

Abstract
A subclone of Huh-7 cells that could be relatively efficiently transfected and infected with hepatitis E virus was identified. Following transfection, infectious virus was produced but remained predominantly cell associated. Intracellular virus, recovered by lysis of transfected cells, infected naïve cells. This in vitro-produced virus appeared to be antigenically identical to virus isolated from clinical samples. Lysates from cells transfected with mutant viral genomes unable to synthesize ORF3 protein contained infectious virions that were similar in number, thermostability, and sedimentation characteristics to those in lysates transfected with wild-type viral genomes. Therefore, in contrast to its requirement in vivo, ORF3 protein is not required for infection of Huh-7 cells or production of infectious virus in vitro.
Hepatitis E was first recognized as a unique disease in 1980 (reviewed in reference 15). The first complete hepatitis E virus (HEV) sequence, obtained in 1991, demonstrated that the virion contained a 7.2-kb positive-strand RNA genome composed of three open reading frames (ORFs), each in a different frame. ORF1 encodes enzymes needed for genomic replication, including an RNA-dependent RNA polymerase and the methyl- and guanylyltransferases (13). Recently, it was demonstrated that ORF2 and ORF3 proteins are encoded by a bicistronic subgenomic RNA that is also capped (9). ORF2 encodes the capsid protein, and ORF3 encodes a protein of unknown function that contains only 114 amino acids (9) rather than the 123 reported previously. The virus does not contain a lipid envelope and is the sole member of the newly created genus Hepevirus (2).
An efficient cell culture system for HEV is not available, and nonhuman primates are the smallest animal models; as a result, much of the molecular biology of this virus remains to be described. Hepatitis E infection is generally acquired by ingestion of water contaminated with sewage (19). The primary site of infection is not known, but virus replicates in the liver and causes acute hepatitis which does not progress to chronicity. Viremia can occur, and infectious virus is shed in feces.
In the absence of a productive cell culture system, attempts to define the molecular biology of HEV have depended on two different approaches. In the first approach, individual viral proteins were expressed at high levels from plasmid vectors. Data from this approach suggested that a subpopulation of the ORF2 protein is glycosylated (19, 22) and that ORF2 protein and ORF3 protein interact with each other (20); ORF3 protein was phosphorylated under these conditions (23). On the basis of other expression studies, ORF3 protein was also proposed to be a regulatory protein that interacts with cellular proteins in the mitogen-activated protein kinase signaling pathway (11). Finally, ORF3 protein was proposed to expedite the export of α1-microglobulin from the liver (21). Because these systems utilized nonphysiological concentrations and combinations of viral proteins, their biological relevance is uncertain at this time.
In the second approach, infectious cDNA clones encoding wild-type viruses have been constructed and transcripts of these clones or derivative mutants have been transfected into cultured cells or have been transfected intrahepatically into nonhuman primates or swine (6, 10). Transfection of HEV genomes into cultured cells permits analysis of genome replication and cis-acting RNA elements under more natural conditions, but since virus does not spread from cell to cell in these cultures, the formation of infectious virions has to be assessed by inoculation of cell lysates into a susceptible host such as a rhesus macaque (5). Alternatively, recombinant genomes have been directly transfected intrahepatically into nonhuman primates or swine to monitor viability; this expensive and lengthy procedure provides a yes or no answer as to viability but provides no insight as to which step is affected in the case of nonviability. Mutant genomes which could not produce ORF2 protein or ORF3 protein (5, 13) replicated in cultured cells, but a mutant lacking ORF3 protein was unable to infect macaques (8). Since one difference between the in vitro and in vivo transfection experiments was that infectious virions needed to be produced in the macaques but not in the cultures to provide an end point, it seemed possible that one function of ORF3 protein might be to enable virion morphogenesis.
In this article, we describe an in vitro system to monitor the production of infectious virions in transfected cells. We have isolated a subclone of human hepatoma cells which can be efficiently transfected with HEV genomic RNA to produce virions that can be harvested and used to infect naïve cultures of the same subclone. We have utilized this system to determine whether ORF3 protein is necessary for the production of infectious virus.
MATERIALS AND METHODS
Cells.
