations of the human disease, with both severe lymphocyte defects and increased IR sensitivity, definitively demonstrating the role of Artemis during DNA repair (6, 7).
The prognosis for RS-SCID patients is poor, with lethality resulting typically within the first year of life. Currently, the only effective treatment consists of bone marrow transplantation (BMT), the success of which depends on the availability of a suitable matched donor (8). Despite the recent serious adverse events associated with retroviral integration into chromosomal DNA (9, 10), gene therapy would still appear to represent an important alternative approach toward the treatment of SCID, because studies by others have unequivocally shown that gene therapy can lead to the complete reconstitution of immune function in SCID patients (11, 12).
In this study, we sought to provide preclinical data in support of the treatment for RS-SCID by gene therapy. The results suggest that a gene therapy approach for RS-SCID is indeed feasible, despite the unique characteristics of the disease that may preclude the use of standard conditioning regimens for the transplantation of genetically modified cells.
Results
Establishment of a Preclinical Gene Therapy Model for Correction of Artemis SCID.
To evaluate the feasibility of a gene therapy for RS-SCID, we chose to make use of a murine KO model that recapitulates the essential features of the human disease (5). Because previous efforts by Li et al. (13) to correct the immunodeficiency in an independently generated Artemis KO strain of mice indicated the need for conditioning of the recipient to obtain significant reconstitution of the B cell compartment after the transplantation of WT congenic cells, we first sought to establish a standard syngeneic BMT model in which highly purified hematopoietic stem cells (HSCs) from the mutant mice were transduced in vitro by lentiviral vectors encoding the Artemis gene product and subsequently transplanted into mutant recipients. Although the sensitivity of Artemis KO fibroblasts to radiation had previously been documented (5, 6), the sensitivity of KO animals to whole-body irradiation had not been addressed in those studies. Accordingly, preliminary experiments were performed to determine whether a suitable radiation dose to enable transplantation of transduced cells could be established. We found that, even at radiation doses considered sublethal for WT animals (i.e., 3, 2.5, or 2 Gy), all KO mice died between 4 and 12 weeks postirradiation (data not shown), suggestive of a nonhematopoietic toxicity. For this reason, we chose to evaluate two BMT models. First, we used Rag-1-deficient mice as the recipients for transplantation of transduced Artemis KO HSCs. Rag-deficient animals have been used previously as recipients for immune rescue studies (14, 15). Although they lack T and B lymphocytes, Rag-1-deficient mice readily tolerate the lethal doses of irradiation necessary to achieve complete myeloablation (unpublished results). To explore a more clinical relevant model, we asked whether transduced Artemis KO HSCs could be effectively introduced into Artemis KO animals using a nonmyeloablative regimen for the conditioning of BMT recipients previously described by others (11, 16–18).
For expression of the Artemis gene product, several lentiviral vectors were constructed in which different internal promoters [CMV, EF1α, and phosphoglycerate kinase (PGK)] were used to drive expression of the transgene (Fig. 1A), because we were interested in determining whether specific levels of Artemis expression or other less well understood characteristics of gene expression afforded by the different promoters might be important for efficient immune reconstitution. Previous studies with similar vectors indicated that the different promoters led to significantly different levels of reporter gene expression in bone marrow and peripheral blood (PB) in mice engrafted with transduced stem cells (19).
Fig. 1.

Lentiviral vector, transduction and expression of human Artemis. (A) Schematic representation of the pHAGE lentiviral construct used to express human Artemis cDNA from three different internal promoters, i.e., CMV, EF1α, and PGK. PBS-K, lysine tRNA-binding site; PSI, packaging signal; RRE, Rev responsive element; CPPT, central polypurine tract; WPRE, Woodchuck hepatitis posttranscriptional regulatory element; dU3, deleted U3. (B) Western blot analysis of lysates purified from 293 HEK cells transduced with lentiviral constructs expressing human Artemis. HEK 293, uninfected control. (C) Southern blot analysis of gDNA purified from bone marrow of transduced/transplanted mice. A representative sample from mice transplanted with cells transduced with lentiviruses expressing human Artemis (CMV, EF1α, and PGK) or GFP is shown. Increasing amounts of known plasmid vector DNA was used for copy number control. (D) RT-PCR of marrow samples as in C. Each group shows the undiluted and 1:3 diluted template reactions. GAPDH amplification serves as loading control.
