

Abstract
The ability of herpes simplex virus type 1 (HSV-1) to activate NF-κB has been well documented. Beginning at 3 to 5 h postinfection, HSV-1 induces a robust and persistent nuclear translocation of an NF-κB-dependent (p50/p65 heterodimer) DNA binding activity, as measured by electrophoretic mobility shift assay. Activation requires virus binding and entry, as well as de novo infected-cell protein synthesis, and is accompanied by loss of both IκBα and IκBβ. In this study, we identified loss of IκBα as a marker of NF-κB activation, and infection with mutants with individual immediate-early (IE) regulatory proteins deleted indicated that ICP27 was necessary for IκBα loss. Analysis of both N-terminal and C-terminal mutants of ICP27 identified the region from amino acids 21 to 63 as being necessary for IκBα loss. Additional experiments with mutant viruses with combinations of IE genes deleted revealed that the ICP27-dependent mechanism of NF-κB activation may be augmented by functional ICP4. We also analyzed two additional markers for NF-κB activation, phosphorylation of the p65 subunit on Ser276 and Ser536. Phosphorylation of both serines was induced upon HSV infection and required functional ICP4 and ICP27. Pharmacological inhibitor studies revealed that both IκBα and Ser276 phosphorylation were dependent on Jun N-terminal protein kinase activity, while Ser536 phosphorylation was not affected during inhibitor treatment. These results demonstrate that there are several layers of regulation of NF-κB activation during HSV infection, highlighting the important role that NF-κB may play in infection.
NF-κB is a cellular integrator of diverse signaling pathways leading to programs of immune, inflammatory, and antiapoptotic gene expression. These pathways are initiated through the engagement of cell surface receptors by a variety of chemical ligands, such as cytokines, phorbol esters, lipopolysaccharide, and virion glycoproteins, or by stresses such as UV irradiation or changes in osmolarity. NF-κB is normally sequestered in the cytoplasm through interactions with its inhibitory binding partner IκB. While a variety of NF-κB activation pathways have been characterized, many ultimately converge on and activate the IκB kinase (IKK) complex, resulting in phosphorylation of IκB. Once IκB is phosphorylated, polyubiquitin-dependent proteolysis of IκB occurs, releasing NF-κB and allowing its translocation to the nucleus, where it participates in transcription activation in conjunction with other transcription factors and coactivators.
Transcription of the herpes simplex virus (HSV) genome during lytic infection is temporally regulated (see reference 62 for a review). Three immediate-early (IE) proteins have important roles in regulating the temporal pattern of gene expression. ICP4 is essential for E and L gene expression and colocalizes with viral DNA (22, 23, 85), RNA polymerase II holoenzyme, and general transcription factors (10, 79). ICP0 can trans-activate viral IE, E, and L promoters alone or in combination with ICP4 and/or ICP27 in transient reporter assays. Studies that evaluated its function in the absence of ICP4 or ICP27 demonstrated a role for ICP0 in efficient transcription of IE and E genes (68). ICP0 affects disruption of ND10 that has relocalized to viral DNA (20-22, 29, 44) and also inhibits the function of a gene silencing complex (28) in order to promote efficient viral transcription.
ICP27, a second essential regulatory protein, affects both transcriptional and posttranscriptional aspects of viral gene expression, as well as the metabolism of host cell mRNA. Specifically, ICP27 contributes to negative regulation of IE gene expression (61, 64) while positively regulating expression of essential E genes for DNA replication except the ICP8 gene (86). ICP27 mutants also express reduced amounts of leaky-late (γ1) genes and fail to express true-late (γ2) genes (35, 45, 64). ICP27 has additional functions dependent on protein-protein interactions as well as protein-RNA interactions, including export of mRNA (11, 38, 50, 57, 69, 80), relocalization of casein kinase 2 (CK2) (39), interaction with splicing factors (8, 31, 43, 72, 76), and stimulation of mRNA 3′-end processing (46).
The ability of HSV type 1 (HSV-1) to activate NF-κB has been well documented. Beginning at 3 to 5 h postinfection (hpi), HSV-1 induces a robust and persistent nuclear translocation of NF-κB and increased p50/p65 heterodimer-dependent DNA binding activity, as measured by electrophoretic mobility shift assay (EMSA) (1, 56, 63). Persistent NF-κB activation requires virus binding and entry, as well as de novo infected-cell protein synthesis, including expression of functional viral IE proteins ICP27 and ICP4 (1, 56). Activation is accompanied by increased IKK activity (1) and loss of both IκBα and IκBβ (56). Interference with NF-κB activation occurs following overexpression of a dominant-negative version of IκBα (DN-IκB) containing alanine substitutions for critical serine residues 32 and 36. The resulting substantial reduction in NF-κB EMSA activity correlates with a reduction in virus yield (1, 56). The latter may be related to the reported role of NF-κB in preventing HSV-1-induced apoptosis (26), though this may be dependent on cell type or on the timing of expression of viral protein US11 (84). Finally, HSV-1 infection of HEp-2 cells transfected with a luciferase reporter plasmid containing three copies of the major histocompatibility complex NF-κB site (3× NF-κB-luc) resulted in a 3.5-fold induction of luciferase activity, which was abolished in the presence of DN-IκB (1).
The foregoing results argue that HSV may have evolved to evade the observed persistent NF-κB activation or to utilize this host response to promote efficient virus replication. With regard to the latter, one way this may occur is through suppression of apoptosis, as mentioned above. Additionally, activation may contribute directly to transcriptional regulation of viral genes. Nucleotide sequences which bind purified NF-κB were identified in intron 1 and exons 2 and 3 of ICP0, as well as upstream of the late UL46 and UL47 genes (63). NF-κB is bound to the ICP0 promoter during infection (2). These sites also bind nuclear factors induced by HSV infection or phorbol myristate acetate treatment. Several examples of HSV-induced expression of cellular genes regulated by NF-κB have been reported. In keratinocytes, association of NF-κB with the IκB promoter is reduced, leading to an impairment in the reconstitution of IκB levels after NF-κB activation (2). In lymphoid cells, such as peripheral blood mononuclear cells treated with gamma interferon and infected by HSV-1, expression of interleukin-6 (IL-6) increased between 2 and 3 hpi (54). While VP16 and IE proteins ICP4, ICP0, and ICP27 were not required for IL-6 mRNA induction, repression of IL-6 induction occurred following infection of a macrophage cell line expressing DN-IκB. The chemokine RANTES/CCL5 was induced in macrophages and fibroblasts by both HSV-1 and HSV-2, dependent on NF-κB and IRF-3 activation and on virus-induced PKR and ICP0 (51, 52). Glucocorticoid receptor expression has been reported to increase during HSV infection (18). Because the anti-inflammatory effects of glucocorticoids act in part by repressing p65 (58), down-regulation of the NF-κB-dependent proinflammatory response may also occur during virus infection. In summary, both increased NF-κB DNA binding activity and NF-κB-dependent gene activation in HSV-1-infected cells have been documented. The role of viral proteins in activation and how NF-κB contributes to infection outcomes have not been clearly defined in all cell types investigated.
The experiments presented here were designed to evaluate the roles of different stages in virus replication for activating NF-κB in CV-1 cells, a cell type often used in HSV infection studies. We assayed for NF-κB nuclear translocation, IκB degradation, and p65 phosphorylation as surrogate markers for NF-κB activation. We determined that virus attachment and introduction of the viral tegument into the cell, without subsequent gene expression, were not sufficient for activating NF-κB. We established that expression of, among the kinetic classes of viral genes, IE genes was sufficient for the activation of NF-κB. Of the IE gene products, ICP27 and ICP4 were required for full NF-κB activation. The results presented here suggest that ICP27 was necessary to activate NF-κB, while the role of ICP4 may be to augment the ICP27-induced activation. We further determined that a small region of the ICP27 amino terminus was required for the activation of NF-κB. This was the same region of ICP27 that we identified as being required for the activation of Jun N-terminal protein kinase (JNK) and p38 (32). The use of a pharmacological inhibitor of JNK suppressed HSV-induced NF-κB activation, suggesting that ICP27 may activate NF-κB indirectly as a downstream target of JNK.
MATERIALS AND METHODS
Cells and viruses.
CV-1 cells were originally obtained from Saul Silverstein (Columbia University) and were grown in Dulbecco's minimal essential medium supplemented with 5% bovine calf serum, 100 U/ml penicillin, 1% streptomycin, and 1% l-glutamine (all from Gibco). Cells were seeded into 100-mm dishes at a density of 2 × 106 cells per plate. Both mock infection and infection with virus were carried out in spent media for one hour at 37°C. The inoculum was then replaced with virus-free spent medium. Spent media were produced by collecting the seeding media from plates prior to infection and were used as inoculum media and overlay media, in order to minimize the introduction of growth factors and other signaling stimulants found in fresh media. We have observed that the background NF-κB activation in mock-infected samples using spent media was lower than that when fresh or low-serum media were used (data not shown). The KOS 1.1 strain of HSV-1 was used in all experiments unless otherwise noted. The mutant tsB7 (6, 37), the parental HFEM strain of HSV-1, and the KOS ICP27 deletion mutant d27-1 (61) were provided by David Knipe (Harvard University). The ICP4 mutants vi13 and n12 (14, 16, 77) were provided by Neal DeLuca (University of Pittsburgh). The IE multiple mutants d100 (ICP0−/ICP4−), d103 (ICP4−/ICP22−/ICP47−), d106 (ICP4−/ICP22−/ICP27−/ICP47−), d107 (ICP4−/ICP27−), and d109 (ICP0−/ICP4−/ICP22−/ICP27−/ICP47−) were also provided by Neal DeLuca (67). The ICP6 mutant ICP6Δ (25) was provided by Sandra Weller (University of Connecticut). The ICP0 mutant 7134 (9) was provided by Priscilla Schaffer (Harvard Medical School). The titer of 7134 used in these experiments was determined on complementing O-28 cells. The ICP27 C-terminal truncation mutants n59r and n504r (61), the in-frame deletion mutants dLeu (41), dAc (41), d5-6 (49), d1-2 (60), d2-3, d6-7 (5), d3-4, and d4-5 (48) and the point mutants m11 (59) and M50T (42) were all provided by Stephen Rice (University of Minnesota). The temperature-sensitive ICP27 mutant vBSLG4 and revertant vBS3-3 (73, 80) were provided by Saul Silverstein (Columbia University). A fuller description of these mutants is provided in the tables.