293T and CaCo-2 cells (HTB37) were purchased from the American Type Culture Collection (Manassas, VA). Huh-7 cells were originally isolated in Japan (14). All cells were grown as monolayers in Dulbecco's modified Eagle's medium supplemented with 10% (Huh-7 and subclones and 293T) or 20% (CaCo-2) fetal bovine serum (ultralow immunoglobulin G; Invitrogen) that had been heat inactivated at 56°C for 30 min. Generally, 0.1 mg gentamicin ml−1, 100 U penicillin ml−1, and 0.1 mg streptomycin ml−1 were included. Stocks were maintained at 37°C; infected or transfected cultures were incubated at 34.5°C. Subclones of Huh-7 cells were obtained by plating highly diluted suspensions of cells in 96-well plates and incubating them at 37°C until the few wells that received cells contained confluent monolayers. Monolayers of cells were trypsinized and individually expanded to provide working stocks. Subclones were screened for their ability to produce green fluorescent protein (GFP) (indirect fluorescence microscopy) or ORF2 protein (immunofluorescence microscopy) following transfection or infection, respectively. A fecal suspension of a genotype 1 clinical isolate was used as the inoculum in infectivity assays of the subclones. The assay was otherwise as described below for the cell lysates. Selected clones were subjected to a second round of cloning and screening.
Plasmids.
Plasmids were all described previously. pSK-E2 (GenBank accession no. AF444002) encodes infectious HEV, Sar55 strain (6). The first 672 nucleotides of ORF2 of pSK-E2 were replaced with the gene for green fluorescent protein to provide the GFP replicon (5). The ORF2-null mutant of pSK-E2 had the first three Met codons of ORF2 mutated to Leu codons and has two in-frame stop codons just downstream of the fourth Met codon (8); the amino acid sequence of ORF3 is not changed. The ORF3-null mutant of pSK-E2 was produced by mutating the second codon in ORF2 to preserve the amino acid in ORF2 but results in a termination codon in ORF3 (8); it encodes authentic ORF2 protein and an ORF3 peptide of five amino acids initiated at the third Met codon in ORF3 (9). In some cases, modified pSK-E2 and ORF3-null plasmids encoding 32 rather than 16 A residues in the poly(A) tail were used; the extra A residues permitted higher replication levels but had no other detectable effect (Judith Graff, unpublished data).
Microscopy.
Cells in eight-well chamber glass slides were fixed with acetone and either air dried or washed in phosphate-buffered saline (PBS). A mixture of rabbit polyclonal antibody specific for ORF3 protein and chimpanzee polyclonal antibody specific for ORF2 protein in a 1:1 mixture of 10% bovine serum albumin and PBS was added, and samples were incubated at room temperature for 20 min. The antibodies were described previously (5). After a wash in PBS, slides were incubated for 20 min at room temperature with a mixture of Alexa Fluor 488-conjugated goat anti-human immunoglobulin G (Molecular Probes) and Alexa Fluor 568-conjugated goat anti-rabbit antibody as described previously (5). Samples were washed in PBS, Vectashield (Vector Laboratories) was added, and the slides were viewed by indirect immunofluorescence microscopy with a fluorescein isothiocyanate filter set for Alexa Fluor 488 (green), a rhodamine filter set for Alexa Fluor 568 (red), and the 25× objective of a Zeiss fluorescence photomicroscope.
For detection of GFP, damp cells were examined for intrinsic fluorescence using the fluorescein isothiocyanate filter set and the 25× objective of the Zeiss fluorescence photomicroscope.
In vitro transcription and transfection.
Plasmids were linearized at a unique BglII site located immediately downstream of the poly(A) tract of the HEV sequence. Capped transcripts were synthesized with the T7 riboprobe in vitro transcription system (Promega) in the presence of cap analog as previously described (5) except that anti-reverse cap analog was substituted for the conventional cap analog. Each 50-μl reaction mixture contained 10 μl of 5× transcription buffer (200 mM Tris-Hcl [pH 7.9], 30 mM MgCl2, 10 mM spermidine, 50 mM NaCl), 5 μl of 100 mM dithiothreitol, 2 μl of 40 U of RNasin/ml, 5 μl of nucleoside triphosphates (5 mM [each] ATP, CTP, and UTP and 0.5 mM GTP), 5 μl of 5 mM 3′-O-Me-m7G(5′) pppG (Ambion), and 2 μl of 20 U of T7 polymerase/μl. The mixtures were incubated at 37°C for 1.5 h. The integrity and yield of transcripts were determined by electrophoresis on a nondenaturing agarose gel. Transcription mixtures were cooled on ice and then mixed with a liposome mixture (25 μl of DMRIE-C [Invitrogen]/ml of OptiMem [Gibco]) for transfection. A total of 25 μl of RNA mixture was diluted with 1 ml of the liposome mixture and added to a T25 flask containing washed cells at 40 to 50% confluence. For six-well plates, 400 μl Optimum, 8 μl DMRIE-C, and 20 μl RNA mixture were mixed. Flasks were incubated at 34.5°C for 5 to 6 h, an additional 1 ml of OptiMem was added, and incubation was continued overnight. Medium was aspirated and replaced with growth medium. Cells were trypsinized and split into two T25 flasks and one or two wells of an eight-well chamber slide 1 or 2 days prior to immunostaining.