For each of the constructed vectors, we were able to generate high-titer virus (>10e9/ml after virus concentration) from the corresponding DNA constructs (data not shown). To assess the ability of the different vectors to drive Artemis expression, each of the viruses were used to infect human 293 cells and subsequently, extracts prepared from the transduced cells were subjected to Western blot analysis. As shown in Fig. 1B, cells transduced with each virus expressed an immunoreactive protein identical in size to that of authentic human Artemis.
To test the ability of the different viruses to correct the Artemis immunodeficiency in vivo, we made use of a transduction protocol previously developed in our laboratory. In this protocol, highly purified murine HSCs are transduced under conditions of minimal in vitro manipulation (19). Those conditions for transduction/transplantation appear to maintain levels of stem cell activity comparable to fresh unmanipulated cells (19) and therefore may be particularly well suited for eventual clinical applications.
Correction of Artemis Deficiency in the Rag-1 KO Model.
In a first series of experiments, purified HSCs derived from Artemis KO mice were transduced by either lenti-CMV-huArtemis, lenti-EF1α-huArtemis, lenti-PGK-huArtemis, or lenti-GFP (negative control), and 2,000 transduced cells were transplanted into lethally irradiated Rag-1 KO recipients (n = 4–6 per group). One group received HSCs purified from WT CD45.1 mice as positive control. As expected from our previous studies, the transduction protocol led to high levels of gene transfer, as evidenced by analysis of genomic DNA purified from the bone marrow cells of transplanted animals (Fig. 1C). This transduction led as well to the expression of specific human Artemis mRNA, as detected by RT-PCR (Fig. 1D). Interestingly, however, there was no apparent consistent correlation between RNA levels observed in total bone marrow and the choice of vector, in contrast to what was observed in our previous studies (ref. 19; also see Discussion). Cells from mice that had engrafted with HSCs transduced with a GFP virus using the same protocol exhibited high levels of GFP expression (70–85%) in PB throughout the duration of the study (data not shown) and, as expected, showed no expression of Artemis RNA (Fig. 1D).
Recovery of B lymphocytes in the PB of BMT recipients engrafted with genetically modified Artemis KO HSCs was measured every 4 weeks by using antibodies against surface B220 and IgM receptors. As shown in Fig. 2A and B, expression of human Artemis resulted in the complete restoration of the B cell population in PB. In contrast to mice transplanted with cells transduced with GFP, which showed complete absence of mature B cells, rescued mice showed the presence of B220+ IgM+ cells at levels almost indistinguishable from those observed in mice transplanted with WT HSCs. Interestingly, only Artemis expression driven by the PGK promoter was able to fully recover the deficiency. Complete B cell rescue was also observed in the spleens of mice receiving lenti-PGK transduced HSCs, which contained normal numbers of B220+ IgM+ splenocytes (Fig. 2B), together with a significant recovery of spleen size (Fig. 2C). In addition, Artemis-expressing transduced HSCs gave rise to B cells capable of undergoing normal V(D)J rearrangements as evidenced by PCR using DNA extracted from splenocytes (Fig. 2D). Accordingly, this immune correction led to the detection of normal levels of both IgM and IgG in serum (Fig. 2 E and F). Interestingly, mice receiving cells transduced with the CMV and EF1α constructs, while having reduced levels of IgG showed normal levels of IgM in serum, despite the relatively low levels of peripheral B cell rescue, a phenomenon that has been described (20).
Fig. 2.

B cell rescue in Rag-1 KO transplanted mice. (A) Rescue of B220+ IgM+ mature B cells in PB of transplanted mice over time. Recovery of mice receiving cells transduced with lenti-CMV (n = 4), lenti-EF1α (n = 6), lenti-PGK (n = 6), lenti-GFP control (n = 6), or cells from WT CD45.1 (n = 4) control is shown. (B) Representative FACS plots of individual mice transplanted with cells as in A. The presence of B220+ IgM+ mature B cells in PB and spleen (SP) is shown. (C) Representative pictures of spleens isolated from mice rescued by using cells transduced with lenti-PGK or -GFP or from mice transplanted with WT CD45.1 cells. (D) PCR analysis of genomic DNA purified from splenocytes was used to evidence the presence of V(D)J rearrangements. Each lane corresponds to an individual mouse. (E) IgM levels in serum of transplanted mice are shown. (F) IgG levels in serum of transplanted mice are shown. For details, see Materials and Methods.