Inhibitor treatment.
Viral gene expression was limited to the IE phase by the treatment of cells with 0.5 μg per ml of cycloheximide for 30 min prior to infection. The same concentration of drug was present in the inoculum and replacement medium. At three hours postinfection, monolayers were rinsed with spent medium and then treated with actinomycin D at a concentration of 0.4 μg per ml in spent medium. To prevent expression of true-late (γ2) viral genes, the viral DNA replication inhibitor phosphonoacetic acid (PAA) (Sigma) was present in the virus inoculum and replacement medium at a concentration of 400 μg per ml (34). When using pharmacological inhibitors of signaling, subconfluent cultures were treated 30 min prior to infection with vehicle control dimethyl sulfoxide (DMSO) (0.5 μl/ml), the p38 inhibitor SB203580, the JNK inhibitor SP100625, or the MEK inhibitor UO126, all from Calbiochem. Inhibitor was maintained in the media throughout infection and was present in the overlay media.
Preparation of whole-cell lysates and immunoblotting.
At the time of harvest, cells were placed on ice to prevent artifactual induction of stress responses. The medium was removed and the monolayers rinsed with ice-cold Dulbecco's phosphate-buffered saline. Cells were scraped directly into 1 × sodium dodecyl sulfate (SDS) sample buffer (3.85 mM Tris base [pH 6.8], 9.1% β-mercaptoethanol, 1.82% SDS, 4.6% glycerol, 0.023% bromophenol blue [in 100% ethanol]) and denatured by boiling. Nuclear and cytoplasmic fractions were prepared as previously described (56). Cell-equivalent amounts of lysate were separated by 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to PolyScreen polyvinylidene difluoride membranes (PerkinElmer Life Sciences) followed by blocking in TBST (150 mM NaCl, 20 mM Tris [pH 7.6], 0.05% Tween 20) with 5% dry milk. All probing and washing of membranes was done with TBST. Rabbit polyclonal antibody for IκBα (C-21, catalog no. SC-371; Santa Cruz Biotechnology) was used at a 1:1,000 dilution overnight at 4°C per the manufacturer's instructions. Rabbit polyclonal antibody for p65 (antibody A, catalog no. SC-109) was used at a 1:1,000 dilution for 1 h at room temperature. Rabbit polyclonal antibody for poly(ADP-ribose) polymerase (PARP) (catalog no. SC-7150) and goat polyclonal antibody for GRP-78 (catalog no. SC-1050) were used at 1:500. Donkey anti-goat secondary antibody (horseradish peroxidase conjugated; Novus Biologicals) was used at 1:3,500. Mouse monoclonal antibody for α-tubulin (B-5-1-2; Sigma) was used at a 1:20,000 dilution. Rabbit polyclonal antibodies for p38 (9219), phospho-p38 (9211), JNK (9252), phospho-JNK (9251), phospho-p65 serine 276 (3037), and phospho-p65 serine 536 (3031) were purchased from Cell Signaling Technology and used at a 1:1,000 dilution overnight at 4°C per the manufacturer's instructions. Mouse monoclonal antibodies for the viral proteins ICP4 (1101), ICP0 (1112), and ICP27 (1113 and 1119) were purchased from the Rumbaugh-Goodwin Institute for Cancer Research (Plantation, FL) and used at 1:800, 1:800, 1:800, and 1:4,000 dilutions, respectively. Rabbit polyclonal antibody for VP16 (Clontech) was used at a 1:5,000 dilution. Rabbit polyclonal antibody against ICP8 (3-83) was a generous gift from David Knipe and used at a 1:20,000 dilution. Rabbit polyclonal antibody against glycoprotein C (gC) (R47), used at a dilution of 1:5,000, was a generous gift of Gary Cohen and Roselyn Eisenberg (University of Pennsylvania). Goat anti-rabbit and anti-mouse secondary antibodies were purchased from Amersham Biosciences. The secondary antibody was detected using the SuperSignal West Pico chemiluminescent substrate agent (Pierce). Films were scanned and images stored as 8-bit grayscale JPEG files. The density of each band was determined using the Image J program (NIH). Relative density values were corrected for average background by subtracting out the density of a blank portion of the film. The corrected values were then used to calculate either average percent change in independent experiments (sample value/mock value ± standard deviation) (see Fig. 3) or the percentage of uninfected cell levels (see Fig. 2, 4, and 6) by using Microsoft Excel. All membranes for IκBα Western blots were stripped and reprobed for α-tubulin. Levels of α-tubulin were used to normalize IκBα band intensities to protein concentrations in the lysate.
FIG. 3.

Immediate-early gene expression is sufficient for loss of IκBα. (A) Replicate cultures of CV-1 cells were mock infected or infected with KOS at an MOI of 5 under the conditions indicated and described in Materials and Methods. Lysates were prepared at 8 hpi, and the accumulations of indicated proteins were determined following fractionation on 12% polyacrylamide gels. The lysates were sequentially probed for IκBα, VP16, and α-tubulin or for ICP0 and ICP8. HSV/Cx rev, infection of cells in the presence of cycloheximide followed by removal of cycloheximide and addition of actinomycin D (ActD) at 5 hpi; HSV+PAA, infection of cells in the presence of PAA. (B) Replicate cultures of CV-1 cells were mock infected or infected with wt KOS or the ICP4 mutant vi13 (MOI = 5). Lysates were prepared at 8 hpi, fractionated on 12% polyacrylamide gels, and sequentially probed for IκBα and α-tubulin or for ICP4 plus ICP0 and ICP8. NS, nonspecific. (C) Relative amounts of IκBα from three independent experiments were determined using Image J and averaged, as described in Materials and Methods.
FIG. 2.

IκBα degradation is dependent on viral gene expression. (A) Replicate CV-1 cultures were mock infected or infected with HSV-1 strain HFEM or mutant tsB7 (MOI = 5) at the indicated temperatures, and lysates were prepared at 8 hpi. Western blot analysis of proteins was performed as described in Materials and Methods. Lysates fractionated on a 12% polyacrylamide gel were sequentially probed for IκBα and α-tubulin, and lysates fractionated on a 6% polyacrylamide gel were simultaneously probed for ICP0 and ICP4. Two additional independent experiments yielded similar results. NS, nonspecific. (B) Band intensities of IκBα from the Western blot in panel A were quantified using Image J as described in Materials and Methods, and the results presented are normalized to the values from mock infection. Filled bars, mock infected; open bars, HFEM infected; hatched bars, tsB7 infected.
FIG. 4.

ICP27 is necessary for loss of IκBα and phosphorylation of p65 after HSV infection. (A) Replicate cultures of CV-1 cells were mock infected or infected with the indicated viruses as described in the text (MOI = 5). Whole-cell lysates were prepared at 6, 8, and 12 hpi, and accumulations of IκBα viral proteins and α-tubulin were determined by Western blotting. Shown are the results for lysates prepared at 12 hpi. Under each panel, the relative amount of protein compared to the mock-infected sample (IκBα) or the wt-KOS-infected sample (ICP4, ICP27, ICP0, or ICP8) as determined by Image J analysis is indicated. (B) Graphical representation of the rate of loss of IκBα over the 12-h course of infection with wt KOS or the indicated mutant viruses. (C) Graphical representation of the phosphorylation of p65 at Ser276 over the 12-h course of infection with wt KOS or the indicated mutant viruses. (D) Graphical representation of the phosphorylation of p65 at Ser536 over the 12-h course of infection with wt KOS or the indicated mutant viruses.
FIG. 6.

The late gene activation function of ICP27 is not required for loss of IκBα. (A) Replicate cultures of CV-1 cells were mock infected or infected (MOI = 5) with either HSV-1 vBS3-3 or vBSLG4 at the indicated temperatures (°C). Whole-cell lysates were prepared at 8 hpi, and Western blotting was performed to detect loss of IκBα or accumulation of the indicated viral proteins. NS, nonspecific. (B) From the results in panel A, IκBα band intensities relative to values from mock-infected cells at the indicated temperatures (°C) were determined. Solid bars, mock infected; open bars, vBS3-3 infected; hatched bars, vBSLG4 infected. (C) Whole-cell lysates were prepared from mock-infected and infected (MOI = 20) CV-1 cells as indicated, and Western blotting was performed to detect IκBα and ICP27.
RESULTS
Loss of IκBα after HSV-1 infection correlates with nuclear translocation of p65.
Amici et al. reported that HSV-1 infection of HEp-2 cells resulted in activation of a 3× NF-κB-luciferase reporter gene and that this activation was abolished in the presence of DN-IκB (1). Similarly, we and others have reported that HSV-1 infection of human cell lines C33-A (56) and A549 (1, 56, 63) induced loss of IκBα and the appearance of NF-κB-dependent EMSA activity. Furthermore, DN-IκB abolished this activation, and loss of IκBα was accompanied by an increase in the amount of nuclear p65 (1, 56, 63). As this result was reported for murine cells, we first determined whether this link also existed in nonhuman-primate cells (Fig. 1). We prepared nuclear and cytoplasmic extracts from mock-infected and HSV-1-infected CV-1 cells at 12 hpi and assayed for the distributions of p65 and IκBα within cytoplasmic and nuclear compartments. In mock-infected cells (lanes 1 and 2), the great majority of p65 (∼95%) was localized in the cytoplasm, while after wild-type (wt) HSV infection (lanes 3 and 4), ∼57% of p65 was localized in the nucleus. Following wt HSV-1 infection, there was a correlation between loss of IκBα and relocalization of a portion of p65 to the nucleus. No residual IκBα was detectable in the wt-KOS-infected cytoplasmic extract. Detection of PARP was used to control for nuclear contamination of the cytoplasmic fraction. A trace amount of cytoplasmic contamination of the nuclear fraction was detected following Western blotting for GRP-78, a mitochondrial protein (data not shown). These results confirm a correlation between loss of IκBα and nuclear translocation of p65, a correlation which supports the use of IκBα loss as a reliable marker for NF-κB activation.
FIG. 1.