Preparation of cell lysates.
Medium was removed from transfected cells 6 to 9 days posttransfection and either discarded or frozen at −80°C for titration. Confluent monolayers of cells in a T25 flask or six-well plate were trypsinized and centrifuged in a 5415C Eppendorf centrifuge in a 1.5- or 2.0-ml Sarstadt tube for 1.5 min at 800 rpm followed by 1.0 min at 13,200 rpm. Supernatant was aspirated, and the cell pellet was stored at −80°C. Frozen pellets were extracted at room temperature by adding 0.9 ml water per T25 pellet and vortexing vigorously until the pellet dispersed and the solution became cloudy. The sample was vortexed once or twice more in the next 10 min, 0.1 ml of 10× concentrated PBS was added, and debris was removed by centrifugation at 13,200 rpm for 2 min. The supernatant was removed and placed on ice, and the pellet was discarded. Solution volumes were halved to extract the cell pellet from a six-well plate.
Infectivity assay.
Confluent monolayers of 10-3 cells were trypsinized and diluted 1 to 4 in growth medium, and 0.1 ml was carefully added to 0.4 ml of growth medium already in a well of an eight-well glass chamber slide (3). Slides were incubated at 37°C for 1 to 2 days until a sparse population of flattened cells was observed. Duplicate 100-μl samples of cell lysate were coded and used to replace the medium in the wells of the chamber slide. Slides were incubated at 34.5°C in a 5% CO2 atmosphere for 1 h; liquid was aspirated and replaced with 0.4 ml of growth medium containing antibiotics. After 5 to 6 days at 34.5°C, cells were fixed and stained for immunofluorescence microscopy, and the number of cells positive for ORF2 protein was tabulated. The code was not broken until all samples in the experiment had been scored.
Thermostability studies were performed by incubating 100 μl of cell lysate at the indicated temperature and time in a circulating water bath, cooling the sample on ice, then coding the samples, and performing the standard infectivity assay. Control samples were kept on ice as 100-μl aliquots during the heating period.
For FAb neutralization assays, 50 μl of cell lysate containing wild-type virus was mixed with 10 μl of PBS or purified FAb in PBS, placed at 4°C overnight, and then diluted with 50 μl of growth medium. The entire 110 μl was inoculated into a well of the chamber slide, and the standard infectivity assay was performed. The FAb preparations were the identical ones described previously (4); all samples were coded and tested in duplicate.
Neutralization assays with primate sera were performed by mixing 100-μl cell lysate with 100-μl (rhesus monkey 387) or 1-μl (chimpanzee 5835) serum sample, and the standard infectivity assay was performed on the entire sample. The reciprocal enzyme-linked immunosorbent assay titers measured against Sar55 recombinant capsid protein were <100 for preinoculation sera and 10,000 and 100,000 for the rhesus monkey and chimpanzee immune sera, respectively (7). The rhesus monkey had been infected with the Sar55 strain of HEV, and the chimpanzee had been vaccinated with the same Sar55 antigen as was used in the enzyme-linked immunosorbent assay.
Velocity gradients.
Cell lysates were prepared as for infectivity assays, except the liquid volumes were halved to provide a 2× concentrated cell lysate. Clarified 2× cell lysate (75 μl) was mixed with 50 μl bovine RNase A (5 Kunitz units/μl; Sigma) and 5 μl of 25% (vol/vol) NP-40 substitute (Fluka) and incubated at 37°C for 60 min. Five microliters of cell culture-grown HAV was added as an internal marker, and the entire mixture was layered over a 5-ml sucrose gradient of 5 to 30% sucrose (wt/vol) in PBS at room temperature. Gradients were centrifuged for 65 min at 30,000 rpm at 20°C in an SW50.1 Beckman rotor. Ten-drop fractions were collected from the bottom of the tube. Two hundred microliters of each fraction was extracted with TRIzol LS (Invitrogen) according to the manufacturer's directions, and RNA was precipitated with isopropanol in the presence of Pellet Paint NF Co-Precipitant (Novagen). One half of the RNA was quantified by Taqman for HAV and HEV, respectively, as described previously (6).
Western blot analysis.