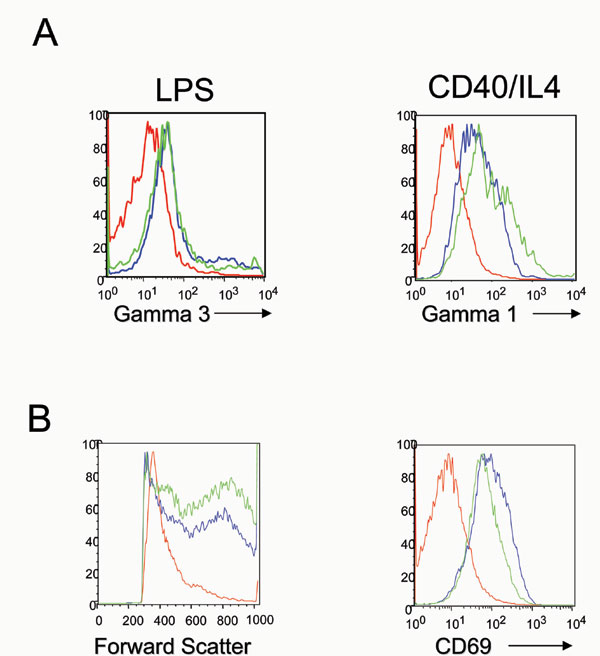
Lastly, we sought to determine the capacity of corrected B cells to undergo proliferation and switching upon cytokine activation. For this purpose, splenocytes isolated from transplanted mice were incubated in vitro in the presence of either LPS (to induce proliferation and switching to IgG3) or CD40-IL4 (to induce proliferation and switching to IgG1). Corrected cells were able to respond normally to stimulation, with robust proliferation, increase in size and class switching to the respective Ig type (Fig. 6A, which is published as supporting information on the PNAS web site).
To study the recovery of the T cell compartment, PB was analyzed for the presence of mature CD4 or CD8 single positive T cells. In contrast to control mice, animals receiving HSCs transduced with Artemis showed significant numbers of mature T lymphocytes in circulation (Fig. 3A and B). As with B cells, the PGK-driven construct showed the most robust T lymphocyte recovery, with numbers of CD4+ and CD8+ T cells that increased over the time course of the experiment. Although the kinetics of T cell recovery were somewhat slower in comparison to mice receiving WT cells, all mice that received PGK—Artemis-transduced cells eventually achieved normal numbers of mature CD4+ T lymphocytes and ≈50% of the number of CD8+ T cells by 16 weeks post-transplantation. In addition, thymuses of treated mice showed normal percentages of CD4+ CD8+ double- and single-positive thymocytes (Fig. 3B), as well as recovery of the macroscopic size of the organ (data not shown). The presence of double positive and CD4+ single-positive cells in thymus as well as low levels of CD4+ cells in PB of GFP transduced/transplanted mice relates to the “leakiness” in the T cell compartment of Artemis KO mice, as reported (5). Finally, when purified splenocytes were stimulated in vitro in the presence of ConA and IL-2, they underwent proliferation, differentiating into mostly CD3+ cells and expressing the activation marker CD69 (Fig. 6B). These data demonstrate that the rescued mature T cells were fully functional.
Fig. 3.

T cell rescue in Rag-1 KO transplanted mice. (A) Rescue of CD4+ mature T cells (Left) and CD8+ mature T cells (Right) in PB of transplanted mice over time. Recovery of mice receiving cells transduced with lenti-CMV (n = 4), lenti-EF1α (n = 6), lenti-PGK (n = 6), lenti-GFP control (n = 6), or cells from WT CD45.1 control (n = 4) is shown. (B) Representative FACS plots of individual mice transplanted with cells as in A. PB cells and thymocytes were stained by using antibodies against CD4 and CD8.
Correction of Artemis Deficiency Using a Nonmyeloablative BMT Model Involving Artemis KO Mice.