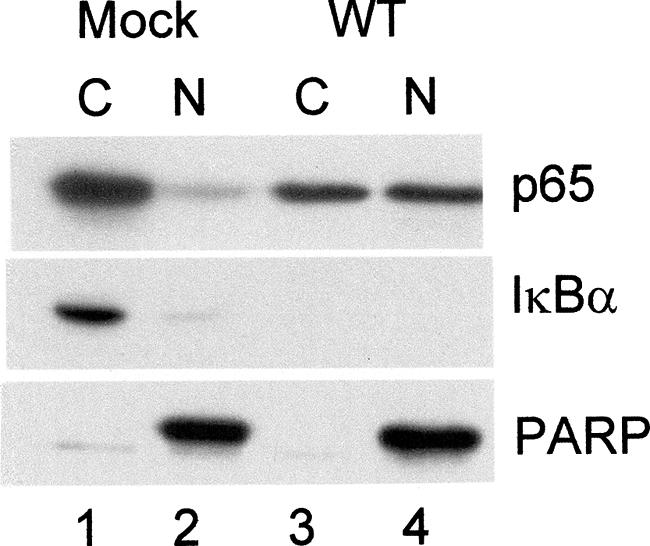
Loss of IκBα after HSV-1 infection correlates with nuclear translocation of p65. Cytoplasmic (C) and nuclear (N) extracts were prepared as previously described (56) from mock-infected and infected (MOI = 5) CV-1 cells and analyzed by Western blotting for p65, IκBα, and PARP.
IκBα degradation depends on viral gene expression.
Because we previously reported that HSV-1 infection triggers loss of IκBα and IκBβ, as well as persistent nuclear translocation of NF-κB (56), we set out to determine if viral protein expression was necessary for activation of the NF-κB pathway. Initially, we carried out infections with tsB7, a mutant that expresses a temperature-sensitive UL36 tegument protein involved in the release of DNA from the nucleocapsid. Thus, at the nonpermissive temperature (NPT), any host cell responses to virus binding and entry should occur normally and allow us to determine if any of these early events in the viral life cycle are important in the activation of the NF-κB pathway. Replicate cultures of CV-1 cells were infected at 33, 37, or 39°C with the parental HFEM strain or mutant tsB7 at a multiplicity of infection (MOI) of 5, and whole-cell extracts were prepared at 8 hpi. Results of Western blot analysis are shown in Fig. 2A, while Fig. 2B shows the results of quantification of IκB following wild-type or mutant infection at each temperature. Results displayed in Fig. 2 are a data set representative of three independent experiments. In each case, tsB7 infection at the NPT was impaired in IκBα loss, though to different extents, compared to the parental HFEM strain at the same temperature. Therefore, the results of the three experiments were not averaged. In the characterization of IκBα, only the lower band of the doublet seen in this experiment was quantified. The upper band was determined to be nonspecific due to our failure to detect it in other Western blot assays (see Fig. 5 and 6). Mock-infected samples displayed constant levels of IκBα at all three temperatures (Fig. 2A, lanes 1 to 3). Wild-type infection induced 70, 56, and 64% decreases in IκBα levels at 33, 37, and 39°C, respectively (lanes 4 to 6), relative to the levels in the corresponding mock-infected cells. Infection with tsB7 resulted in a 73% decrease in IκBα at the permissive temperature (PT) of 33°C but no decrease at 37 and 39°C (Fig. 2A, lanes 7 to 9, and B) relative to the mock-infected cells at the corresponding temperature. To ensure that tsB7 inhibited the expression of viral genes at elevated temperatures, we performed Western blot analysis for the IE proteins ICP0 and ICP4 and determined the levels of α-tubulin as a loading control. Viral proteins were expressed in all wild-type infections and at 33°C after tsB7 infection (Fig. 2A, lanes 4 to 7). Levels of viral proteins were severely inhibited in tsB7 infection at 37°C and were undetectable at 39°C (lanes 8 and 9). The residual level of viral protein detected at 37°C was insufficient to induce IκBα degradation, as the level of IκBα detected was comparable to the level found in mock-infected cells at 37°C. Band densities were quantified as described in Materials and Methods and normalized to levels of α-tubulin in each lane.
FIG. 5.

ICP27 is necessary for IκBα loss. (A) Replicate cultures of CV-1 cells were mock infected or infected with wt KOS, d100, d103, d106, d107, or d109 at the indicated MOI (2.5, 5, or 10). Whole-cell lysates were prepared at 8 hpi, and Western blotting was performed to detect IκBα; the membranes were then stripped and reprobed for α-tubulin. Amounts of IκBα normalized to α-tubulin are indicated under the appropriate lanes (see Materials and Methods). (B) CV-1 cells were mock infected or infected (MOI = 5) with wt KOS, d27-1, or d100 and harvested at 16 hpi. Lane 5 represents a d100 lysate prepared at 24 hpi. Western blot analyses for IκBα, ICP27, and α-tubulin were performed through subsequent stripping and reprobing of the same membrane.
A recent report demonstrate that UV-irradiated virus was able to transiently activate the NF-κB pathway during entry but was unable to support the sustained NF-κB activation we observe, which begins around 3 hpi (2). We confirmed these observations by infecting cells with equivalent amounts of unirradiated or UV-irradiated virus and assaying for IκBα loss. As expected, unirradiated virus induced efficient loss of IκBα when assayed at 8 hpi, while UV-irradiated virus failed to display levels of IκBα comparable to those seen in mock-infected cells (data not shown). We conclude that the events prior to viral genome entry into the nucleus are not sufficient for the sustained induction of the NF-κB pathway.
Immediate-early gene expression is sufficient for IκBα degradation.
Previously we demonstrated degradation of IκBα as early as 6 hpi (56). At that time, there was significant expression of the IE and E proteins, but there were lower levels of L proteins. Therefore, we hypothesized that either IE or E proteins controlled the degradation of IκBα. We took advantage of drugs that selectively inhibit protein synthesis (cycloheximide), mRNA synthesis (actinomycin D), or viral DNA replication (PAA) to define the requirements for viral gene expression in NF-κB activation. Whole-cell lysates were prepared and analyzed by Western blot for all three classes of viral genes (Fig. 3A). No viral proteins were detected in mock-infected cells (lane 1), but we detected ICP0, ICP8, and VP16 in lysates from infected, untreated cells (lane 2). Following the reversal of cycloheximide and the addition of actinomycin D, cells accumulated ICP0 but not ICP8 and VP16 (lane 3). Finally, infection in the presence of PAA resulted in the accumulation of both IE and E proteins but not VP16 (lane 4). Probing for IκBα revealed its loss from all infected-cell lysates regardless of drug treatment, suggesting that only immediate-early gene expression was required for the activation of the NF-κB pathway.
To confirm this observation, we infected CV-1 cells with either wt KOS or the ICP4 mutant vi13 (77). This mutant contains a two-amino-acid (aa) substitution in the DNA binding domain of ICP4 with a resulting protein synthesis phenotype marked by overexpression of IE genes and absence of E or L gene expression. We probed whole-cell extracts prepared at 8 hpi for accumulation of ICP4, ICP0, and ICP8. Figure 3B illustrates a typical result: wt virus and vi13 both expressed ICP4 and ICP0 (lanes 1 and 2) while only the wt virus expressed ICP8 (lane 3). The overaccumulation of ICP4 and ICP0 in vi13-infected cells was consistent with the results of other reports using this mutant (56, 77). The level of IκBα was undetectable in the wt-KOS-infected lysate (lane 2) and was reduced in the lysate from vi13-infected cells compared to that from mock-infected cells (lanes 1 and 3). Panel C of Fig. 3 shows the quantification of band intensities relative to those of mock-infected cells from three independent experiments, while the Western blots from panels A and B are representative of one of the repeats. From the results presented in panels A and B, we conclude that immediate-early gene expression is required and sufficient for the IκBα degradation seen during HSV infection. Of note, however, is that while IE proteins were overexpressed during vi13 infection, the mutant was not as efficient as wt virus in causing nuclear translocation of p65 or loss of IκBα (Fig. 1 and 3B).
ICP27 is necessary for loss of IκBα.
Having demonstrated that IE protein synthesis was sufficient for loss of IκBα to be observed, we conducted a time course infection with several mutants defective in IE protein expression to determine which was necessary for this effect. This experiment was conducted twice with similar results. The panel of immediate-early mutants included (i) n12, a nonsense mutant which expresses a ∼41-kDa ICP4 protein of aa 1 to 251 (15); (ii) 7134, which contains a lacZ substitution for the ICP0 open reading frame (9); (iii) ICP6Δ, from which the ICP6 open reading frame is deleted (25); and (iv) d27-1, from which the entire ICP27 gene is deleted (61). The n12 mutant was reported to express IE proteins and the E protein ICP6 only (14, 15). Whole-cell lysates were prepared at 6, 8, and 12 hpi, separated by SDS-PAGE, and analyzed by Western blotting for IκBα. Western blots of IκBα, IE proteins ICP4, ICP27, and ICP0, and E protein ICP8 in the 12-hpi lysates are shown in Fig. 4A, and the quantification of IE protein accumulations from all three time points is summarized in Table 1. Western blots of viral proteins shown in panel A were from purposefully overexposed autoradiograms to confirm the viral mutant phenotypes, while all quantifications, using Image J, were performed with exposures which were in the linear range. Figure 4B summarizes the levels of IκBα throughout the 12-h time course of the wt and mutant infections. Levels of IκBα in mock-infected cells remained constant throughout the time course, while levels in wt virus-infected cells gradually decreased over the 12 h of the analysis. Following infection with the ICP4 mutant n12, the level of IκBα remained similar to those in mock-infected cells at 6 and 8 hpi and was reduced to 55% of the level in mock-infected cells by 12 hpi (Fig. 4A, lane 3, and B). Analysis of IκBα in 7134- and ICP6Δ-infected cells revealed a delay in loss of protein relative to that of mock-infected cells until 12 hpi, when 80% and 88% losses, respectively, were detected (Fig. 4A, lanes 4 and 5, and B). In contrast, over the entire time course of infection, d27-1-infected cells retained IκBα levels near those of the mock-infected cells (Fig. 4A, lane 6, and B). This result is consistent with our previous finding that d27-1 was impaired in nuclear translocation of NF-κB in C33-A cells (56).
TABLE 1.
