

Abstract
Expression of Tumor Necrosis Factor alpha (TNF-α), a pleiotropic cytokine, is elevated during stroke and cerebral ischemia. TNF-α regulates arterial diameter, although mechanisms mediating this effect are unclear. Here, we tested the hypothesis that TNF-α regulates the diameter of resistance-size (∼150 μm diameter) cerebral arteries by modulating local and global intracellular calcium (Ca2+) signals in smooth muscle cells. Laser-scanning confocal imaging revealed that TNF-α increased Ca2+ spark and Ca2+ wave frequency, but reduced global Ca2+ concentration ([Ca2+]i) in smooth muscle cells of intact arteries. TNF-α elevated reactive oxygen species (ROS) in smooth muscle cells of intact arteries and this was prevented by apocynin or diphenyleneiodonium (DPI), NAD(P)H oxidase blockers, but was unaffected by inhibitors of other ROS generating enzymes. In voltage-clamped (-40 mV) cells, TNF-α increased the frequency and amplitude of Ca2+ spark-induced large-conductance Ca2+-activated potassium (KCa) channel transients ∼1.7-fold and ∼1.4-fold, respectively. TNF-α-induced transient KCa current activation was reversed by apocynin or MnTMPyP, a membrane-permeant antioxidant, and prevented by intracellular dialysis of catalase. TNF-α induced reversible and similar amplitude dilations in either endothelium-intact or-denuded pressurized (60 mm Hg) cerebral arteries. MnTMPyP, thapsigargin, a sarcoplasmic reticulum Ca2+ ATP-ase blocker that inhibits Ca2+ sparks, and iberiotoxin, a KCa channel blocker, reduced TNF-α-induced vasodilations to between 15 and 33% of control. In summary, data indicate that TNF-α activates NAD(P)H oxidase, resulting in an increase in intracellular H2O2 that stimulates Ca2+ sparks and transient KCa currents, leading to a reduction in global [Ca2+]i, and vasodilation.
Keywords: cerebrovascular circulation, ryanodine-sensitive calcium release channel, calcium-activated potassium channel, reactive oxygen species, vasodilation
INTRODUCTION
Stroke and transient brain ischemia are vascular-born brain pathologies accompanied by inflammatory changes in blood vessels and parenchyma tissues (15; 46). Tumor Necrosis Factor-alpha (TNF-α), a pleiotropic cytokine, is a pro-inflammatory agent generated by glial cells, neurons, macrophages and vascular endothelium during brain injury. Clinical data demonstrate a correlation between TNF-α levels and the severity of ischemic brain damage, but controversy exists regarding the functional role of TNF-α, with both beneficial and detrimental roles proposed (3; 8; 9; 15; 36; 46). A chronic TNF-α elevation is associated with risk factors for other vascular pathologies, including coronary artery disease and atherosclerosis (26). During stroke, TNF-α may cause neuronal damage directly and by accelerating ischemia-induced local inflammatory responses. In contrast, studies also indicate that TNF-α may be cytoprotective (8; 15; 36; 46).
TNF-α acts in a paracrine manner by binding to two membrane receptors, p55 and p75. Besides its actions on neurons, astrocytes and microglia, TNF-α also regulates arterial contractility (15), an effect that may modulate injury by altering local blood supply. However, the regulation of arterial diameter and blood flow by TNF-α is complex. In vivo, intracranial injection of TNF-α constricted pial arterioles and reduced cerebral blood flow (29; 38; 44). TNF-α also induced pro-contractile effects in coronary arteries (23; 51). In contrast, TNF-α dilated cerebral, mesenteric and cremaster muscle arterioles, and relaxed endothelium-denuded aorta (4; 7; 16; 22; 32; 37). In bronchial arteries, TNF-α initially induced dilation followed by constriction 2 hours later (45). TNF-α-mediated vasoregulation can also occur through both endothelium-dependent and endothelium-independent mechanisms (4; 7; 16; 22; 23; 29; 32; 37; 38; 44; 45; 51). Collectively, data suggest TNF-α can induce both vasoconstriction and vasodilation, and effects may be time-dependent. Regardless, mechanisms mediating vasoregulation by TNF-α are poorly understood.
A major regulator of arterial diameter is the smooth muscle cell global intracellular Ca2+ concentration ([Ca2+]i). In smooth muscle cells, global [Ca2+]i is regulated by extracellular Ca2+ influx and intracellular Ca2+ release (19). An elevation in global [Ca2+]i induces constriction, whereas a decrease in global [Ca2+]i leads to vasodilation. Another Ca2+ signal, termed a “Ca2+ spark ”, is a rapid, localized intracellular Ca2+ transient that occurs due the activation of several ryanodine-sensitive Ca2+ release (RyR) channels on the sarcoplasmic reticulum (SR) (19; 33). In arterial smooth muscle cells, Ca2+ sparks occur close to the plasma membrane and activate several large-conductance Ca2+-activated K+ (KCa) channels, resulting in a transient KCa current. The resulting membrane hyperpolarization reduces voltage-dependent Ca2+ channel activity, leading to a decrease in Ca2+ entry, a reduction in global [Ca2+]i and vasodilation (19; 33). In contrast, Ca2+ spark inhibition leads to vasoconstriction due to depolarization-induced voltage-dependent Ca2+ channel activation (19). Alterations in Ca2+ sparks and effective coupling to KCa channels have been implicated in hypertension and diabetes (1; 6; 19; 31). Another intracellular Ca2+ signal, termed a “Ca2+ wave ”, is a propagating [Ca2+]i elevation that occurs due to the activation of SR RyR channels and/or inositol-trisphosphate-gated Ca2+ channels. Although the physiological functions of Ca2+ waves are less well described, when stimulated, Ca2+ waves contribute Ca2+ for contraction (35).
The goal of this study was to investigate whether TNF-α regulates cerebral artery diameter by modulating smooth muscle Ca2+ signaling and if so, to determine the underlying mechanisms. Such information would improve our understanding of cytokine-mediated changes in arterial diameter during vascular pathologies, including stroke.
METHODS
Tissue preparation and single cell isolation
Sprague-Dawley rats (∼250 g) of either sex were euthanized by peritoneal injection of a sodium pentobarbital overdose (150 mg kg-1) in accordance with the Animal Care and Use Committee policies and procedures at the University of Tennessee. The brain was removed and placed into ice-cold (4 °C), oxygenated (95 % O2-5 % CO2), physiological saline solution (PSS) containing (mM): 119 NaCl, 4.7 KCl, 24 NaHCO3, 1.2 KH2PO4, 1.6 CaCl2, 1.2 MgSO4, 0.023 EDTA and 11 glucose. Posterior cerebral, middle-cerebral and cerebellar arteries (50-200 μm diameter) were removed, cleaned of connective tissue and maintained in ice-cold PSS. Where appropriate, the endothelium was removed by introducing an air bubble into the artery lumen for 2 minutes followed by a 30 s wash with H2O. Individual smooth muscle cells were dissociated from cerebral arteries using enzymes and an isolation solution of the following composition (in mM): 55 NaCl, 80 sodium glutamate, 5.6 KCl, 2 MgCl2, 10 HEPES, and 10 glucose (pH 7.3 with NaOH), a previously described (18). Cells were stored on ice and used for experiments between 1 and 6 hours after isolation.