Although the results obtained in the Rag-1 KO model clearly establish the ability of gene modification to correct the Artemis defect, an important objective of our studies was to establish the practical feasibility of correction of the disease in RS-SCID human patients. Based on our inability to develop a lethal-irradiation BMT model for correction of the disease, because of the extreme radiosensitivity of Artemis KO mice, we next asked whether nonmyeloablative conditioning regimens could be used to enable effective transplantation of gene-corrected Artemis KO stem cells. To this purpose, we used a nonmyeloablative protocol that makes use of Busulfan, a reagent that has been extensively used as a conditioning agent not only in mice but also in human patients (11, 16–18). In contrast to whole-body irradiation, Busulfan seems to specifically affect nondividing stem cells in bone marrow (21, 22), and doses that are relatively non toxic are still possible while allowing for long-term engraftment of transplanted HSCs (23). Indeed, all treated mice responded well to the dose used in these studies (2 × 10 mg/kg). Artemis KO mice were conditioned with Busulfan and then transplanted with 2,000 HSCs that were transduced with either the lenti-PGK vector (n = 7) or a GFP lentiviral vector control (n = 3). One group received 2,000 HSCs purified from WT CD45.1 donors (n = 3) as a positive control. Similar to what we observed in the Rag-1 KO model, all mice receiving Artemis-transduced HSCs showed rescue of mature B and T lymphocytes in their circulation, in sharp contrast to mice transplanted with GFP-transduced cells. The rescue was detectable already at 4 weeks after BMT and steadily increased over time (Fig. 4A and B). This finding correlated well with the recovery in the absolute number of lymphocytes in blood (Fig. 4C). Importantly, Artemis-transduced HSCs performed almost identically to HSCs purified from WT CD45.1 mice, confirming that the expression of human Artemis driven by the PGK promoter is capable of fully correcting the Artemis defects. The overall level of B cell recovery in mice receiving PGK—Artemis-transduced HSCs as well as mice transplanted with CD45.1 HSCs did not reach WT values (Fig. 4A). This finding contrasts the complete B cell rescue obtained in Rag-1 KO transplanted mice and could be attributed to the lower donor engraftment obtained with the dose of Busulfan used in these studies, compared with the engraftment observed in Rag-1-deficient transplanted mice undergoing whole-body irradiation (Fig. 7, which is published as supporting information on the PNAS web site). The correction of both lymphoid populations was also evident in the spleen (Fig. 4B), in which the level of mature B and T cells obtained after lentiviral gene transfer was almost identical from that of mice receiving WT CD45.1 cells.
Fig. 4.

Rescue of the immune system of Artemis-deficient mice. (A) Rescue of B220+ IgM+ mature B cells and CD4+/CD8+ mature T cells in PB of transplanted mice over time. Recovery of mice receiving cells transduced with lenti-PGK (n = 7), lenti-GFP (n = 3) or cells from WT CD45.1 (n = 3) control is shown. (B) Representative FACS plots of individual mice transplanted with cells as in A. The presence of B220+ IgM+ mature B cells and CD4+/CD8+ mature T cells in PB and spleen is shown. (C) Total number of lymphocytes in PB of transplanted mice 16 weeks after BMT compared with unmanipulated C57BL/6 mice (WT B6).
To demonstrate that these lymphocytes were functionally normal, we performed an in vivo immune challenge against a T cell-dependent antigen, keyhole limpets hemocyanin (KLH). As shown in Fig. 5A, Artemis-corrected mice showed a specific IgM, and more importantly, a robust IgG anti-KLH response, similar to what was observed in the positive control mice. Furthermore, splenocytes purified from corrected mice were also stimulated in vitro as described above, with proliferation and expression of the activation marker CD69 when using T cell conditions (Con-A + IL-2) or proliferation and switching when using B cell conditions (LPS or CD40 + IL-4) (data not shown).
Fig. 5.

In vivo challenge and recovery of lymphocyte progenitors in Artemis KO rescued mice. (A) Relative levels of anti-keyhole limpets hemocyanin IgM (Left) or IgG (Right) antibodies in serum of mice receiving cells transduced with lenti-PGK or -GFP or cells from CD45.1 (n = 3 in all groups). Values represent levels of antibodies detected at 1:1,250 dilution of serum obtained either before (“pre”) or 28 days after challenge (“d28”). Data are expressed as mean values (±SEM) of triplicate samples. (B) Representative FACS plots of individual mice from the different groups. Bone marrow samples were analyzed for the presence of different B cell progenitors by using antibodies against IgM, B220, and CD43. Pregated IgM− cells were further defined as B220+ CD43+ (pro-B cells) or B220+ CD43− (pre-B cells). The red square denotes the percentage of pre-B cells in all of the different mice. (C) Representative FACS plots of individual mice transplanted as in B. Thymuses were stained by using antibodies against CD4 and CD8.