Relative levels of infected-cell polypeptides (ICPs) in wt- and mutant-virus-infected cellsa
| Virus (product of affected gene) | Relative level of ICP at indicated hpi
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ICP4
|
ICP0
|
ICP27
|
ICP8
|
|||||||||
| 6 | 8 | 12 | 6 | 8 | 12 | 6 | 8 | 12 | 6 | 8 | 12 | |
| wt KOS | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| n12 (ICP4) | — | — | — | 4.60 | 1.09 | 1.07 | 1.20 | 1.00 | 1.42 | — | — | — |
| 7134 (ICP0) | 1.44 | 1.12 | 0.89 | — | — | — | 0.79 | 0.86 | 0.79 | 1.61 | 0.86 | 0.96 |
| ICP6Δ (ICP6) | 1.56 | 0.68 | 0.60 | 2.60 | 0.70 | 0.81 | 0.71 | 0.41 | 0.67 | 1.5 | 0.7 | 0.9 |
| d27-1 (ICP27) | 1.23 | 0.59 | 0.6 | 3.11 | 0.74 | 0.83 | — | — | — | 1.02 | 0.49 | 0.88 |
Values represent levels of ICPs at each time postinfection relative to wt-virus-infected cells. Accumulations of proteins in whole-cell lysates prepared at 6, 8, and 12 hpi were determined by Western blotting and quantified as described in Materials and Methods. Dashes indicate no detectable protein.
To confirm the status of viral protein synthesis under conditions of wt or mutant virus infection, lysates prepared at 6, 8, and 12 hpi were also analyzed for ICP4, ICP27, ICP0, and ICP8. In Fig. 4A, the results of Western blotting for viral proteins and values for accumulation at 12 hpi relative to wt KOS are shown. As expected, each IE null mutant failed to express the product of its cognate deleted gene but expressed the remaining IE proteins. Relative levels of viral protein accumulation at 6-, 8-, and 12-hpi time points are presented in Table 1. Infection with n12 resulted in a large relative overexpression of ICP0 at 6 h similar to what we observed with another ICP4 mutant, vi13 (Fig. 3) (47). Levels of ICP27 in cells infected with n12 differed little from those observed in wt-infected cells at earlier times and were somewhat elevated at 12 h. Under our conditions of 7134 infection (a MOI of 5 based on the titer obtained on complementing O-28 cells), there were slightly reduced levels of ICP4, ICP27, and ICP8 accumulation relative to wt KOS, while infection with ICP6Δ resulted in reduced relative levels of ICP4 and ICP27 at the 8- and 12-h time points only. These results, combined with those in Fig. 3A and B, indicate that ICP27, and to a lesser extent ICP4, is necessary for maximum loss of IκBα during HSV infection. The residual IκBα in ICP0 and ICP6 mutant-infected cells can be explained at least in part by reduced ICP27 and ICP4 accumulation. This in turn is consistent with delayed protein expression phenotypes of ICP0 mutants (12, 19, 83) and the slower kinetics of ICP6Δ virus production on confluent, resting cell monolayers or under conditions of serum starvation (24, 25). Alternatively, it might reflect a role for these viral proteins in modulating aspects of ICP27 and ICP4 intracellular localization or posttranslational modification and, thus, function.
The p65 subunit of NF-κB is phosphorylated on serines 276 and 536 during HSV infection.
Activation of NF-κB is regulated in part through phosphorylation of p65 by distinct kinase activities. Ser276 can be phosphorylated by protein kinase A or mitogen- and stress-activated protein kinase 1 (MSK-1) and is important for interactions with CREB-binding protein (CBP)/p300 during transcription activation (87, 89-91). Ser536 is phosphorylated by IKK after tumor necrosis factor (65, 66, 78) or lipopolysaccharide treatment (88), increasing its transcriptional activity. To characterize p65 phosphorylation during infection, we performed Western blot analyses using antibodies specific for phosphorylated Ser276 and phosphorylated Ser536. The protein lysates analyzed were from the wt and mutant time course infections that we had analyzed for IκBα (Fig. 4A and B). Following the use of phospho-specific antibodies, the membranes were stripped and reprobed for total p65. The resulting Western blots were quantified using Image J (Fig. 4C and D). For each sample, a phosphorylated p65-to-total p65 ratio was calculated and the data were expressed as the change in activation over that in mock-infected cells (n-fold) at the same time point. Strikingly, wt infection induced increasing amounts of Ser276 phosphorylation over the 12-h time course. ICP0 and ICP6 mutants also induced increasing amounts of Ser276 phosphorylation. The slower kinetics of phosphorylation was consistent with the slower degradation of IκBα also seen with these mutants (compare panels B and D), which we attribute to delayed replication kinetics of these viruses. Consistent with the previous finding that ICP4 and ICP27 were necessary for NF-κB activation, Ser276 phosphorylation in ICP4 and ICP27 mutant infected cells was consistently between 5 and 33% of wt virus induction, suggesting that expression of both of these IE proteins is necessary for p65 phosphorylation at Ser276. At 6 hpi, wt HSV induced an 8.4-fold activation of Ser536 phosphorylation, but this was not observed with any of the IE mutants (Fig. 4D). At later times postinfection, HSV infection resulted in only a 2- to 2.3-fold induction. By 12 hpi, all of the IE mutants induced levels of Ser536 phosphorylation that were similar to that induced by wt virus.
In order to determine the role of ICP27 on the stability of IκBα during infection, we investigated a panel of viral mutants expressing various combinations of IE genes as described in Table 2. Three independent sets of infections with d100, d103, d106, d107, and d109 mutants were conducted, and the levels of IκBα from a representative experiment at 8 hpi are shown in Fig. 5A. Infection with wt virus (lane 2) resulted in a 95% loss of IκBα compared to the outcome with mock infection. Infection with the d103 mutant, which expresses only ICP0 and ICP27, resulted in a 33% reduction of IκBα (lane 6). We have reported that this mutant was capable of activating both p38 and JNK (32). The d100 mutant expresses only ICP22, ICP27, and ICP47 among the IE proteins but failed to reduce IκBα levels at any MOI tested (lanes 3 to 5). Infection with this mutant resulted in phosphorylation of p38 as early as 8 hpi, though activation of JNK was not detected until 16 hpi (32). Additionally, we observed that lysates from all infections with mutants that failed to express ICP27 (d106, d107, and d109) had IκBα levels similar to or higher than the level in lysates with mock infection (lanes 7 to 9). Since the d107 mutant, which expresses ICP22 and ICP47 in addition to ICP0, retained more IκBα after infection than the d106 mutant, which expresses only ICP0, we conclude that ICP22 and ICP47 have no significant role in the degradation of IκBα during HSV infection.
TABLE 2.
IE deletion mutants
| Mutant | α gene product(s) expressed | α gene product(s) deleted | Reference |
|---|---|---|---|
| d27-1 | ICP0, ICP4, ICP22, ICP47 | ICP27 | 61 |
| d100 | ICP22, ICP27, ICP47 | ICP0, ICP4 | 67 |
| d103 | ICP0, ICP27 | ICP4, ICP22, ICP47 | 67 |
| d106 | ICP0 | ICP4, ICP22, ICP27, ICP47 | 67 |
| d107 | ICP0, ICP22, ICP47 | ICP4, ICP27 | 67 |
| d109 | ICP0, ICP4, ICP22, ICP27, ICP47 | 67 |
Because d100 infection was capable of activating both JNK and p38 only at late times during infection (32), we compared the levels of IκB of mock-infected, wt-infected, d27-1-infected, and d100-infected cells at 16 hpi, as well as d100-infected cells at 24 hpi (Fig. 5B). IκBα was not detectable in the lysate of wt-infected cells at 16 h but IκBα levels were reduced in the d27-1 and d100 lysates compared to that in the mock-infected sample. IκBα was not detectable in the d100-infected lysate at 24 hpi (Fig. 5B), though we could detect IκBα in d27-1-infected lysates at 24 hpi in other experiments (data not shown). To determine whether the IκBα degradation we observed in d100 infections at 24 hpi was due to induction of apoptosis, levels of caspase 3 and 7 cleavage were determined. We did not observe caspase cleavage with wt virus or d100 at either 16 or 24 hpi (data not shown). Cleavage of caspase proteins was detected in d27-1 infections, consistent with findings that infection with an ICP27 deletion mutant was abortive and induced apoptosis (3-5). Collectively, these results were consistent with the model that ICP27 expression may be sufficient for the degradation of IκB in the context of viral infection.
Late gene transactivation function of ICP27 is not required for loss of IκBα.
Because of the requirement for ICP27 in IκBα loss, we began a more detailed analysis of this protein. To determine the importance of the transactivation functions of ICP27 and consequent expression of true-late (γ2) viral genes for IκBα loss, we compared the ICP27 mutant vBSLG4, which at the NPT of 39°C is incapable of stimulating γ2 gene expression, with revertant vBS3-3 (70, 71, 80). The results presented in Fig. 6A are representative of the results from three independent experiments. Significant loss of IκBα relative to the mock-infected controls was detected at both the PT and NPT in both mutant vBSLG4- and revertant vBS3-3-infected cells (compare lanes 1, 3, and 5 with lanes 2, 4, and 6). At the same time, ICP27 accumulation was reduced at 33°C compared to that at 39°C because of the slower kinetics of the infection at the lower temperature. Figure 6B presents the quantification of IκBα levels under the various conditions of infection. Relative to the IκBα levels in mock-infected cells, reductions of 40 to 60% in IκBα levels were seen at 33°C and reductions of 80 to 90% were observed at 39°C, correlating with the amounts of ICP27 accumulated (Fig. 6A). We analyzed levels of VP16 and gC, the latter a γ2 gene transactivation target of ICP27, in order to confirm loss of ICP27 transactivation activity at the NPT in vBSLG4-infected cells. At 12 hpi, we detected comparable amounts of VP16 and gC in the lysates from vBS3-3-infected cells at either temperature (lanes 3 and 4). While both L proteins accumulated at the PT in the lysate from vBSLG4-infected cells (lane 5), considerably less gC and a reduced amount of VP16 accumulated at the NPT (lane 6).
We also monitored the status of IκBα in cells infected with the ICP27 point mutant m11 (48), which is also impaired in late gene expression. We have consistently observed that infection with the m11 mutant results in less ICP27 accumulation than infection with wt virus. Therefore, to increase the amount of m11 ICP27, infections were conducted at an MOI of 20. While the amount of IκBα in mock- and d27-1-infected cells remained high (Fig. 6C, lanes 1 and 3), the amount of IκBα was greatly reduced in wt-KOS- and m11-infected cell lysates (lanes 2 and 4). Therefore, we conclude that the transactivation function of ICP27 and γ2 viral gene expression are not necessary for the activation of the NF-κB pathway, as evidenced by the loss of IκBα.
Residues 21 to 63 of ICP27 are required for the loss of IκBα.