Laser scanning confocal Ca2+ imaging
Artery segments (1-2 mm in length) were placed into HEPES-buffered PSS of composition (mM): 134 NaCl, 6 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES and 10 glucose (pH 7.4, NaOH) containing fluo-4 AM (10 μM) and pluronic F-127 (0.05 %) for 60 min at 22 °C. To allow indicator de-esterification, arteries were subsequently placed into HEPES-buffered PSS for 30 min at 22 °C. Smooth muscle cells within the artery wall were imaged using a Noran Oz laser scanning confocal microscope (Noran Instruments, Middleton, WI, USA) and a ×60 water immersion objective (NA = 1.2) attached to a Nikon TE300 microscope. Cells were illuminated with a krypton-argon laser at 488 nm and emitted light greater than 500 nm was collected. Planar images (56.3 μm × 52.8 μm) were recorded every 16.7 ms, i.e. 60 images s-1. To compare confocal Ca2+ imaging data with electrophysiological recordings performed in this study, Ca2+ sparks in smooth muscle cells of artery segments were measured in an extracellular solution containing 30 mM K+, which depolarizes smooth muscle cells to ∼-40 mV. This membrane potential is similar to that of cerebral arteries pressurized to 60 mmHg (see (20) for similar procedure). The 30 mM K+ bath solution contained (mM): NaCl 110; KCl 30; HEPES 10; CaCl2 2; MgCl2 1; and glucose 10 (pH 7.4, NaOH). At least two different representative areas of the each artery were scanned for at least 10 s under each condition. The same area of artery was scanned only once to avoid any laser-induced changes in Ca2+ signaling and the effects of drugs were measured in paired experiments. Ca2+ sparks were detected in smooth muscle cells using custom analysis software written using IDL 5.2 (Research Systems Inc., Boulder, CO, USA) kindly provided by Drs M. T. Nelson and A. D. Bonev (University of Vermont, VT, USA). Automated and manual detection of Ca2+ sparks were performed by dividing an area 1.54 μm (7 pixels) × 1.54 μm (7 pixels) (i.e. 2.37 μm2) in each image (F) by a baseline (F0) that was determined by averaging 10 images without Ca2+ spark activity. The entire area of each image was analyzed to detect Ca2+ sparks. A Ca2+ spark was defined as a localized increase in F/F0 that was greater than 1.2 (10). Artery Ca2+ spark frequency (Hz) was calculated by averaging values from at least two different areas. Ca2+ waves were defined as an elevation in F/F0 greater than 1.2 that propagated for at least 20 μm. Global Ca2+ fluorescence was calculated from the same images used for Ca2+ spark analysis and was the mean pixel value of 100 different images acquired over 10 s. To calculate whether TNF-α altered global Ca2+, global Ca2+ fluorescence in TNF-α was divided by the corresponding control value.
Patch-clamp electrophysiology
K+ currents were measured using either the conventional whole-cell or perforated-configuration of the patch-clamp technique with an Axopatch 200B amplifier (Axon Instruments, Union City, CA, USA). Bath solution was 6 mM K+ HEPES-buffered PSS (composition described above). For perforated-patch experiments, the pipette solution contained (mM): 110 potassium aspartate, 30 KCl, 10 NaCl, 1 MgCl2, 10 HEPES, 0.05 EGTA (pH 7.2 with KOH). For conventional whole-cell experiments, the pipette solution contained (mM): 140 KCl, 1.9 MgCl2, 0.037 CaCl2, 0.1 EGTA, 10 HEPES, 2 Na2ATP (pH 7.2 with KOH); the calculated free Ca2+ and free Mg2+ concentrations of this solution are 100 nM and 1 mM, respectively (WEBMAXC, Stanford University, CA, USA). In some experiments, catalase (200 U/ml) was included in the whole-cell pipette solution. Where appropriate, catalase was heat-inactivated by incubation at 92 °C for 25 min. TNF-α was applied to the bath solution 10 minutes after formation of the whole-cell configuration, to ensure exchange of the pipette solution with the cytosol. All experiments were performed using a holding potential of-40 mV. Membrane currents were recorded with a sample rate of 2.5 kHz and filtered at 1 kHz. Transient KCa current analysis was performed off-line. A transient KCa current was defined as the simultaneous opening of three or more KCa channels.
Reactive Oxygen Species (ROS) measurements
1) Dichlorofluorescein (DCF) imaging
ROS were measured in isolated intact artery segments using H2DCFDA (2`7`-dichlorodihydrofluorescein diacetate). Isolated endothelium-denuded cerebral artery segments (1-2 mm) were incubated for 45 min at room temperature in 10μM H2DCFDA. H2DCFDA is hydrolyzed by intracellular esterases to the nonfluorescent derivative H2DCF. ROS oxidize H2DCF to fluorescent DCF. DCF fluorescence was excited with 488 nm light and emitted light greater than 525 nm was collected. Planar images were acquired using a Zeiss LSM 5 laser-scanning confocal microscope. Images were recorded 10 minutes after TNF-α or H2O2 application. Time controls were also performed, wherein agents were not applied. Since DCF fluorescence increases spontaneously upon exposure to excitation light (12), artery regions were imaged only once. Images were background corrected. ImageJ software (NIH) was used for analysis.
2) Dihydroethidium (DHE) imaging
Cerebral artery segments were incubated for 20 min in HEPES-buffered PSS without (time-matched control) or with TNF-α. Arteries were then embedded in OTC and snap frozen in liquid N2. 30 μm thick transverse sections were cut, mounted on glass slides, and incubated with DHE (10 μM) in a 100 % N2 atmosphere in the dark for 30 min at 37°C. Arterial sections were excited with 488 nm light and emitted light >590 was collected using a Noran Oz laser-scanning confocal microscope. Custom written software was used to measure DHE fluorescence intensity. Fluorescence intensity in diazoxide was divided by that in time-matched controls and multiplied by 100 to determine effects of diazoxide on DHE intensity. Tissue processing did not elevate ROS, because DHE fluorescence intensity of arteries pre-incubated with MnTMPyP were 1.04 ± 0.06 of those that were untreated (P>0.05, n=5).
Pressurized artery diameter measurements
Cerebral artery segments (100-200 μm diameter) were placed in a temperature-controlled perfusion chamber (Living Systems, Vermont) and cannulated with glass pipettes at each end. The chamber was continuously perfused with physiological saline solution equilibrated with 5% CO2, 21% O2 and 74% N2 to pH 7.4 and maintained at 37°C. Arteries were observed with a charge-coupled device (CCD) camera attached to an inverted microscope (Nikon TS 100-F). Artery diameter was monitored using edge-detection software (IonOptix; Milton, MA). Artery intraluminal pressure was elevated to 60 mmHg, which resulted in the development of myogenic tone. After diameter stabilization, tested agents were applied by chamber perfusion. Artery diameter was digitized at 1 Hz using the edge-detection function of Ionwizard (Ionoptix). Endothelium-denudation was confirmed by the absence of a dilatory response to carbachol (10-5 M), an endothelium-dependent cerebral vasodilator. At the end of each experiment, passive artery diameter was determined using a Ca2+ -free bath solution containing 1 mM EGTA.