Proteins in cell lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis in a 10 to 20% Novex tricine gel (Invitrogen) and electrophoretically transferred onto a polyvinylidene difluoride membrane (0.2 μm; Invitrogen) for analysis by Western blotting. The membrane was pretreated for 3 min in methanol and 5 min in Novex Tris-glycine transfer buffer (Invitrogen) and blocked with 3% nonfat milk and 0.25% Tween 20 in Tris-buffered saline. The membrane was incubated with rabbit anti-ORF3 antibody for 1 h at room temperature and then with an anti-rabbit peroxidase-conjugated secondary antibody (1:50,000) (Jackson ImmunoResearch); bands were detected with the Visualizer Western blot detection kit (Upstate, Lake Placid, NY) as specified by the manufacturer.
RESULTS
Identification of a highly infectible subclone of Huh-7 cells.
Previously, it was shown that transfected HEV genomic RNA replicated in Huh-7 cells and that a lysate obtained by freeze/thawing the cells could infect rhesus macaques (5). However, transfection levels were limited to approximately 10% of the cells, and the virus did not spread within the cell culture even after 160 days. Also, only a small number of Huh-7 cells were infected by exogenously added clinical isolates of virus, suggesting that the number of functional viral receptors might be low or unevenly expressed throughout the population of cells. This possibility prompted us to clone the Huh-7 cells and compare subclones for their ability to be infected by HEV and to support replication of transfected genomes. Significant differences in both these parameters, but especially in infectibility by clinical strains, were found among the subclones (Table 1). Subclone 10-3, which exhibited both superior transfectability (as monitored by production of GFP) and infectibility (as monitored by production of ORF2 protein) was expanded and used for subsequent experiments. Approximately 50% of the 10-3 cells routinely were transfected with wild-type, ORF3-null, or ORF2-null viral genomes, respectively (Fig. 1A). The staining pattern of wild-type proteins was identical following transfection or infection of 10-3 cells, but only one cell or a very small cluster of cells was stained for each infectious virus (Fig. 1G and H).
TABLE 1.
Comparison of Huh-7 subclones for transfectability and infectibility by HEV
| Subclone | Transfection efficiencya (GFP intensity) | No. of cells infectedb |
|---|---|---|
| 10-3 | 4+ (3+) | 273 |
| 10-7 | 1+ (2+) | 5 |
| 10-8 | 1+ (2+) | 17 |
| 10-9 | 2+ (2+) | 48 |
| 10-10 | 4+ (4+) | 42 |
| 10-11 | 1+ (2+) | 49 |
| 13-1 | 3+ (3+) | 9 |
Cells were transfected with an HEV replicon encoding GFP, and the proportion of cells producing GFP and the GFP fluorescence intensity were estimated on a scale of 0 to 4.
Five days after cultures were incubated with a human stool suspension containing HEV, the cells were stained with anti-ORF2 protein for immunofluorescence microscopy, and the number of cells containing ORF2 capsid protein was tallied.
FIG. 1.
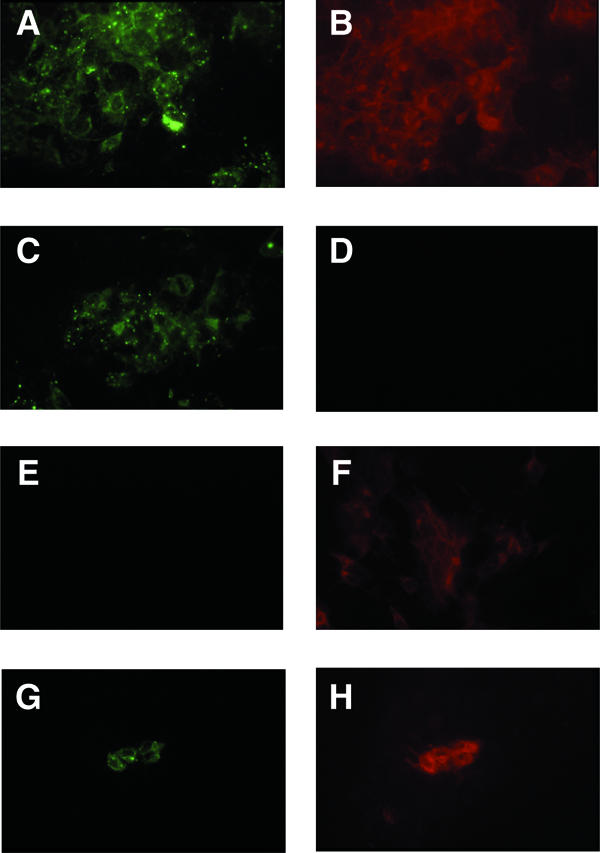
Immunofluorescence microscopy of transfected or infected 10-3 cells. Cells were stained for ORF2 protein (green) or ORF3 protein (red) after transfection (A to F) or infection (G and H). Wild-type (A, B, G, and H), ORF3-null (C and D), and ORF2-null (E and F) viruses were used. Each pair of panels represents the same field as photographed through the 25× objective, and each field contains a confluent monolayer of cells.