We analyzed a series of ICP27 truncation and in-frame deletion mutants to identify which domain of ICP27 was required for the degradation of IκBα. Table 3 lists the mutants, the deleted or mutated regions, and their replication capacities in Vero cells. CV-1 cells were mock infected or infected with wt HSV-1 or the panel of ICP27 mutants at an MOI of 5. Triplicate whole-cell lysates were prepared at 8 hpi, separated by SDS-PAGE, and analyzed by Western blotting for IκBα and ICP27 (Fig. 7A to C, which were representative of the results). Mock-infected cells contained high levels of IκBα, as did cells infected with the null mutant d27-1, the truncation mutant n59r (panel A, lanes 1, 3, and 4), and the in-frame deletion mutant d1-2 (panel B, lane 4). Reduced amounts of IκBα were observed in wt-virus-infected cells (lane 2 of all panels) and also following infection with the truncation mutant n504r (panel A, lane 5) and the in-frame deletion mutants d2-3 (panel B, lane 5), d4-5, d5-6, and d6-7 (panel C, lanes 3 to 5). Intermediate levels of IκBα were observed with the in-frame deletion mutants dLeu and d3-4 (panel B, lanes 3 and 6), suggesting that they may play an auxiliary role in ICP27-induced loss of IκBα. Because the monoclonal antibody H1119 used to probe for ICP27 is directed at an epitope missing from the dLeu mutant, the presence of this protein was verified using the monoclonal antibody H1113 (data not shown).
TABLE 3.
ICP27 mutants
| Mutant | Affected region (aa)a | Replicationb | Reference(s) |
|---|---|---|---|
| Deletion mutant (d27-1) | 1-512 | D | 61 |
| C-terminal truncation mutant | |||
| n59r | 60-512 | D | 61 |
| n504r | 505-512 | D | 61 |
| In-frame deletion mutant | |||
| dLeu | 6-19 | I | 41 |
| dAc | 21-63 | I | |
| d1-2 | 12-63 | I | 60 |
| d2-3 | 64-109 | C | 5 |
| d3-4 | 109-138 | C | 48 |
| d4-5 | 139-153 | D | 48 |
| d5-6 | 154-173 | C | 49 |
| d6-7 | 174-200 | C | 5 |
| Point mutant | |||
| M50T | 50 | I | |
| m11 | 340, 341 | D | 58 |
| vBSLG4 | 480 | Cc | 73, 80 |
FIG. 7.

A domain of ICP27 encompassing aa residues 12 to 63 is necessary for loss of IκBα. Replicate cultures of CV-1 cells were mock infected or infected with the indicated viruses at an MOI of 5. Whole-cell lysates were prepared at 8 hpi, fractionated on a 12% polyacrylamide gel, and transferred to a membrane for Western blot analysis as described in Materials and Methods. Membranes were sequentially probed for IκBα and ICP27. (A) Analysis of wt virus, the null mutant d27-1, and N-terminal truncation mutants n59r and n504r. Results of probing the n504r lysate for IκBα and ICP27 were from a separate part of the same autoradiograph containing the results in lanes 1 to 4. (B) Analysis of wt virus and in-frame deletion mutants dLeu, d1-2, d2-3, and d3-4. Because the monoclonal antibody H1119 epitope is missing in the dLeu mutant, the presence of ICP27 was verified using the monoclonal antibody H1113 (data not shown). Results of probing mock-infected, wt-infected, and dLeu-infected lysates for IκBα and ICP27 were from separate parts of the same autoradiograph containing the results in lanes 4 to 6. (C) Analysis of wt virus and in-frame deletion mutants d4-5, d5-6, and d6-7.
Recently, we reported that d1-2 (aa 12 to 63 deleted) was incapable of activating JNK and p38, while dLeu (aa 6 to 19 deleted) retained the ability to activate these stress kinases (41). Since d1-2 was as impaired as d27-1 for loss of IκBα, two additional mutants within the domain deleted from d1-2 were characterized. The dAc mutation deletes amino acids 21 to 63 of ICP27, and the expressed protein was primarily nuclear, though cytoplasmic staining was detectable (41). The affected domain of ICP27 was termed the export control sequence because of its ability to restrict shuttling (81). The second mutant tested was M50T, which effects an amino acid substitution conferring leptomycin B resistance to HSV replication (53). Leptomycin B is an antifungal antibiotic originally isolated from Streptomyces sp. (30) and was reported to be a potent inhibitor of nuclear export signal-dependent and Crm-1 dependent export of proteins into the cytoplasm (40). Several studies have described mechanisms of ICP27-mediated RNA export by a Crm1-dependent pathway (69, 81) and an Aly/REF-dependent pathway (11, 38, 50, 57, 69, 80).
To test the ability of dAc and M50T to activate stress-activated protein kinases and degrade IκBα, we compared them to wt HSV and d27-1. Three independent infections were performed, two with an MOI of 5 and one with an MOI of 10, and cells were harvested at 8 hpi. Western blots from one of the experiments with infection at an MOI of 5 are shown in Fig. 8. The data presented were representative of the outcomes observed in all three experiments. Infection with wt HSV (lane 2) resulted in activation of both p38 and JNK, and as we previously reported (32), d27-1 showed little to no activation of either p38 or JNK (lane 3), equivalent to the background activation seen in mock infection (lane 1). Infection with both dAc and M50T resulted in little to no activation (lanes 4 and 5). These results suggested that amino acids 21 to 63 were required for the ICP27-induced activation of the stress-activated protein kinase and NF-κB pathways (as detected by IκBα loss) and that the methionine at position 50 was important in mediating the induction.
FIG. 8.

Loss of IκBα correlates with JNK and p38 phosphorylation during infection with ICP27 mutants affecting aa residues 21 to 63. Replicate cultures of CV-1 cells were mock infected or infected with the indicated viruses at an MOI of 5. Whole-cell lysates were prepared at 8 hpi, fractionated on a 12% polyacrylamide gel, and transferred to a membrane for Western blot analysis as described in Materials and Methods. Replicate membranes were sequentially probed for IκBα, ICP27, and α-tubulin or for phosphorylated (p) and total JNK and p38.
JNK activation is upstream of IκBα loss and Ser276 p65 phosphorylation.
To understand if either p38 or JNK activation was upstream of NF-κB activation, we monitored IκB levels and p65 phosphorylation after infection in the presence of pharmacological inhibitors of signaling. Replicate cultures of CV-1 cells were treated with DMSO or inhibitors for p38 (SB203580), JNK (SP600125), and MEK1 (UO126) for 30 min prior to infection. Cells were then mock infected or infected with wt KOS at an MOI of 5 in the presence of inhibitor and harvested at 8 hpi. Western blot analysis for IκB or phosphorylated-p65 Ser276 or Ser536 was performed (Table 4). The membranes were then stripped and reprobed for α-tubulin or total p65. Normalized IκBα and phospho-p65 levels were calculated as described in Materials and Methods. The results from two independent experiments could not be averaged because of differences in scale, derived during background correction but showed similar trends in IκBα and phospho-p65 levels due to inhibitor treatment. Table 4 displays the results of one representative experiment. When the levels of IκBα were compared across the inhibitors used, consistent degradation of IκBα was observed in cells with DMSO- and UO126-treated infections, as well as at low concentrations of SB203580. The use of higher concentrations of SB203580 and all concentrations of SP600125 resulted in levels of IκBα similar to those resulting from mock infection (values in italics). Higher levels of p38 inhibitor may also affect JNK activity. Therefore, JNK activity, but not p38, may be required for ICP27-induced IκBα loss. While HSV infection resulted in levels of Ser276 phosphorylation 2- to 10.8-fold higher than those in mock-infected DMSO-, SB203580-, and UO126-treated cells, exposure to increasing concentrations of SP600125 resulted in increasingly lower levels of phosphorylated Ser276 (values in italics). Though these levels were not reduced to levels shown by mock-infected cells, decreased phosphorylation in the presence of the inhibitor indicated a role for JNK activity in Ser276 phosphorylation. On the other hand, neither SP600125 nor any of the other inhibitors prevented the modest induction of phospho-Ser536 seen during HSV infection. Inhibition of p38 kinase activity by SB203580 resulted in increased phospho-Ser536 levels during infection, suggesting that p38 activity may negatively regulate the ability of the virus to phosphorylate p65 on Ser536. Taken together, these results support a model in which the loss of IκBα and phosphorylation of p65 at Ser276 are positively regulated by JNK activity, while phosphorylation of p65 at Ser536 is negatively regulated by p38 kinase activity.
TABLE 4.
Effects of pharmacological inhibitors of p38, JNK, and extracellular signal-regulated kinase on IκBα accumulation and NF-κB p65 phosphorylation after HSV infectiona
| Inhibitorb | Dosec | % IκBαd | Increase (n-fold)e
|
|
|---|---|---|---|---|
| Ser276 | Ser536 | |||
| DMSO | 0.25 μl | 37.9 | 6.7 | 1.5 |
| 0.5 μl | 35.9 | 4.9 | 2.6 | |
| 1.0 μl | 34.9 | 4.8 | 1.4 | |
| 1.25 μl | 32.5 | 5.2 | 2.2 | |
| 2.5 μl | 24.6 | 5.3 | 1.2 | |
| SB203580 | 2.5 μM | 37.8 | 3.2 | 1.5 |
| 5 μM | 4.5 | 2.9 | 3.1 | |
| 10 μM | 6.6 | 3.1 | 5.3 | |
| 25 μM | 165 | 6.4 | 7.4 | |
| 50 μM | 192 | 2.2 | 8.6 | |
| SP600125 | 5 μM | 110 | 11.1 | 2.4 |
| 10 μM | 143 | 8.6 | 5.2 | |
| 25 μM | 85.5 | 7.8 | 2.9 | |
| 50 μM | 122 | 3.0 | 2.2 | |
| 100 μM | 99 | 3.5 | 2.0 | |
| UO126 | 2.5 μM | 39.9 | 2.6 | 2.4 |
| 5 μM | 49.4 | 5.0 | 1.9 | |
| 10 μM | 26.5 | 10.8 | 1.5 | |
| 25 μM | 13.1 | 4.0 | 1.6 | |
| 50 μM | 12.3 | 2.1 | 2.3 | |
Infections were performed with wt HSV-1 KOS at an MOI of 5 and lysates prepared at 8 hpi.
Vehicle or inhibitor present for 30 min prior to and throughout infection.