Data analysis
Values are expressed as mean ± standard error of the mean (SEM). Students t-test was used for comparing paired and unpaired data from two populations, and ANOVA and Student-Newman-Keuls test were used for multiple group comparisons. Evaluation of whether distributions were Gaussian was by the method of Kolmogorov and Smirnov. P<0.05 was considered significant.
Chemicals
Unless stated otherwise, all chemicals used in this study were obtained from Sigma or Calbiochem (USA). Papain was purchased from Worthington Biochemical Co. (Lakewood, NJ), fluo-4 AM and H2DCFDA from Molecular Probes (Eugene, OR) and rat recombinant TNF-α from BD Pharmingen (CA).
RESULTS
TNF-α activates Ca2+ sparks and Ca2+ waves and reduces global [Ca2+]i in smooth muscle cells of intact cerebral arteries
The regulation of local and global Ca2+ signals in smooth muscle cells by TNF-α was measured in cerebral artery segments. TNF-α (50 ng/ml) increased mean Ca2+ spark frequency from 1.53 ± 0.16 Hz to 2.6 ± 0.51 Hz, or ∼1.7 -fold (Fig. 1A, B). TNF-α did not change Ca2+ spark amplitude (F/F0; control, 1.36 ± 0.01, n=184 sparks; TNF-α, 1.34 ± 0.01, n=329 sparks; P>0.05). In the same cells, TNF-α also elevated mean Ca2+ wave frequency from 0.32 ± 0.02 Hz to 0.48 ± 0.03 Hz, or ∼1.5 -fold (Fig. 1C). In contrast, TNF-α decreased mean global [Ca2+]i to 0.85 ± 0.02 of control (n=6 arteries, p<0.05). These experiments suggest TNF-α elevates Ca2+ spark and wave frequency and reduces global [Ca2+]i in cerebral artery smooth muscle cells.
Fig. 1.
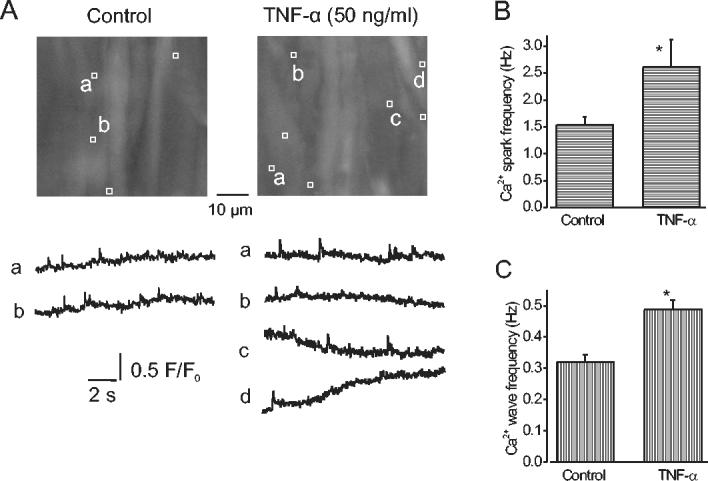
TNF-α activates Ca2+ sparks and Ca2+ waves in smooth muscle cells of intact cerebral arteries. A, average fluorescence (100 of 600 images) over 10 s of two different areas of the same cerebral artery in control and TNF-α. The locations of Ca2+ sparks that occurred during 10 s are indicated by white boxes (1.54 μm×1.54 μm) and representative localized F/F0 changes with time are illustrated below respective images and labeled accordingly. In control, 8 sparks were identified in traces a and b. In TNF-α, 11 sparks and 2 waves were detected in traces a-d. B, Mean effects of TNF-α (50 ng/ml) on Ca2+ spark frequency (n=6 arteries). C, Mean effects on Ca2+ wave frequency. * P<0.05, when compared with control.
TNF-α activates NAD(P)H oxidase leading to a ROS elevation in smooth muscle cells of intact cerebral arteries
TNF-α activates ROS generation in smooth muscle cells (13). Since RyR channels are redox-sensitive (42), we tested the hypothesis that TNF-α regulates local Ca2+ signaling by elevating intracellular ROS. Several enzymes generate ROS including NAD(P)H oxidase, xanthine oxidase, cytochrome P450, and mitochondrial electron transport chain complexes (13; 28; 41; 49). The DCF fluorescence method was used to determine pathways by which TNF-α elevates ROS in smooth muscle cells of small cerebral arteries. TNF-α increased DCF fluorescence intensity 2.7 ± 0.32-fold, and DHE intensity 1.51 ± 0.03-fold, in smooth muscle cells of intact arteries (Fig. 2A-D). Over the same time period, H2O2 elevated DCF fluorescence 4.7 ± 0.68-fold, whereas in control (absence of applied agents) DCF fluorescence did not change (Fig. 2B). In the presence of apocynin or diphenyleneiodonium (DPI), NAD(P)H oxidase blockers, TNF-α did not elevate DCF fluorescence (Fig. 2B). MnTMPyP, a membrane-permeant superoxide and catalase mimetic, also prevented the TNF-α-induced DCF fluorescence elevation (Fig. 2B). Apocynin (1.07 ± 0.23 of control, n=5), DPI (0.85 ± 0.1, n=4), and MnTMPyP (1.14 ± 0.17, n=4) did not change ROS when applied alone (P>0.05 for each). Oxypurinol, a xanthine oxidase inhibitor, 17-octadecanoic acid, a cytochrome P-450 blocker, or lonidamine, a mitochondrial permeability transition pore (PTP) opener did not reduce TNF-α-induced DCF fluorescence elevations (Fig. 2B). Oxypurinol (0.98 ± 0.13 of control, n=4), 17-octadecanoic acid (1.09 ± 0.12, n=5), and lonidamine (0.86 ± 0.13, n=4) did not change DCF fluorescence when applied alone (P>0.05 for each). Data indicate that TNF-α activates NAD(P)H oxidase, leading to a ROS elevation in cerebral artery smooth muscle cells.
Fig. 2.

TNF-α elevates ROS in smooth muscle cells of intact cerebral arteries via NAD(P)H oxidase activation. A, representative confocal images of DCF fluorescence in two different areas of the same cerebral artery in control (left) and after TNF-α (50 ng/ml) (right). B, mean effects of TNF-α on DCF fluorescence. TNF-α (50 ng/ml, n= 10 arteries), H2O2 (100 μM, n=6), TNF-α + apocynin (Apo, 25 μM, n=10), TNF-α + DPI (10 μM, n=7), TNF-α + MnTMPyP, (MnT, 10 μM, n=10), TNF-α + oxypurinol (Oxy, 10 μM, n=6), TNF-α + 17-octadecanoic acid (Oda, 10 μM, n=10), TNF-α + lonidamine (Lon, 100 μM, n=5), Time control (Cntrl, n=5). C, TNF-α (50 ng/ml) elevated DHE fluorescence in intact cerebral arteries. D, Average effects of TNF-α (50 ng/ml) on DHE fluorescence (n=5). * P<0.05 when compared with control. # P<0.05 when compared with TNF-α.