Finally, we sought to analyze bone marrow and thymus of treated mice to study the recovery of the progenitor populations. In untreated Artemis-deficient mice, B cell development is stalled at the IgM− B220+ CD43+ (pro-B) progenitor stage in bone marrow (5). Staining of bone marrow samples showed the rescue of the pre-B cell compartment (B220+ CD43−), which was completely absent in mice receiving GFP-transduced HSCs (Fig. 5B). Interestingly, the rescue of this pre-B cell population in Artemis-treated mice was not as efficient as observed in Rag-1 KO transplanted animals (Fig. 8, which is published as supporting information on the PNAS web site; also see Discussion). In addition, thymuses extracted from rescued mice showed normal ratios of all thymocyte populations (Fig. 5C), mirroring the profile observed in thymuses extracted from mice transplanted with WT CD45.1 HSCs.
Adverse Effects Associated with the Gene Transfer/BMT Protocol.
During the course of the studies, 3 of 24 Rag-1 KO transplanted mice developed moist dermatitis as a result of lethal irradiation. In addition, one of the mice receiving lenti-PGK-transduced cells had an enlarged spleen (splenomegaly) that was discovered during tissue analysis. However, this finding was not accompanied by the appearance of abnormal blood cell counts or abnormal cell populations in bone marrow or thymus of the animal (data not shown). We did not observe any abnormalities in Artemis-transplanted mice.
Discussion
The studies reported here clearly demonstrate the ability to fully correct the immunodeficiency manifest in Artemis KO mice through the transduction of mutant cells by lentiviral vectors encoding the Artemis gene product. Our results therefore suggest that, like other forms of SCID, RS-SCID may be an appropriate candidate for treatment by gene therapy. In contrast to previous efforts to establish preclinical models for the treatment of other forms of SCID by gene therapy, which generally involved the use of standard BMT protocols, our studies were complicated by the extreme radiosensitivity of the Artemis KO mice, which precluded the conditioning of BMT recipients by lethal doses of irradiation. For this reason, we chose to establish both a BMT model in which corrected Artemis KO cells were transferred into Rag-1-deficient hosts and a true syngeneic model in which corrected cells were introduced into Artemis KO recipients conditioned by nonmyeloablative doses of Busulfan. Although in the case of this latter model, the conditioning regimen used did not result in complete donor-cell-derived reconstitution, effective correction of the functional immunological defects manifest in Artemis KO mice was readily observed. Because Busulfan has already been used as part of a conditioning regimen for RS-SCID patients undergoing BMT (24), our studies provide an important proof of principle for the use of such nonmyeloablative regimens in future gene therapy protocols involving RS-SCID patients.
Before our studies, Li et al. (13) had described the generation of a distinct murine Artemis KO strain and evaluated the ability to correct the immunodeficiency manifest in this strain by transplantation of WT cells. An important finding from those studies was that, whereas transplantation of enriched stem cell populations into nonconditioned Artemis recipients led to a modest restoration of the T lymphoid cell compartment, little engraftment of the B lymphoid compartment could be achieved. Sublethal irradiation of recipients led to significantly improved T and B cell reconstitution. Our inability to define sublethal doses of irradation in our Artemis KO strain contrasts the studies of Li et al. (13). Whether this apparent differential sensitivity to irradiation between the two KO strains is truly related to strain or mutation differences or simply to technical differences in the manner the BMT protocols were performed is unclear.
The findings of Li et al. (13) are consistent with clinical studies of RS-SCID patients transplanted in the absence of conditioning, which have shown that B cell reconstitution is most often poor (24, 25). Recently, Liu et al. (26) have proposed that the extent of B cell reconstitution observed in both murine and clinical studies in which BMT is performed in the absence of conditioning likely depends upon whether the genetic lesion underlying the immunodeficiency results in a normal pro-B cell compartment. The incomplete restoration of the B cell compartment observed in our studies of Artemis recipients transplanted with WT or gene-modified mutant cells after Busulfan treatment therefore likely reflects the inability of our Busulfan regimen to completely ablate endogenous HSCs and/or the pro-B cell compartment known to be present in Artemis mice.
An interesting vector-related finding from our studies was that, whereas each of the different vectors used resulted in the transduction of HSCs at a comparable (and high) efficiency, only the lentiviral vector that made use of the PGK promoter led to a complete correction of the immunodeficiency. The explanation for this result is not entirely clear, because our previous BMT studies, which made use of a reporter transgene, had suggested that the CMV promoter should lead to the highest levels of transgene expression relative to the other promoters used (ref. 19 and unpublished results). More detailed studies of Artemis expression levels in different cell types may shed light on this important issue. At a minimum, our studies with the different vectors underscore the importance of tailoring the expression characteristics of the vector system to the specific disease being treated, as well as the utility of a preclinical animal model for choosing an appropriate vector for eventual clinical studies.