Concentrations of DMSO are equivalent to concentrations used to achieve indicated doses of inhibitors dissolved in DMSO.
Percent residual IκBα relative to the amount in mock-infected cells treated with the maximum volume of DMSO or at the maximum drug concentration.
Increase (n-fold) in phosphorylated-p65 Ser276 or Ser536 relative to mock-infected cells treated with the maximum volume of DMSO or at the maximum drug concentration.
DISCUSSION
In this report, we established a correlation between NF-κB activation, namely p65 nuclear translocation, and the loss of IκBα after HSV infection. Using IκBα as a surrogate marker, we characterized requirements for the HSV-induced activation of NF-κB. Virus binding and entry into the cell were not sufficient for IκBα degradation, based on infections with the uncoating mutant tsB7 or with UV-irradiated virus. Using strategies to limit viral gene expression, we determined that IE gene expression was sufficient for degradation of IκB. We confirmed the sufficiency of IE gene expression with the ICP4 mutant vi13, which expresses IE genes only. We previously reported that extracts prepared from both ICP4 mutant vi13- and ICP27 mutant d27-1-infected C33-A cells did not display increased NF-κB DNA binding activity, as measured by EMSA (56), while infection with wt HSV or an ICP8 mutant resulted in large increases in NF-κB DNA binding activity in C33-A or U2-OS cells. IκB degradation was not evaluated for either cell type (56). We speculate that the IκBα degradation observed here following vi13 infection in CV-1 cells is regulated separately from NF-κB DNA binding activity. In this model, ICP4 may play a role in the efficient or sustained activation of NF-κB during HSV infection through regulation of DNA binding activity, while ICP27 activity is required for loss of IκB and subsequent nuclear translocation of NF-κB (Fig. 3). The effects of ICP4 on NF-κB may be associated with the ICP27 effects, since the vi13 mutant was not as efficient as wt virus in causing loss of IκBα. ICP4 and ICP27 are known to physically interact (55), and if complexed during NF-κB activation, loss of one protein from the complex could affect the complex structure and therefore its activities. Additional experiments to determine which viral proteins interact with which cellular factors in this pathway are needed to better define the relationship between ICP4 and ICP27 during NF-κB activation.
We further investigated the individual contributions of ICP4 and ICP27 in a panel of immediate-early deletion mutants. Over a 12-h time course, wt-HSV infection resulted in IκB degradation. ICP4, ICP0, or ICP6 mutant infection also resulted in degradation, although at a reduced rate, suggesting that complete degradation of IκB would eventually be observed in these mutants. On the other hand, ICP27 mutant-infected cells retained 99% of the level of IκB in mock-infected cells even out to 12 hpi. In addition, infection with the d100 mutant, which expresses only ICP27, ICP22, and ICP47, eventually resulted in decreased levels of IκB, demonstrating that ICP4 was not absolutely required for the degradation of IκBα during the activation of the NF-κB pathway. This result was consistent with IκB degradation seen in vi13 mutant-infected CV-1 cells.
We also characterized the phosphorylation status of p65 during HSV infection. The phosphorylation of p65 on Ser276 leads to increased NF-κB transcriptional activation (87) and may lead to more efficient recruitment of CBP (91). Constitutive phosphorylation of Ser536 was detected in mock-infected lysates (data not shown), with a higher level of phosphorylation seen after 6 hpi, which dropped to twofold at 8 hpi. As the peak of Ser536 phosphorylation occurred as degradation of IκBα (56) and phosphorylation of Ser276 were beginning, phosphorylation at Ser536 may be an earlier and transient event in the NF-κB activation cascade. Since Ser536 phosphorylation was less sensitive to ICP4 and ICP27 deletion than was Ser276 phosphorylation, especially at 12 hpi, the mechanism of Ser536 phosphorylation may require other viral factors or events in the viral life cycle. Ser536 is located in the transactivation domain of p65 (75), can be phosphorylated in an IκBα-independent manner (75) and, when phosphorylated, mediates increased transcriptional activity (65, 66, 78). We are continuing our analysis of posttranslational modifications to p65 in wt- and mutant-infected cells.
A recent report presented findings indicating that ICP0, when overexpressed from a plasmid, could directly bind and ubiquitylate IκB in vitro (17). Additionally, polyubiquitin chains were not formed when a RING finger point mutant for bovine herpesvirus 1 ICP0 (13G/51A) was expressed, suggesting that ICP0-dependent IκB degradation and subsequent NF-κB activation was dependent on this domain. Results of our immediate-early mutant time course led us to conclude that ICP0 is not required for IκBα loss. We have tested a similar HSV-1 ICP0 mutant (C116G/C156A) (12, 55) for activation of p38, shown here to be an upstream activator of NF-κB, and found that this RING finger domain mutant was not impaired for activation of p38 (data not shown). We are currently studying the ability of this RING finger mutant to degrade IκB. Our studies evaluated the impact of deleting ICP0 while delivering the rest of the viral genome. The difference between these two studies may suggest redundant mechanisms for the degradation of IκB. Alternatively, it may suggest that during infection, ICP0 may be localized to areas where it does not interact with IκB, demonstrating that the ICP0-dependent ubiquitylation of IκB may reflect altered localization of ICP0 in the absence of other viral proteins.
Using a panel of ICP27 mutant viruses, we identified a N-terminal region (amino acids 12 to 63) previously implicated in nuclear-cytoplasmic shuttling (50), as required for the ICP27-dependent degradation of IκB in CV-1 cells. Viruses expressing ICP27 with deletions from this region are deficient in HSV-induced apoptosis suppression (5). Other regions of ICP27 involved in apoptosis suppression were C-terminal of residue 263 or point mutations involved in ICP27 transactivation activity or ICP27 nuclear-cytoplasmic shuttling (5) in human cells. Since ICP27 mutants do not induce significant apoptosis in cells of nonhuman primate origin, we chose to evaluate ICP27 mutants for NF-κB activation in CV-1 cells. Our observations on the importance of the N-terminal region of ICP27 in NF-κB activation are consistent with previous reports demonstrating (i) a requirement for NF-κB activation for apoptosis suppression in HSV infections (24), (ii) a requirement for IE gene expression but not VP16 in the induction of apoptosis during HSV infection (74), and (iii) correlations between ICP27 (3) and E and leaky-late (5) gene expression and the ability to suppress apoptosis during HSV infection.
As other regions of ICP27 have been identified as being important for apoptosis suppression, functions of ICP27 in addition to NF-κB activation may be required for complete suppression. In one model, sustained NF-κB activation could be mediated by ICP27-regulated E or leaky-late proteins, perhaps by phosphorylation of one or both upstream activators IKK and IκB during infection (1, 56). The need for E and L genes for sustained NF-κB activity could also explain the potential role for ICP4, as it is required for expression of all E and L genes. In another model, ICP27 could serve as the nexus between a cellular factor, e.g., CK2 (7, 39), and key apoptosis effectors.
In this report, we also demonstrated that IκB degradation was inhibited in the presence of the JNK inhibitor SP100625, suggesting that JNK is upstream of NF-κB activation. Though an IKK-independent activation of NF-κB mediated by a p38-CK2-IκB phosphorylation pathway has been described previously (36), NF-κB activation during HSV infection was dependent on the IKKs (2, 27). Therefore, JNK-induced IκB degradation would have to target IKK/IκB or a further upstream activator. A possible mechanism for JNK-dependent IκB phosphorylation during HSV infection could involve IκB kinase complex-associated protein (IKAP). IKAP interacts with JNK through a C-terminal domain (33) and was reported to induce JNK activation after stimulation of HeLa or 293 cells with tumor necrosis factor alpha and UV irradiation (33). IKAP can be detected in a complex that includes NIK, IKKα, IKKβ, IκB, and NF-κB (13). IKK may be required for IKAP-IκB complex formation, explaining its role in HSV-induced NF-κB activation. JNK may interact with other proteins, such as β-transducin repeat containing protein, to cause IκB phosphorylation (82). Further characterization of HSV-induced IκB phosphorylation is required to test these models.
An additional JNK-mediated mechanism for regulating NF-κB involves phosphorylation of p65. MSK-1 was reported to phosphorylate p65 on Ser276 (87), the same serine residue phosphorylated after lipopolysaccharide treatment (90, 91). MSK-1 phosphorylation of p65 can be mediated via either p38 or extracellular signal-regulated kinase (87). While JNK has not previously been demonstrated to activate MSK, interactions with viral proteins may affect the ability of JNK to phosphorylate a substrate and, therefore, cannot be ruled out as a mechanism at this time.
Acknowledgments
This work was supported by NIH grant AI43314.
We thank Jessica Prince for technical support and helpful discussions of the data and Devon Gregory for technical advice. We thank Neal DeLuca, David Knipe, Priscilla Schaffer, and Saul Silverstein for providing some of the HSV mutants and David Knipe, Gary Cohen, and Roz Eisenberg for some of the antibodies used in these studies.
Footnotes
Published ahead of print on 23 August 2006.