TNF-α activates transient KCa currents in isolated arterial smooth muscle cells
In arterial smooth muscle cells, a Ca2+ spark activates several KCa channels, resulting in a transient KCa current that induces membrane hyperpolarization (19). To investigate the effects of TNF-α-induced changes in Ca2+ signaling, transient KCa current regulation by TNF-α was measured in isolated, voltage-clamped (-40 mV) arterial smooth muscle cells. When using the perforated-patch clamp configuration, TNF-α evoked a gradual increase in transient KCa current frequency and amplitude, that reached a steady-state elevation of ∼1.7-fold and ∼1.4-fold, respectively in ∼10 minutes (Fig. 3A, C, D). Similar observations were made when using the conventional whole-cell configuration (Fig. 3B-D). Apocynin did not alter transient KCa current frequency (0.89 ± 0.12 of control, n=6) or amplitude (1.0 ± 0.06, n=6) when applied alone (P>0.05 for each), but reversed the TNF-α-induced increase in transient KCa current frequency and amplitude (Fig. 3B-D). Data suggest that TNF-α stimulates transient KCa currents by activating NAD(P)H oxidase.
Fig. 3.

TNF-α stimulates transient KCa currents in isolated cerebral artery smooth muscle cells due to NAD(P)H oxidase activation. A, B, original traces illustrating transient KCa currents in cells voltage-clamped at -40 mV. A, TNF-α activated transient KCa currents when using the perforated-patch clamp configuration. B, TNF-α activated transient KCa currents when using the conventional-whole cell configurations, and this was reversed by apocynin. C, D, Mean effects on transient KCa current frequency and amplitude of: TNF-α (50 ng/ml) perforated-patch (p-p, n=6); TNF-α (50 ng/ml) whole-cell (w-c, n=6); TNF-α + apocynin (Apo, 25 μM, n=6), TNF-α washout (Wash, n=6); ceramide (Cer, 10 μM, n=6), atractyloside (Atr, 100 μM, n=4). * P<0.05 when compared with control. # P<0.05 when compared with TNF-α.
TNF-α-induced intracellular signaling may involve mitochondrial PTP activation and ceramide generation (14; 22; 28; 30; 39; 41; 52). However, ceramide did not alter transient KCa currents, indicating that it is unlikely to mediate TNF-α-induced activation of these events (Fig. 3C, D). Lonidamine decreased mean Ca2+ spark frequency from 1.45 ± 0.16 Hz to 0.73 ± 0.18 Hz, or to ∼0.5 of control (n=4, P<0.05), and inhibited transient KCa currents (10). Similarly, atractyloside, another mitochondrial PTP opener, decreased transient KCa current frequency and amplitude to ∼0.4 -fold and ∼0.6 of control, respectively (Fig. 3C, D). Data demonstrate that PTP openers inhibit transient KCa currents, indicating that such an effect cannot mediate TNF-α-induced transient KCa current activation.
To determine if TNF-α-induced transient KCa current activation occurs due to a ROS elevation, transient KCa current regulation by MnTMPyP, and by catalase, that converts H2O2 to H2O, was measured. MnTMPyP reversed TNF-α-induced elevations in transient KCa current frequency and amplitude (Fig. 4A-C). In contrast, MnTMPyP did not change transient KCa current frequency (1.0 ± 0.1 of control, n=5) or amplitude (1.0 ± 0.07) when applied alone (P>0.05 for each). Similarly, when using the conventional whole-cell configuration, inclusion of catalase in the pipette solution (to allow intracellular access), prevented TNF-α-induced transient KCa current activation (Fig. 4B, C). In contrast, when supplementing the pipette solution with heat-inactivated catalase TNF-α increased transient KCa current frequency and amplitude ∼1.8 and ∼1.4-fold, respectively (Fig. 4B, C). In summary, data indicate that TNF-α activates Ca2+ sparks and transient KCa currents by inducing an NAD(P)H oxidase-mediated ROS elevation.
Fig. 4.
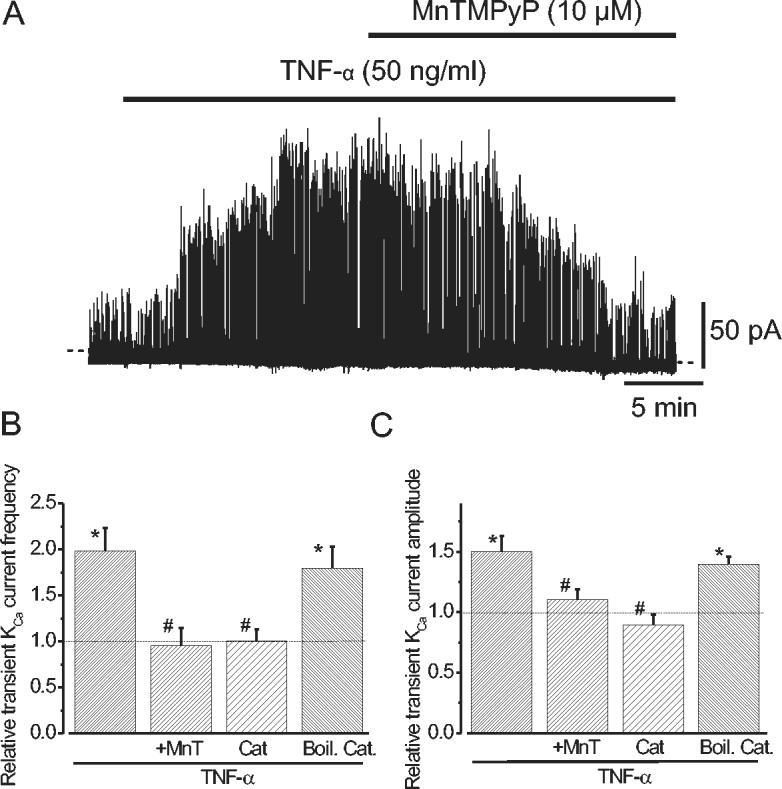
TNF-α induced transient KCa current activation occurs due a ROS elevation. A, TNF-α (50 ng/ml)-induced transient KCa current activation was reversed by MnTMPyP. B, C, Mean transient KCa current frequency and amplitude. TNF-α (50 ng/ml, n=12), TNF-α + MnTMPyP (10 μM, n=6), TNF-α with catalase (Cat, 200 U/ml, n=6), TNF-α with boiled catalase (Boil. catalase, 200 U/ml, n=6). * P<0.05 when compared with control. # P<0.05 when compared with TNF-α.