Materials and Methods
Mice.
Artemis-deficient mice were previously described (5). Rag-1-deficient and CD45.1 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All animals were housed in a specific-pathogen-free animal facility at Harvard Medical School. All animal procedures were approved by the Standing Committee on Animals of Harvard Medical School.
Lentiviral Constructs and Viral Production.
Human Artemis cDNA was amplified from existing cDNA sequences by using the following primers: forward 5′-ATGAGTTCTTTCGAGGGGCAGATG-3′ and reverse 5′-TTAGGTATCTAAGAGTGAGCATTTTCTTTTTTTG-3′. The amplified product was cloned into the pHAGE lentiviral vector by using NotI and BamHI. pHAGE is a third-generation self-inactivating lentiviral vector whose detailed structure will be described elsewhere (A. B. Balazs and R.C.M., unpublished work). Production of lentiviruses were done in 293T cells as described (19).
HSC Isolation, Viral Transduction, and Transplantations.
Purification of HSCs, transduction, and BMT were performed as described (19). Rag-1 KO recipient mice were lethally irradiated with two doses of 7 Gy, 3 h apart, 1 day before BMT and maintained under antibiotic-supplemented water for 15 days. Artemis KO recipient mice received two doses of 10 mg/kg of Busulfan (Busulfex; ESP Pharma, Edison, NJ) on day −3 and −2 before transplant (23, 27) and maintained under antibiotic-supplemented water. Transduced cells were injected retroorbitally into recipient mice under isoflurane anesthesia.
Flow Cytometry, in Vitro Proliferation Assays, and in Vivo Challenge.
PB was obtained from the retroorbital plexus every 4 weeks and stained by using fluorescently conjugated anti-IgM-PE and B220-APC antibodies to detect B lymphocytes and anti-CD8-PE and CD4-APC antibodies to detect T lymphocytes. Sixteen to 20 weeks after BMT, single-cell suspensions were prepared from spleen, thymus, and bone marrow of transplanted mice and stained by using the same antibodies as above. For bone marrow samples, anti-CD43-FITC was also used. All antibodies were purchased from BD Biosciences Pharmingen (San Diego, CA). Cells were incubated with antibodies for 30 min on ice, washed once, and resuspended in PBS/1% BSA for analysis. Dead cells were excluded by using propidium iodide stain. Samples were analyzed in a FACScalibur machine (Beckton Dickinson, Fullerton, CA) and data processed by FlowJo software (Tree Star, Ashland, OR). For in vitro assays, 1 × 106 splenocytes were incubated in RPMI medium 1640 containing 15% FCS, 100 μM 2-mercaptoethanol, 10 mM Hepes, and antibiotics (all from Gibco, Carlsbad, CA) and supplemented with either 25 ng/ml LPS (for IgG3 switching), 500 ng/ml anti-CD40 (Pharmingen, Franklin Lakes, NJ), and 25 ng/ml mouse recombinant IL-4 (for IgG1 switching), as described (28), or 5 μg/ml ConA with 40 units/ml rhIL-2 (for T cell proliferation; ref. 29). Four days later, cells were stained for switching and activation by using anti IgG1-FITC, IgG3-PE, CD3-FITC, and CD69-PE (all from BD Biosciences Pharmingen) and analyzed by FACS. In vivo challenge was performed as described (30).
Western Blot Analysis.
Immunoblotting of cell lysates was performed by using standard procedures. Human Artemis protein was detected by using a rabbit anti-human polyclonal antibody (Bethyl Laboratories, Montgomery, TX).
PCR for V(D)J Rearrangement and Southern Blot Analysis.
PCR to detect V(D)J rearrangements was performed as described (31). Southern blot analysis was performed as described (19).
ELISA for Serum IgM and IgG.
ELISA 96-well plates (Corning, Corning, NY) were coated with 2 μg/ml goat anti-mouse IgM or goat anti-mouse IgG and incubated with serial dilutions of serum samples for 2 h at 37°C. After washing, plates were incubated with alkaline phosphatase-conjugated anti-IgM or -IgG antibodies for 45 min at 37°C, and detection was performed by using alkaline phosphatase substrate (Sigma-Aldrich, St. Louis, MO). All antibodies were purchased from SouthernBiotech (Birmingham, AL).