REFERENCES
- 1.Amici, C., G. Belardo, A. Rossi, and M. G. Santoro. 2001. Activation of IκB kinase by herpes simplex virus type 1. A novel target for anti-herpetic therapy. J. Biol. Chem. 276:28759-28766. [DOI] [PubMed] [Google Scholar]
- 2.Amici, C., A. Rossi, A. Costanzo, S. Ciafre, B. Marinari, M. Balsamo, M. Levrero, and M. G. Santoro. 2006. Herpes simplex virus disrupts NF-κB regulation by blocking its recruitment on the IκBα promoter and directing the factor on viral genes. J. Biol. Chem. 281:7110-7117. [DOI] [PubMed] [Google Scholar]
- 3.Aubert, M., and J. A. Blaho. 1999. The herpes simplex virus type 1 regulatory protein ICP27 is required for the prevention of apoptosis in infected human cells. J. Virol. 73:2803-2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aubert, M., J. O'Toole, and J. A. Blaho. 1999. Induction and prevention of apoptosis in human HEp-2 cells by herpes simplex virus type 1. J. Virol. 73:10359-10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aubert, M., S. A. Rice, and J. A. Blaho. 2001. Accumulation of herpes simplex virus type 1 early and leaky-late proteins correlates with apoptosis prevention in infected human HEp-2 cells. J. Virol. 75:1013-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Batterson, W., D. Furlong, and B. Roizman. 1983. Molecular genetics of herpes simplex virus. VIII. Further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J. Virol. 45:397-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bryant, H. E., D. A. Matthews, S. Wadd, J. E. Scott, J. Kean, S. Graham, W. C. Russell, and J. B. Clements. 2000. Interaction between herpes simplex virus type 1 IE63 protein and cellular protein p32. J. Virol. 74:11322-11328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bryant, H. E., S. E. Wadd, A. I. Lamond, S. J. Silverstein, and J. B. Clements. 2001. Herpes simplex virus IE63 (ICP27) protein interacts with spliceosome-associated protein 145 and inhibits splicing prior to the first catalytic step. J. Virol. 75:4376-4385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai, W. Z., and P. A. Schaffer. 1989. Herpes simplex virus type 1 ICP0 plays a critical role in the de novo synthesis of infectious virus following transfection of viral DNA. J. Virol. 63:4579-4589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carrozza, M. J., and N. A. DeLuca. 1996. Interaction of the viral activator protein ICP4 with TFIID through TAF250. Mol. Cell. Biol. 16:3085-3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen, I.-H. B., K. S. Sciabica, and R. M. Sandri-Goldin. 2002. ICP27 interacts with the RNA export factor Aly/REF to direct herpes simplex virus type 1 intronless mRNAs to the TAP export pathway. J. Virol. 76:12877-12889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen, J., and S. Silverstein. 1992. Herpes simplex viruses with mutations in the gene encoding ICP0 are defective in gene expression. J. Virol. 66:2916-2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen, L., W. J. Henzel, and P. A. Baeuerle. 1998. IKAP is a scaffold protein of the IκB kinase complex. Nature 395:292-296. [DOI] [PubMed] [Google Scholar]
- 14.DeLuca, N. A., A. M. McCarthy, and P. A. Schaffer. 1985. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J. Virol. 56:558-570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeLuca, N. A., and P. A. Schaffer. 1988. Activities of herpes simplex virus type 1 (HSV-1) ICP4 genes specifying nonsense peptides. Nucleic Acids Res. 15:4491-4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.DeLuca, N. A., and P. A. Schaffer. 1988. Physical and functional domains of the herpes simplex virus transcriptional regulatory protein ICP4. J. Virol. 62:732-743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diao, L., B. Zhang, J. Fan, X. Gao, S. Sun, K. Yang, D. Xin, N. Jin, Y. Geng, and C. Wang. 2005. Herpes virus proteins ICP0 and BICP0 can activate NF-κB by catalyzing IκBα ubiquitination. Cell Signal. 17:217-229. [DOI] [PubMed] [Google Scholar]
- 18.Erlandsson, A. C., L. G. Bladh, P. Stierna, T. Yucel-Lindberg, O. Hammarsten, T. Modeer, J. Harmenberg, and A. C. Wikstrom. 2002. Herpes simplex virus type 1 infection and glucocorticoid treatment regulate viral yield, glucocorticoid receptor and NF-kappaB levels. J. Endocrinol. 175:165-176. [DOI] [PubMed] [Google Scholar]
- 19.Everett, R. D. 1989. Construction and characterization of herpes simplex virus type 1 mutants with defined lesions in immediate early gene 1. J Gen. Virol. 70:1185-1202. [DOI] [PubMed] [Google Scholar]
- 20.Everett, R. D., C. Boutell, and A. Orr. 2004. Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J. Virol. 78:1763-1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Everett, R. D., and G. G. Maul. 1994. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 13:5062-5069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Everett, R. D., G. Sourvinos, C. Leiper, J. B. Clements, and A. Orr. 2004. Formation of nuclear foci of the herpes simplex virus type 1 regulatory protein ICP4 at early times of infection: localization, dynamics, recruitment of ICP27, and evidence for the de novo induction of ND10-like complexes. J. Virol. 78:1903-1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Everett, R. D., G. Sourvinos, and A. Orr. 2003. Recruitment of herpes simplex virus type 1 transcriptional regulatory protein ICP4 into foci juxtaposed to ND10 in live, infected cells. J. Virol. 77:3680-3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goldstein, D. J., and S. K. Weller. 1988. Factor(s) present in herpes simplex virus type 1-infected cells can compensate for the loss of the large subunit of the viral ribonucleotide reductase: characterization of an ICP6 deletion mutant. Virology 166:41-51. [DOI] [PubMed] [Google Scholar]
- 25.Goldstein, D. J., and S. K. Weller. 1988. Herpes simplex virus type 1-induced ribonucleotide reductase activity is dispensable for virus growth and DNA synthesis: isolation and characterization of an ICP6 lacZ insertion mutant. J. Virol. 62:196-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Goodkin, M. L., A. T. Ting, and J. A. Blaho. 2003. NF-κB is required for apoptosis prevention during herpes simplex virus type 1 infection. J. Virol. 77:7261-7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gregory, D., D. Hargett, D. Holmes, E. Money, and S. L. Bachenheimer. 2004. Efficient replication by herpes simplex virus type 1 involves activation of the IκB kinase-IκB-p65 pathway. J. Virol. 78:13582-13590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu, H., Y. Liang, G. Mandel, and B. Roizman. 2005. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. USA 102:7571-7576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gu, H., and B. Roizman. 2003. The degradation of promyelocytic leukemia and Sp100 proteins by herpes simplex virus 1 is mediated by the ubiquitin-conjugating enzyme UbcH5a. Proc. Natl. Acad. Sci. USA 100:8963-8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hamamoto, T., S. Gunji, H. Tsuji, and T. Beppu. 1983. Leptomycins A and B, new antifungal antibiotics. I. Taxonomy of the producing strain and their fermentation, purification and characterization. J. Antibiot. (Tokyo) 36:639-645. [DOI] [PubMed] [Google Scholar]
- 31.Hardy, W. R., and R. M. Sandri-Goldin. 1994. Herpes simplex virus inhibits host cell splicing, and regulatory protein ICP27 is required for this effect. J. Virol. 68:7790-7799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hargett, D., T. McLean, and S. L. Bachenheimer. 2005. Herpes simplex virus ICP27 activation of stress kinases JNK and p38. J. Virol. 79:8348-8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holmberg, C., S. Katz, M. Lerdrup, T. Herdegen, M. Jaattela, A. Aronheim, and T. Kallunki. 2002. A novel specific role for IκB kinase complex-associated protein in cytosolic stress signaling. J. Biol. Chem. 277:31918-31928. [DOI] [PubMed] [Google Scholar]
- 34.Honess, R. W., and D. H. Watson. 1977. Herpes simplex virus resistance and sensitivity to phosphonoacetic acid. J. Virol. 21:584-600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jean, S., K. M. LeVan, B. Song, M. Levine, and D. M. Knipe. 2001. Herpes simplex virus 1 ICP27 is required for transcription of two viral late (gamma 2) genes in infected cells. Virology 283:273-284. [DOI] [PubMed] [Google Scholar]
- 36.Kato, T., Jr., M. Delhase, A. Hoffmann, and M. Karin. 2003. CK2 is a C-terminal IkappaB kinase responsible for NF-κB activation during the UV response. Mol. Cell 12:829-839. [DOI] [PubMed] [Google Scholar]
- 37.Knipe, D. M., W. Batterson, C. Nosal, B. Roizman, and A. Buchan. 1981. Molecular genetics of herpes simplex virus. VI. Characterization of a temperature-sensitive mutant defective in the expression of all early viral gene products. J. Virol. 38:539-547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koffa, M. D., J. B. Clements, E. Izaurralde, S. Wadd, S. A. Wilson, I. W. Mattaj, and S. Kuersten. 2001. Herpes simplex virus ICP27 protein provides viral mRNAs with access to the cellular mRNA export pathway. EMBO J. 20:5769-5778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koffa, M. D., J. Kean, G. Zachos, S. A. Rice, and J. B. Clements. 2003. CK2 protein kinase is stimulated and redistributed by functional herpes simplex virus ICP27 protein. J. Virol. 77:4315-4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kudo, N., B. Wolff, T. Sekimoto, E. P. Schreiner, Y. Yoneda, M. Yanagida, S. Horinouchi, and M. Yoshida. 1998. Leptomycin B inhibition of signal-mediated nuclear export by direct binding to CRM1. Exp. Cell Res. 242:540-547. [DOI] [PubMed] [Google Scholar]
- 41.Lengyel, J., C. Guy, V. Leong, S. Borge, and S. A. Rice. 2002. Mapping of functional regions in the amino-terminal portion of the herpes simplex virus ICP27 regulatory protein: importance of the leucine-rich nuclear export signal and RGG box RNA-binding domain. J. Virol. 76:11866-11879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lengyel, J., A. K. Strain, K. D. Perkins, and S. A. Rice. 2006. ICP27-dependent resistance of herpes simplex virus type 1 to leptomycin B is associated with enhanced nuclear localization of ICP0 and ICP4. Virology 352:368-379. [DOI] [PubMed] [Google Scholar]
- 43.Lindberg, A., and J. P. Kreivi. 2002. Splicing inhibition at the level of spliceosome assembly in the presence of herpes simplex virus protein ICP27. Virology 294:189-198. [DOI] [PubMed] [Google Scholar]
- 44.Maul, G. G., A. M. Ishov, and R. D. Everett. 1996. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 217:67-75. [DOI] [PubMed] [Google Scholar]