TNF-α dilates pressurized cerebral arteries by elevating ROS, SR Ca2+ release and KCa channel activity
TNF-α has been demonstrated to induce both vasoconstriction and vasodilation by endothelial-dependent and -independent pathways (4; 7; 16; 22; 23; 29; 32; 37; 38; 44; 51). Here, data suggest TNF-α activates a dilatory Ca2+ signaling pathway in smooth muscle cells of small, resistance size cerebral arteries. Therefore, vasoregulation by TNF-α was investigated using resistance-size pressurized (60 mm Hg) cerebral arteries. In endothelium-intact arteries, from a mean active diameter of 142 ± 3 μm (0.64 ± 0.01 of passive diameter) TNF-α induced a mean dilation of 22 ± 1 μm (Fig. 5A-C). In endothelium-denuded arteries, TNF-α increased mean diameter by 18 ± 3 μm from 130 ± 9 μm (0.57 ± 0.02 of passive diameter, Fig. 5C). TNF-α-induced dilations were not significantly different in endothelium-intact or endothelium-denuded arteries (P>0.05). TNF-α-induced dilations were fully reversible (Fig. 5A) and consecutive TNF-α applications resulted in dilations of similar magnitude (first application, 19 ± 2 μm; second application 21 ± 2 μm (n=2).
Fig. 5.

TNF-α dilates pressurized (60 mm Hg) cerebral arteries by elevating ROS and by activating SR Ca2+ release and KCa channels. A, TNF-α reversibly dilated a pressurized (60 mmHg) endothelium-intact cerebral artery. B, TNF-α (10 ng/ml)-induced vasodilation was inhibited by ibertiotoxin (IbTX, 100 nM). C, mean TNF-α (10 ng/ml)-induced dilations in endothelium-intact arteries in control (Cntrl, n=29), MnTMPyP (MnT, 10 μM, n=4), thapsigargin (+Tg., 100 nM, n=7), and iberiotoxin (+IbTX, 100 nM, n=9), and in endothelium-denuded arteries (Denud, n=6). MnTMPyP reduced arterial diameter by 4 ± 1 μm (n=4, P<0.05). Thapsigargin and iberiotoxin reduced mean arterial diameter by 9 ± 3 μm and 9 ± 1 μm, respectively (P<0.05 for each), consistent with Ca2+ spark and KCa channel inhibition (19). * P < 0.05 when compared with control. # P<0.05 when compared with TNF-α in endothelium-intact arteries.
To investigate the importance of ROS generation in TNF-α-induced vasodilation, responses to TNF-α were measured in control and then in the presence of MnTMPyP (10 μM) in the same artery. MnTMPyP reduced mean TNF-α-induced vasodilations to 7 ± 2 μm, or to 0.34 ± 0.03 of control dilations (Fig. 5C). The same protocol was also performed with thapsigargin (100 nM), a SR Ca2+ ATP-ase blocker that inhibits Ca2+ sparks, or iberiotoxin (100 nM), a selective KCa channel blocker (19). Thapsigargin reduced mean TNF-α-induced vasodilations to 4 ± 2 μm, or 0.15 ± 0.01 of those obtained in control in the same arteries (Fig. 5C). Similarly, iberiotoxin reduced mean TNF-α-induced vasodilations to 6 ± 1 μm or 0.29 ± 0.05 of control (Fig. 5B, C). Data indicate that TNF-α dilates cerebral arteries by generating ROS that stimulate Ca2+ sparks and KCa channels in smooth muscle cells.
DISCUSSION
The mechanisms by which TNF-α, a pleiotropic cytokine, regulates the diameter of small cerebral arteries was investigated. Data indicate that TNF-α activates smooth muscle cell NAD(P)H oxidase, leading to the generation of ROS that activate Ca2+ sparks, and thus, transient KCa currents. The ensuing decrease in global [Ca2+]i leads to vasodilation. These findings show for the first time that an inflammatory mediator central in a variety of vascular diseases induces vasodilation through Ca2+ spark activation.
TNF-α is produced in the brain during ischemia and is implicated in the etiology of several cardiovascular diseases, including stroke, cardiac failure, coronary heart disease and atherosclerosis (15; 46). TNF-α is reported to induce both vasoconstriction and vasodilation. Topical application of TNF-α dilated cerebral arterioles (7; 37), whereas intracranial injection of TNF-α induced cerebrovascular constriction and reduced blood flow (29; 38; 44). TNF-α also dilated mesenteric and cremaster muscle arterioles (4; 32; 45). Earlier studies suggest TNF-α promotes vasodilation by elevating NO and prostaglandin production in the arterial wall and perivascular tissues (4; 7; 37). In contrast, in pulmonary artery rings, a hypoxia-induced increase in TNF-α mRNA was associated with contraction (43). TNF-α also reduced bradykinin- and acetylcholine (ACh)-induced vasodilation in rat mesenteric arteries (48), reduced bradykinin- and A23187-induced vasodilation in bovine coronary arteries (51), blunted ACh-induced vasodilation in cat carotid arteries (2) and evoked endothelium-dependent vasoconstriction in human and rat coronary arteries (17; 23). In humans, intrabrachial TNF-α injection impaired bradykinin- and ACh-induced vasodilation (11). Data also suggest that TNF-α inhibits vasodilation by triggering ROS that scavenge endothelial NO (51). It is unclear what underlies differential regulation of arterial contractility by TNF-α, but several possibilities exist.
Conceivably, vasoregulatory actions of TNF-α may be vascular bed specific. Different experimental protocols employed in studies may also explain the diversity of observations. In addition, the effects of TNF-α on arterial contractility may be time-dependent. For example, in sheep bronchial arteries, TNF-α initially induced dilation, followed by constriction two hours later (45). Data also suggest differential effects on contractility may occur when TNF-α acts primarily on either endothelial or smooth muscle cells. Conceivably, TNF-α-induced vasoconstriction may occur primarily due to an endothelium-dependent mechanism, whereas vasodilation may be mediated by a direct effect on smooth muscle cells. Here, we show that in isolated, pressurized, myogenic rat cerebral arteries, acute TNF-α application (<30 minutes) induced vasodilation that was endothelium-independent and was markedly attenuated by an antioxidant and inhibitors of SR Ca2+ release and KCa channels.
TNF-α activates several signaling pathways in vascular smooth muscle cells. TNF-α stimulated NAD(P)H oxidase leading to ROS production in rat aortic smooth muscle cells (13). TNF-α relaxed endothelium-denuded, phenylephrine-constricted aortic rings by PLA2 activation, implicating ceramide as a mediator (22). In cardiac and endothelial cells, signaling mechanisms for TNF-α involve mitochondria 20, 23-25, 35, 36. TNF-α disrupted the mitochondrial electron transport chain, caused release of cytochrome c, induced mitochondrial ROS generation and triggered apoptosis (14; 28; 30; 41; 52). Although the downstream intramitochondrial target for TNF-α is unclear, in endothelial cells PTP involvement was proposed (52). The PTP is also a downstream target for TNF-α-induced signaling in other cell types, and PTP opening may be triggered by electron transport chain inhibition and mitochondrial depolarization (5; 10; 39). Ceramide had no effect on transient KCa currents and PTP openers inhibited these events, consistent with a previous report in arterial smooth muscle cells (10). Therefore, TNF-α is unlikely to activate Ca2+ sparks and transient KCa currents by elevating ceramide or by inducing PTP opening. In some cell types, including neutrophils, pre-incubation with apocynin is required to block association of NAD(P)H oxidase subunits (40). Here, we show that in cerebral artery smooth muscle cells, apocynin reverses TNF-α-induced transient KCa current activation within ∼10 minutes. This effect may reflect rapid subunit turnover in the presence of TNF-α in these cells. Data indicate that TNF-α stimulates NAD(P)H oxidase, generating ROS that activate Ca2+ sparks and transient KCa currents in cerebral artery smooth muscle cells. These findings are consistent with a recent report that mitochondria-derived ROS activate Ca2+ sparks and transient KCa currents in arterial smooth muscle cells (50). Thus, ROS derived from different sources induce vasodilation through a common pathway, namely Ca2+ spark activation.