Supplementary Material
Acknowledgments
We thank David Jung, Ali Zarrin, Raul Mostoslavsky, Shan Zha, and Sonia Franco (all from Department of Genetics, Harvard Medical School) for technical assistance and reagents; Peggy Russell for invaluable work in tissue culture; and Alejandro Balazs, George Murphy, and Ittai Ben-Porath for critical reading of the manuscript. This work was supported by National Institutes of Health Grants 5P0-HL54785 (to R.C.M.) and AI35714 (to F.W.A.) and by a grant from L'Association Française contre les Myopathies. F.W.A. is an Investigator of the Howard Hughes Medical Institute.
Abbreviations
- SCID
severe combined immunodeficiency
- RS-SCID
radiation-sensitive SCID
- KO
knockout
- BMT
bone marrow transplantation
- HSC
hematopoietic stem cell
- PB
peripheral blood
- PGK
phosphoglycerate kinase.
Footnotes
The authors declare no conflict of interest.
References
- 1.Fischer A, Cavazzana-Calvo M, De Saint Basile G, DeVillartay JP, Di Santo JP, Hivroz C, Rieux-Laucat F, Le Deist F. Annu Rev Immunol. 1997;15:93–124. doi: 10.1146/annurev.immunol.15.1.93. [DOI] [PubMed] [Google Scholar]
- 2.Moshous D, Callebaut I, de Chasseval R, Corneo B, Cavazzana-Calvo M, Le Deist F, Tezcan I, Sanal O, Bertrand Y, Philippe N, et al. Cell. 2001;105:177–186. doi: 10.1016/s0092-8674(01)00309-9. [DOI] [PubMed] [Google Scholar]
- 3.Li L, Moshous D, Zhou Y, Wang J, Xie G, Salido E, Hu D, de Villartay JP, Cowan MJ. J Immunol. 2002;168:6323–6329. doi: 10.4049/jimmunol.168.12.6323. [DOI] [PubMed] [Google Scholar]
- 4.Cavazzana-Calvo M, Le Deist F, De Saint Basile G, Papadopoulo D, De Villartay JP, Fischer A. J Clin Invest. 1993;91:1214–1218. doi: 10.1172/JCI116282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rooney S, Sekiguchi J, Zhu C, Cheng HL, Manis J, Whitlow S, DeVido J, Foy D, Chaudhuri J, Lombard D, Alt FW. Mol Cell. 2002;10:1379–1390. doi: 10.1016/s1097-2765(02)00755-4. [DOI] [PubMed] [Google Scholar]
- 6.Rooney S, Alt FW, Lombard D, Whitlow S, Eckersdorff M, Fleming J, Fugmann S, Ferguson DO, Schatz DG, Sekiguchi J. J Exp Med. 2003;197:553–565. doi: 10.1084/jem.20021891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma Y, Pannicke U, Schwarz K, Lieber MR. Cell. 2002;108:781–794. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 8.Buckley RH, Schiff SE, Schiff RI, Markert L, Williams LW, Roberts JL, Myers LA, Ward FE. N Engl J Med. 1999;340:508–516. doi: 10.1056/NEJM199902183400703. [DOI] [PubMed] [Google Scholar]
- 9.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, Lim A, Osborne CS, Pawliuk R, Morillon E, et al. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 10.Kohn DB, Sadelain M, Glorioso JC. Nat Rev Cancer. 2003;3:477–488. doi: 10.1038/nrc1122. [DOI] [PubMed] [Google Scholar]
- 11.Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A, Morecki S, Andolfi G, Tabucchi A, Carlucci F, et al. Science. 2002;296:2410–2413. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 12.Hacein-Bey-Abina S, Le Deist F, Carlier F, Bouneaud C, Hue C, De Villartay JP, Thrasher AJ, Wulffraat N, Sorensen R, Dupuis-Girod S, et al. N Engl J Med. 2002;346:1185–1193. doi: 10.1056/NEJMoa012616. [DOI] [PubMed] [Google Scholar]