- 45.McCarthy, A. M., L. McMahan, and P. A. Schaffer. 1989. Herpes simplex virus type 1 ICP27 deletion mutants exhibit altered patterns of transcription and are DNA deficient. J. Virol. 63:18-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McLauchlan, J., A. Phelan, C. Loney, R. M. Sandri-Goldin, and J. B. Clements. 1992. Herpes simplex virus IE63 acts at the posttranscriptional level to stimulate viral mRNA 3′ processing. J. Virol. 66:6939-6945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McLean, T. I., and S. L. Bachenheimer. 1999. Activation of cJUN N-terminal kinase by herpes simplex virus type 1 enhances viral replication. J. Virol. 73:8415-8426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mears, W., V. Lam, and S. Rice. 1995. Identification of nuclear and nucleolar localization signals in the herpes simplex virus regulatory protein ICP27. J. Virol. 69:935-947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mears, W., and S. Rice. 1996. The RGG box motif of the herpes simplex virus ICP27 protein mediates an RNA-binding activity and determines in vivo methylation. J. Virol. 70:7445-7453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mears, W. E., and S. A. Rice. 1998. The herpes simplex virus immediate-early protein ICP27 shuttles between nucleus and cytoplasm. Virology 242:128-137. [DOI] [PubMed] [Google Scholar]
- 51.Melchjorsen, J., and S. R. Paludan. 2003. Induction of RANTES/CCL5 by herpes simplex virus is regulated by nuclear factor kappa B and interferon regulatory factor 3. J. Gen. Virol. 84:2491-2495. [DOI] [PubMed] [Google Scholar]
- 52.Melchjorsen, J., F. S. Pedersen, S. C. Mogensen, and S. R. Paludan. 2002. Herpes simplex virus selectively induces expression of the CC chemokine RANTES/CCL5 in macrophages through a mechanism dependent on PKR and ICP0. J. Virol. 76:2780-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murata, T., F. Goshima, T. Koshizuka, H. Takakuwa, and Y. Nishiyama. 2001. A single amino acid substitution in the ICP27 protein of herpes simplex virus type 1 is responsible for its resistance to leptomycin B. J. Virol. 75:1039-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paludan, S. R. 2001. Requirements for the induction of interleukin-6 by herpes simplex virus-infected leukocytes. J. Virol. 75:8008-8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Panagiotidis, C. A., E. K. Lium, and S. J. Silverstein. 1997. Physical and functional interactions between herpes simplex virus immediate-early proteins ICP4 and ICP27. J. Virol. 71:1547-1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Patel, A., J. Hanson, T. I. McLean, J. Olgiate, M. Hilton, W. E. Miller, and S. L. Bachenheimer. 1998. Herpes simplex type 1 induction of persistent NF-κB nuclear translocation increases the efficiency of virus replication. Virology 247:212-222. [DOI] [PubMed] [Google Scholar]
- 57.Phelan, A., J. Dunlop, and J. B. Clements. 1996. Herpes simplex virus type 1 protein IE63 affects the nuclear export of virus intron-containing transcripts. J. Virol. 70:5255-5265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ray, A., and K. E. Prefontaine. 1994. Physical association and functional antagonism between the p65 subunit of transcription factor NF-kappa B and the glucocorticoid receptor. Proc. Natl. Acad. Sci. USA 91:752-756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rice, S., and V. Lam. 1994. Amino acid substitution mutations in the herpes simplex virus ICP27 protein define an essential gene regulation function. J. Virol. 68:823-833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rice, S., V. Lam, and D. Knipe. 1993. The acidic amino-terminal region of herpes simplex virus type 1 alpha protein ICP27 is required for an essential lytic function. J. Virol. 67:1778-1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rice, S. A., and D. M. Knipe. 1990. Genetic evidence for two distinct transactivation functions of the herpes simplex virus alpha protein ICP27. J. Virol. 64:1704-1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roizman, B., and D. Knipe. 2001. Herpes simplex viruses and their replication, p. 2399-2459. In B. N. Fields, D. M. Knipe, P. M. Howley, and D. E. Griffin (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams & Wilkins, Philadelphia, Pa. [Google Scholar]
- 63.Rong, B. L., T. A. Libermann, K. Kogawa, S. Ghosh, L. X. Cao, D. Pavan-Langston, and E. C. Dunkel. 1992. HSV-1-inducible proteins bind to NF-κB-like sites in the HSV-1 genome. Virology 189:750-756. [DOI] [PubMed] [Google Scholar]
- 64.Sacks, W. R., C. C. Greene, D. P. Aschman, and P. A. Schaffer. 1985. Herpes simplex virus type 1 ICP27 is an essential regulatory protein. J. Virol. 55:796-805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sakurai, H., H. Chiba, H. Miyoshi, T. Sugita, and W. Toriumi. 1999. IκB kinases phosphorylate NF-κB p65 subunit on serine 536 in the transactivation domain. J. Biol. Chem. 274:30353-30356. [DOI] [PubMed] [Google Scholar]
- 66.Sakurai, H., S. Suzuki, N. Kawasaki, H. Nakano, T. Okazaki, A. Chino, T. Doi, and I. Saiki. 2003. Tumor necrosis factor-alpha-induced IKK phosphorylation of NF-κB p65 on serine 536 is mediated through the TRAF2, TRAF5, and TAK1 signaling pathway. J. Biol. Chem. 278:36916-36923. [DOI] [PubMed] [Google Scholar]
- 67.Samaniego, L. A., L. Neiderhiser, and N. A. DeLuca. 1998. Persistence and expression of the herpes simplex virus genome in the absence of immediate-early proteins. J. Virol. 72:3307-3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Samaniego, L. A., N. Wu, and N. A. DeLuca. 1997. The herpes simplex virus immediate-early protein ICP0 affects transcription from the viral genome and infected-cell survival in the absence of ICP4 and ICP27. J. Virol. 71:4614-4625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sandri-Goldin, R. M. 1998. ICP27 mediates HSV RNA export by shuttling through a leucine-rich nuclear export signal and binding viral intronless RNAs through an RGG motif. Genes Dev. 12:868-879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sandri-Goldin, R. M., A. L. Goldin, L. E. Holland, J. C. Glorioso, and M. Levine. 1983. Expression of herpes simplex virus beta and gamma genes integrated in mammalian cells and their induction by an alpha gene product. Mol. Cell. Biol. 3:2028-2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sandri-Goldin, R. M., A. L. Goldin, M. Levine, and J. Glorioso. 1983. High-efficiency transfer of DNA into eukaryotic cells by protoplast fusion. Methods Enzymol. 101:402-411. [DOI] [PubMed] [Google Scholar]
- 72.Sandri-Goldin, R. M., M. K. Hibbard, and M. A. Hardwicke. 1995. The C-terminal repressor region of herpes simplex virus type 1 ICP27 is required for the redistribution of small nuclear ribonucleoprotein particles and splicing factor SC35; however, these alterations are not sufficient to inhibit host cell splicing. J. Virol. 69:6063-6076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sandri-Goldin, R. M., M. Levine, and J. C. Glorioso. 1981. Method for induction of mutations in physically defined regions of the herpes simplex virus genome. J. Virol. 38:41-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sanfilippo, C. M., F. N. Chirimuuta, and J. A. Blaho. 2004. Herpes simplex virus type 1 immediate-early gene expression is required for the induction of apoptosis in human epithelial HEp-2 cells. J. Virol. 78:224-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sasaki, C. Y., T. J. Barberi, P. Ghosh, and D. L. Longo. 2005. Phosphorylation of RelA/p65 on serine 536 defines an IκBα-independent NF-κB pathway. J. Biol. Chem. 280:34538-34547. [DOI] [PubMed] [Google Scholar]
- 76.Sciabica, K. S., Q. J. Dai, R. M. Sandri-Goldin, and I. H. Chen. 2003. ICP27 interacts with SRPK1 to mediate HSV splicing inhibition by altering SR protein phosphorylation. EMBO J. 22:1608-1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shepard, A. A., and N. A. DeLuca. 1991. Activities of heterodimers composed of DNA-binding- and transactivation-deficient subunits of the herpes simplex virus regulatory protein ICP4. J. Virol. 65:299-307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sizemore, N., N. Lerner, N. Dombrowski, H. Sakuria, and G. R. Stark. 2002. Distinct roles of the IκB kinase α and β subunits in liberating NFκB from IκB and in phosphorylating the p65 subunit of NFκB. J. Biol. Chem. 277:3863-3869. [DOI] [PubMed] [Google Scholar]
- 79.Smith, C. A., P. Bates, R. Rivera-Gonzalez, B. Gu, and N. A. DeLuca. 1993. ICP4, the major transcriptional regulatory protein of herpes simplex virus type 1, forms a tripartite complex with TATA-binding protein and TFIIB. J. Virol. 67:4676-4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Soliman, T. M., R. M. Sandri-Goldin, and S. J. Silverstein. 1997. Shuttling of the herpes simplex virus type 1 regulatory protein ICP27 between the nucleus and cytoplasm mediates the expression of late proteins. J. Virol. 71:9188-9197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Soliman, T. M., and S. J. Silverstein. 2000. Identification of an export control sequence and a requirement for the KH domains in ICP27 from herpes simplex virus type 1. J. Virol. 74:7600-7609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Spiegelman, V. S., P. Stavropoulos, E. Latres, M. Pagano, Z. Ronai, T. J. Slaga, and S. Y. Fuchs. 2001. Induction of beta-transducin repeat-containing protein by JNK signaling and its role in the activation of NF-κB. J. Biol. Chem. 276:27152-27158. [DOI] [PubMed] [Google Scholar]
- 83.Stow, N. D., and E. C. Stow. 1986. Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J. Gen. Virol. 67:2571-2585. [DOI] [PubMed] [Google Scholar]
- 84.Taddeo, B., T. R. Luo, W. Zhang, and B. Roizman. 2003. Activation of NF-κB in cells productively infected with HSV-1 depends on activated protein kinase R and plays no apparent role in blocking apoptosis. Proc. Natl. Acad. Sci. USA 100:12408-12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tang, Q., L. Li, A. M. Ishov, V. Revol, A. L. Epstein, and G. G. Maul. 2003. Determination of minimum herpes simplex virus type 1 components necessary to localize transcriptionally active DNA to ND10. J. Virol. 77:5821-5828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Uprichard, S. L., and D. M. Knipe. 1996. Herpes simplex ICP27 mutant viruses exhibit reduced expression of specific DNA replication genes. J. Virol. 70:1969-1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vermeulen, L., G. De Wilde, P. V. Damme, W. Vanden Berghe, and G. Haegeman. 2003. Transcriptional activation of the NF-κB p65 subunit by mitogen- and stress-activated protein kinase-1 (MSK1). EMBO J. 22:1313-1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang, F., E. Tang, K. Guan, and C. Y. Wang. 2003. IKK beta plays an essential role in the phosphorylation of RelA/p65 on serine 536 induced by lipopolysaccharide. J. Immunol. 170:5630-5635. [DOI] [PubMed] [Google Scholar]
- 89.Zhong, H., M. J. May, E. Jimi, and S. Ghosh. 2002. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell 9:625-636. [DOI] [PubMed] [Google Scholar]
- 90.Zhong, H., H. SuYang, H. Erdjument-Bromage, P. Tempst, and S. Ghosh. 1997. The transcriptional activity of NF-κB is regulated by the IκB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell 89:413-424. [DOI] [PubMed] [Google Scholar]
- 91.Zhong, H., R. E. Voll, and S. Ghosh. 1998. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell 1:661-671. [DOI] [PubMed] [Google Scholar]