TNF-α-induced ROS may activate Ca2+ sparks and transient KCa currents due to a direct or indirect effect on RyR and KCa channels. Cardiac and skeletal muscle RyR channels are redox-sensitive and are activated by oxidizing agents, including H2O2 (42). ROS also activate a number of other signal transduction pathways that may regulate RyR channel activity (42; 49). Although superoxide (O2-•) is the initial product of NAD(P)H oxidase, O2-• has a short half-life and rapidly dismutates to H2O2, a relatively stable oxidant. Superoxide is unlikely to mediate effects of TNF-α on Ca2+ sparks, particularly since catalase blocked TNF-α-induced transient KCa current activation. Data suggest H2O2, formed from NAD(P)H oxidase-generated O2-•, activates RyR channels, leading to the Ca2+ spark frequency elevation. Indeed, exogenous H2O2 activates transient KCa currents in cerebral artery smooth muscle cells (10). Another potential ROS mediating Ca2+ spark activation in this study is hydroxyl radical (OH•), a highly reactive species produced in a transition metal catalyzed reaction (Fenton reaction) between H2O2 and O2-•. Conventional whole-cell experiments used a pipette solution absent in transition metals, and dialysis of the pipette solution within the cell interior should wash out endogenous cytosolic transition metals. OH• generation in these experiments should have been minimal, arguing against a significant role for OH• in TNF-α-induced Ca2+ spark and KCa channel activation.
ROS production or inhibition of ROS degradation leads to vasoconstriction, high blood pressure, and vascular hypertrophy (24). However, ROS also stimulate vasodilation. For example, flow-mediated vasodilation occurs due to the generation of mitochondria-derived H2O2 (27). Furthermore, topical application of ROS dilates cerebral arteries in vivo (47). Thus, ROS induce both vasoconstriction and vasodilation. Conceivably, there may be a delicate balance between ROS concentrations that regulate physiological functions and higher ROS concentrations that lead to pathologies (24). Data here indicate that TNF-α regulates Ca2+ sparks by generating ROS from a single source, NAD(P)H oxidase. During disease, vasoconstriction may result from ROS produced in higher concentrations, from additional enzymes, from exogenous sources, and for prolonged periods. For example, oxyhemoglobin, that may contribute to vasoconstriction following subarachnoid hemorrhage, inhibits Ca2+ sparks in cerebral artery smooth muscle cells and this can be blocked by antioxidants (21). TNF-α receptors were recently proposed to spatially confine NAD(P)H oxidase-mediated redox signaling in human microvascular endothelial cells (25). TNF-α may also activate Ca2+ sparks by elevating ROS in the local vicinity of Ca2+ spark sites. Through restriction, global effects of ROS would be lessened and dilatory signaling could be maintained. Since Ca2+ spark sites are close to sarcolemmal KCa channels, local NAD(P)H oxidase-derived ROS should also regulate KCa channel activity. TNF-α did not alter Ca2+ spark amplitude, but increased transient KCa current amplitude. NAD(P)H oxidase inhibitors and antioxidants reduced the TNF-α-induced transient KCa current amplitude elevation, indicating the effect was mediated by NAD(P)H oxidase-derived ROS. Thus, TNF-α also enhances the effective coupling of Ca2+ sparks to KCa channels, presumably by elevating KCa channel Ca2+-sensitivity within the micromolar concentration range generated by these Ca2+ transients (34). The combined effects of elevated Ca2+ spark frequency and transient KCa current amplitude would significantly increase KCa channel activity, leading to vasodilation (19; 33).
TNF-α elevated Ca2+ wave frequency ∼ 1.5-fold, but reduced global [Ca2+]i. In the absence of receptor agonists, Ca2+ wave frequency in arterial smooth muscle cells is low, and Ca2+ waves contribute little to global [Ca2+]i (18). When stimulated to high frequencies, Ca2+ waves can contribute to global [Ca2+]i and thus, induce smooth muscle contraction (35). Although TNF-α stimulated Ca2+ waves, the frequency elevation was insufficient to contribute significantly to global Ca2+. In contrast, TNF-α-induced Ca2+ spark activation significantly elevated KCa channel activity. The resulting membrane hyperpolarization would reduce voltage-dependent Ca2+ channel activity, leading to the observed decrease in global [Ca2+]i (19). Therefore, the net effect of TNF-α-induced Ca2+ spark and Ca2+ wave activation is a reduction in global [Ca2+]i, leading to vasodilation.
In summary, this study defines a novel vasodilatory signaling pathway for an inflammatory cytokine mediated by Ca2+ spark and KCa channel activation in smooth muscle cells. Following ischemia and stroke, TNF-α-induced Ca2+ spark activation and cerebrovascular dilation would be important in regulating local blood supply in the affected brain region.
ACKNOWLEDGEMENTS
This project was supported by grants from the NIH and American Heart Association National Center to J.H.J.. S.Y.C is a recipient of a Postdoctoral Fellowship from the Southeast Affiliate of the American Heart Association.