- 13.Li L, Salido E, Zhou Y, Bhattacharyya S, Yannone SM, Dunn E, Meneses J, Feeney AJ, Cowan MJ. J Immunol. 2005;174:2420–2428. doi: 10.4049/jimmunol.174.4.2420. [DOI] [PubMed] [Google Scholar]
- 14.Klein C, Nguyen D, Liu CH, Mizoguchi A, Bhan AK, Miki H, Takenawa T, Rosen FS, Alt FW, Mulligan RC, et al. Blood. 2003;101:2159–2166. doi: 10.1182/blood-2002-05-1423. [DOI] [PubMed] [Google Scholar]
- 15.Soudais C, Shiho T, Sharara LI, Guy-Grand D, Taniguchi T, Fischer A, Di Santo JP. Blood. 2000;95:3071–3077. [PubMed] [Google Scholar]
- 16.Andersson BS, Kashyap A, Gian V, Wingard JR, Fernandez H, Cagnoni PJ, Jones RB, Tarantolo S, Hu WW, Blume KG, et al. Biol Blood Marrow Transplant. 2002;8:145–154. doi: 10.1053/bbmt.2002.v8.pm11939604. [DOI] [PubMed] [Google Scholar]
- 17.Down JD, Ploemacher RE. Exp Hematol. 1993;21:913–921. [PubMed] [Google Scholar]
- 18.Yeager AM, Shinn C, Pardoll DM. Blood. 1991;78:3312–3316. [PubMed] [Google Scholar]
- 19.Mostoslavsky G, Kotton DN, Fabian AJ, Gray JT, Lee J-S, Mulligan RC. Mol Ther. 2005;11:932–940. doi: 10.1016/j.ymthe.2005.01.005. [DOI] [PubMed] [Google Scholar]
- 20.Yates F, Malassis-Seris M, Stockholm D, Bouneaud C, Larousserie F, Noguiez-Hellin P, Danos O, Kohn DB, Fischer A, de Villartay JP, et al. Blood. 2002;100:3942–3949. doi: 10.1182/blood-2002-03-0782. [DOI] [PubMed] [Google Scholar]
- 21.Botnick LE, Hannon EC, Vigneulle R, Hellman S. Cancer Res. 1981;41:2338–2342. [PubMed] [Google Scholar]
- 22.Fried W, Kedo A, Barone J. Cancer Res. 1977;37:1205–1209. [PubMed] [Google Scholar]
- 23.Yeager AM, Shinn C, Shinohara M, Pardoll DM. Transplantation. 1993;56:185–190. doi: 10.1097/00007890-199307000-00034. [DOI] [PubMed] [Google Scholar]
- 24.O'Marcaigh AS, DeSantes K, Hu D, Pabst H, Horn B, Li L, Cowan MJ. Bone Marrow Transplant. 2001;27:703–709. doi: 10.1038/sj.bmt.1702831. [DOI] [PubMed] [Google Scholar]
- 25.Haddad E, Landais P, Friedrich W, Gerritsen B, Cavazzana-Calvo M, Morgan G, Bertrand Y, Fasth A, Porta F, Cant A, et al. Blood. 1998;91:3646–3653. [PubMed] [Google Scholar]
- 26.Liu A, Vosshenrich CA, Lagresle-Peyrou C, Malassis-Seris M, Hue C, Fischer A, Di Santo JP, Cavazzana-Calvo M. Blood. 2006;108:1123–1128. doi: 10.1182/blood-2006-01-0061. [DOI] [PubMed] [Google Scholar]
- 27.Andersson G, Illigens BM, Johnson KW, Calderhead D, LeGuern C, Benichou G, White-Scharf ME, Down JD. Blood. 2003;101:4305–4312. doi: 10.1182/blood-2002-06-1649. [DOI] [PubMed] [Google Scholar]
- 28.Manis JP, Dudley D, Kaylor L, Alt FW. Immunity. 2002;16:607–617. doi: 10.1016/s1074-7613(02)00306-0. [DOI] [PubMed] [Google Scholar]
- 29.Sleckman BP, Bardon CG, Ferrini R, Davidson L, Alt FW. Immunity. 1997;7:505–515. doi: 10.1016/s1074-7613(00)80372-6. [DOI] [PubMed] [Google Scholar]
- 30.Lagresle-Peyrou C, Yates F, Malassis-Seris M, Hue C, Morillon E, Garrigue A, Liu A, Hajdari P, Stockholm D, Danos O, et al. Blood. 2006;107:63–72. doi: 10.1182/blood-2005-05-2032. [DOI] [PubMed] [Google Scholar]
- 31.Ehlich A, Martin V, Muller W, Rajewsky K. Curr Biol. 1994;4:573–583. doi: 10.1016/s0960-9822(00)00129-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}