REFERENCES
- 1.Amberg GC, Bonev AD, Rossow CF, Nelson MT, Santana LF. Modulation of the molecular composition of large conductance, Ca2+ activated K+ channels in vascular smooth muscle during hypertension. J Clin Invest. 2003;112:717–724. doi: 10.1172/JCI18684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aoki N, Siegfried M, Lefer AM. Anti-EDRF effect of tumor necrosis factor in isolated, perfused cat carotid arteries. Am J Physiol. 1989;256:H1509–H1512. doi: 10.1152/ajpheart.1989.256.5.H1509. [DOI] [PubMed] [Google Scholar]
- 3.Barone FC, Arvin B, White RF, Miller A, Webb CL, Willette RN, Lysko PG, Feuerstein GZ. Tumor necrosis factor-α. A mediator of focal ischemic brain injury. Stroke. 1997;28:1233–1244. doi: 10.1161/01.str.28.6.1233. [DOI] [PubMed] [Google Scholar]
- 4.Baudry N, Vicaut E. Role of nitric oxide in effects of tumor necrosis factor-α on microcirculation in rat. J Appl Physiol. 1993;75:2392–2399. doi: 10.1152/jappl.1993.75.6.2392. [DOI] [PubMed] [Google Scholar]
- 5.Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- 6.Brenner R, Perez GJ, Bonev AD, Eckman DM, Kosek JC, Wiler SW, Patterson AJ, Nelson MT, Aldrich RW. Vasoregulation by the β1 subunit of the calcium-activated potassium channel. Nature. 2000;407:870–876. doi: 10.1038/35038011. [DOI] [PubMed] [Google Scholar]
- 7.Brian JE, Jr., Faraci FM. Tumor necrosis factor-α-induced dilatation of cerebral arterioles. Stroke. 1998;29:509–515. doi: 10.1161/01.str.29.2.509. [DOI] [PubMed] [Google Scholar]
- 8.Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat Med. 1996;2:788–794. doi: 10.1038/nm0796-788. [DOI] [PubMed] [Google Scholar]
- 9.Castellanos M, Castillo J, Garcia MM, Leira R, Serena J, Chamorro A, Davalos A. Inflammation-mediated damage in progressing lacunar infarctions: a potential therapeutic target. Stroke. 2002;33:982–987. doi: 10.1161/hs0402.105339. [DOI] [PubMed] [Google Scholar]
- 10.Cheranov SY, Jaggar JH. Mitochondrial modulation of Ca2+ sparks and transient KCa currents in smooth muscle cells of rat cerebral arteries. J Physiol. 2004;556:755–771. doi: 10.1113/jphysiol.2003.059568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chia S, Qadan M, Newton R, Ludlam CA, Fox KA, Newby DE. Intra-arterial tumor necrosis factor-α impairs endothelium-dependent vasodilatation and stimulates local tissue plasminogen activator release in humans. Arterioscler Thromb Vasc Biol. 2003;23:695–701. doi: 10.1161/01.ATV.0000065195.22904.FA. [DOI] [PubMed] [Google Scholar]
- 12.Chignell CF, Sik RH. A photochemical study of cells loaded with 2′,7′-dichlorofluorescin: implications for the detection of reactive oxygen species generated during UVA irradiation. Free Radic Biol Med. 2003;34:1029–1034. doi: 10.1016/s0891-5849(03)00022-4. [DOI] [PubMed] [Google Scholar]
- 13.De Keulenaer GW, Alexander RW, Ushio-Fukai M, Ishizaka N, Griendling KK. Tumour necrosis factor α activates a p22phox-based NADH oxidase in vascular smooth muscle. Biochem J. 1998;329(Pt 3):653–657. doi: 10.1042/bj3290653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Engel D, Peshock R, Armstong RC, Sivasubramanian N, Mann DL. Cardiac myocyte apoptosis provokes adverse cardiac remodeling in transgenic mice with targeted TNF overexpression. Am J Physiol. 2004;287:H1303–H1311. doi: 10.1152/ajpheart.00053.2004. [DOI] [PubMed] [Google Scholar]
- 15.Hallenbeck JM. The many faces of tumor necrosis factor in stroke. Nat Med. 2002;8:1363–1368. doi: 10.1038/nm1202-1363. [DOI] [PubMed] [Google Scholar]
- 16.Hollenberg SM, Cunnion RE, Parrillo JE. The effect of tumor necrosis factor on vascular smooth muscle. In vitro studies using rat aortic rings. Chest. 1991;100:1133–1137. doi: 10.1378/chest.100.4.1133. [DOI] [PubMed] [Google Scholar]
- 17.Iversen PO, Nicolaysen A, Kvernebo K, Benestad HB, Nicolaysen G. Human cytokines modulate arterial vascular tone via endothelial receptors. Pflugers Arch. 1999;439:93–100. doi: 10.1007/s004249900149. [DOI] [PubMed] [Google Scholar]
- 18.Jaggar JH. Intravascular pressure regulates local and global Ca2+ signaling in cerebral artery smooth muscle cells. Am J Physiol. 2001;281:C439–C448. doi: 10.1152/ajpcell.2001.281.2.C439. [DOI] [PubMed] [Google Scholar]
- 19.Jaggar JH, Porter VA, Lederer WJ, Nelson MT. Calcium sparks in smooth muscle. Am J Physiol. 2000;278:C235–C256. doi: 10.1152/ajpcell.2000.278.2.C235. [DOI] [PubMed] [Google Scholar]
- 20.Jaggar JH, Stevenson AS, Nelson MT. Voltage dependence of Ca2+ sparks in intact cerebral arteries. Am J Physiol. 1998;274:C1755–C1761. doi: 10.1152/ajpcell.1998.274.6.C1755. [DOI] [PubMed] [Google Scholar]
- 21.Jewell RP, Saundry CM, Bonev AD, Tranmer BI, Wellman GC. Inhibition of Ca++ sparks by oxyhemoglobin in rabbit cerebral arteries. J Neurosurg. 2004;100:295–302. doi: 10.3171/jns.2004.100.2.0295. [DOI] [PubMed] [Google Scholar]
- 22.Johns DG, Webb RC. TNF-α-induced endothelium-independent vasodilation: a role for phospholipase A2-dependent ceramide signaling. Am J Physiol. 1998;275:H1592–H1598. doi: 10.1152/ajpheart.1998.275.5.H1592. [DOI] [PubMed] [Google Scholar]
- 23.Klemm P, Warner TD, Hohlfeld T, Corder R, Vane JR. Endothelin 1 mediates ex vivo coronary vasoconstriction caused by exogenous and endogenous cytokines. Proc Natl Acad Sci U S A. 1995;92:2691–2695. doi: 10.1073/pnas.92.7.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lassegue B, Griendling KK. Reactive oxygen species in hypertension; An update. Am J Hypertens. 2004;17:852–860. doi: 10.1016/j.amjhyper.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 25.Li JM, Fan LM, Christie MR, Shah AM. Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascular endothelial cells: role of p47phox phosphorylation and binding to TRAF4. Mol Cell Biol. 2005;25:2320–2330. doi: 10.1128/MCB.25.6.2320-2330.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135–1143. doi: 10.1161/hc0902.104353. [DOI] [PubMed] [Google Scholar]
- 27.Liu Y, Zhao H, Li H, Kalyanaraman B, Nicolosi AC, Gutterman DD. Mitochondrial sources of H2O2 generation play a key role in flow-mediated dilation in human coronary resistance arteries. Circ Res. 2003;93:573–580. doi: 10.1161/01.RES.0000091261.19387.AE. [DOI] [PubMed] [Google Scholar]
- 28.Machida Y, Kubota T, Kawamura N, Funakoshi H, Ide T, Utsumi H, Li YY, Feldman AM, Tsutsui H, Shimokawa H, Takeshita A. Overexpression of tumor necrosis factor-α increases production of hydroxyl radical in murine myocardium. Am J Physiol. 2003;284:H449–H455. doi: 10.1152/ajpheart.00581.2002. [DOI] [PubMed] [Google Scholar]
- 29.Megyeri P, Abraham CS, Temesvari P, Kovacs J, Vas T, Speer CP. Recombinant human tumor necrosis factor alpha constricts pial arterioles and increases blood-brain barrier permeability in newborn piglets. Neurosci Lett. 1992;148:137–140. doi: 10.1016/0304-3940(92)90823-p. [DOI] [PubMed] [Google Scholar]
- 30.Moe GW, Marin-Garcia J, Konig A, Goldenthal M, Lu X, Feng Q. In vivo TNF-α inhibition ameliorates cardiac mitochondrial dysfunction, oxidative stress, and apoptosis in experimental heart failure. Am J Physiol. 2004;287:H1813–H1820. doi: 10.1152/ajpheart.00036.2004. [DOI] [PubMed] [Google Scholar]
- 31.Mokelke EA, Dietz NJ, Eckman DM, Nelson MT, Sturek M. Diabetic dyslipidemia and exercise affect coronary tone and differential regulation of conduit and microvessel K+ current. Am J Physiol Heart Circ Physiol. 2005;288:H1233–H1241. doi: 10.1152/ajpheart.00732.2004. [DOI] [PubMed] [Google Scholar]
- 32.Naziri W, Joshua IG. Role of tumor necrosis factor-α in small intestinal microcirculation. Am Surg. 1998;64:203–209. [PubMed] [Google Scholar]
- 33.Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- 34.Perez GJ, Bonev AD, Nelson MT. Micromolar Ca2+ from sparks activates Ca2+-sensitive K+ channels in rat cerebral artery smooth muscle. Am J Physiol. 2001;281:C1769–C1775. doi: 10.1152/ajpcell.2001.281.6.C1769. [DOI] [PubMed] [Google Scholar]
- 35.Ruehlmann DO, Lee CH, Poburko D, van Breemen C. Asynchronous Ca2+ waves in intact venous smooth muscle. Circ Res. 2000;86:E72–E79. doi: 10.1161/01.res.86.4.e72. [DOI] [PubMed] [Google Scholar]
- 36.Sairanen T, Carpen O, Karjalainen-Lindsberg ML, Paetau A, Turpeinen U, Kaste M, Lindsberg PJ. Evolution of cerebral tumor necrosis factor-α production during human ischemic stroke. Stroke. 2001;32:1750–1758. doi: 10.1161/01.str.32.8.1750. [DOI] [PubMed] [Google Scholar]
- 37.Shibata M, Parfenova H, Zuckerman SL, Leffler CW. Tumor necrosis factor-α induces pial arteriolar dilation in newborn pigs. Brain Res Bull. 1996;39:241–247. doi: 10.1016/0361-9230(95)02142-6. [DOI] [PubMed] [Google Scholar]
- 38.Sibson NR, Blamire AM, Perry VH, Gauldie J, Styles P, Anthony DC. TNF-α reduces cerebral blood volume and disrupts tissue homeostasis via an endothelin- and TNFR2-dependent pathway. Brain. 2002;125:2446–2459. doi: 10.1093/brain/awf256. [DOI] [PubMed] [Google Scholar]
- 39.Soriano ME, Nicolosi L, Bernardi P. Desensitization of the permeability transition pore by cyclosporin a prevents activation of the mitochondrial apoptotic pathway and liver damage by tumor necrosis factor-α. J Biol Chem. 2004;279:36803–36808. doi: 10.1074/jbc.M405297200. [DOI] [PubMed] [Google Scholar]
- 40.Stolk J, Hiltermann TJ, Dijkman JH, Verhoeven AJ. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am J Respir Cell Mol Biol. 1994;11:95–102. doi: 10.1165/ajrcmb.11.1.8018341. [DOI] [PubMed] [Google Scholar]
- 41.Suematsu N, Tsutsui H, Wen J, Kang D, Ikeuchi M, Ide T, Hayashidani S, Shiomi T, Kubota T, Hamasaki N, Takeshita A. Oxidative stress mediates tumor necrosis factor-α-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation. 2003;107:1418–1423. doi: 10.1161/01.cir.0000055318.09997.1f. [DOI] [PubMed] [Google Scholar]
- 42.Suzuki YJ, Ford GD. Redox regulation of signal transduction in cardiac and smooth muscle. J Mol Cell Cardiol. 1999;31:345–353. doi: 10.1006/jmcc.1998.0872. [DOI] [PubMed] [Google Scholar]
- 43.Tsai BM, Wang M, Pitcher JM, Meldrum KK, Meldrum DR. Hypoxic pulmonary vasoconstriction and pulmonary artery tissue cytokine expression are mediated by protein kinase C. Am J Physiol. 2004;287:L1215–L1219. doi: 10.1152/ajplung.00179.2004. [DOI] [PubMed] [Google Scholar]
- 44.Tureen J. Effect of recombinant human tumor necrosis factor-α on cerebral oxygen uptake, cerebrospinal fluid lactate, and cerebral blood flow in the rabbit: role of nitric oxide. J Clin Invest. 1995;95:1086–1091. doi: 10.1172/JCI117755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wagner EM. TNF-α induced bronchial vasoconstriction. Am J Physiol. 2000;279:H946–H951. doi: 10.1152/ajpheart.2000.279.3.H946. [DOI] [PubMed] [Google Scholar]
- 46.Wang CX, Shuaib A. Involvement of inflammatory cytokines in central nervous system injury. Prog Neurobiol. 2002;67:161–172. doi: 10.1016/s0301-0082(02)00010-2. [DOI] [PubMed] [Google Scholar]
- 47.Wei EP, Kontos HA, Beckman JS. Mechanisms of cerebral vasodilation by superoxide, hydrogen peroxide, and peroxynitrite. Am J Physiol. 1996;271:H1262–H1266. doi: 10.1152/ajpheart.1996.271.3.H1262. [DOI] [PubMed] [Google Scholar]
- 48.Wimalasundera R, Fexby S, Regan L, Thom SA, Hughes AD. Effect of tumour necrosis factor-α and interleukin 1β on endothelium-dependent relaxation in rat mesenteric resistance arteries in vitro. Br J Pharmacol. 2003;138:1285–1294. doi: 10.1038/sj.bjp.0705168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wolin MS. Interactions of oxidants with vascular signaling systems. Arterioscler Thromb Vasc Biol. 2000;20:1430–1442. doi: 10.1161/01.atv.20.6.1430. [DOI] [PubMed] [Google Scholar]
- 50.Xi Q, Cheranov SY, Jaggar JH. Mitochondria-Derived Reactive Oxygen Species Dilate Cerebral Arteries by Activating Ca2+ Sparks. Circ Res. 2005;97(4):354–362. doi: 10.1161/01.RES.0000177669.29525.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang DX, Yi FX, Zou AP, Li PL. Role of ceramide in TNF-α-induced impairment of endothelium-dependent vasorelaxation in coronary arteries. Am J Physiol. 2002;283:H1785–H1794. doi: 10.1152/ajpheart.00318.2002. [DOI] [PubMed] [Google Scholar]
- 52.Zhang R, Al Lamki R, Bai L, Streb JW, Miano JM, Bradley J, Min W. Thioredoxin-2 inhibits mitochondria-located ASK1-mediated apoptosis in a JNK-independent manner. Circ Res. 2004;94:1483–1491. doi: 10.1161/01.RES.0000130525.37646.a7. [DOI] [PubMed] [Google Scholar]