Recovery of infectious HEV from transfected 10-3 cells.
Unfortunately, even though the 10-3 cells could be infected with exogenously added virus, there was still no evidence of cell-to-cell spread of virus after either transfection or infection. Possible explanations for the lack of virus spreading were that the numbers of infectious virions produced were not sufficient to permit detection of infected cells or, alternatively, those produced were unable to exit the cells. In an attempt to distinguish between these hypotheses, we separated the medium and cells from a culture transfected with wild-type viral genomes and concentrated the cells by trypsinization and centrifugation. The cell pellet was resuspended and lysed by adding water and vortexing the sample vigorously. The salt concentration was then readjusted to the physiological concentration by adding 10× PBS, and debris was removed by centrifugation. Aliquots (100 μl) of the medium and cell lysate were inoculated onto naïve cultures of 10-3 cells. After 5 days, the cells were prepared for immunofluorescence microscopy, and the number of infected cells was determined by manually counting cells that were stained for ORF2 protein. The culture inoculated with the medium contained only four ORF2-positive cells compared to 145 ORF2-positive cells in the culture inoculated with the cell lysate. Adjustment of the numbers to account for the total volume of each preparation indicated that there was 12 times more infectious virus in the cell lysate than in the medium (1,450 versus 122, respectively). It should be noted that the number of cells infected varied from experiment to experiment, but the pattern was reproducible. In numerous experiments, we never detected more than a few, if any, infectious viruses in the medium, while we always detected a significant number in the cell lysate. Therefore, infectious virus apparently was produced, but the vast majority was retained within the cells.
ORF2 protein is required for passage to naïve cells.
In order to confirm that the cells were being infected with virus rather than being transfected with viral RNA in the lysate, cell cultures were transfected in parallel with wild-type viral genomes, a null mutant of ORF2 unable to express ORF2 capsid protein because of point mutations (8), and an HEV replicon that was unable to synthesize ORF2 protein because most of the ORF2 gene was deleted and replaced with the green fluorescent protein gene (5). Immunofluorescence microscopy (for wild-type or ORF2-null mutant) or fluorescence microscopy (for GFP replicon) confirmed that equivalent numbers of cells were transfected by each of the three genome preparations and that the ORF2-null mutant made ORF3 protein but not ORF2 protein (data not shown). Cell lysates were prepared from each of the three cultures in parallel, and duplicate samples containing the wild-type viral genome or the ORF2-null mutant were coded and inoculated onto 10-3 cells in a chamber slide. Duplicate samples of the GFP replicon-transfected cell lysate and the corresponding culture medium were coded and plated on a separate chamber slide. Six days later, slides were prepared for immunofluorescence microscopy or direct fluorescence microscopy, and the number of fluorescent cells in each well was determined prior to breaking the code. As expected, cultures inoculated with the lysate from cells transfected with wild-type viral genomes contained fluorescent cells, whereas those inoculated with a lysate from cells transfected with either the ORF2-null mutant or the GFP replicon did not (Table 2). Therefore, the ORF2 capsid protein was absolutely required to transfer the HEV genome from the cell lysate into new cells, and infectivity signified the production of virions.
TABLE 2.
Infection assay for wild-type HEV and mutants
| Inoculuma | No. of positive cellsb |
|---|---|
| Wild-type virus | 34, 32 |
| ORF2-null mutant virus | 0, 0 |
| HEV replicon encoding GFP | 0, 0 |
Inocula were prepared by transfecting 10-3 cells in parallel with the indicated genomes and harvesting the cells on day 9.
Cultures were inoculated with identical amounts of cell lysate, and the number of cells per well exhibiting green fluorescence due to immunofluorescence staining of ORF2 (wild-type virus) or ORF3 protein (ORF2-null mutant) with Alexa Fluor 488 or due to production of GFP (replicon) were tallied. The values for duplicate samples are given.
Previously we showed that transfected HEV genomes replicated in some nonhepatic primate cell lines derived from intestine, kidney, or lung origin and that the intestinal (CaCo-2) and lung (A549) cells could also be infected with HEV, albeit with a very low efficiency (5). In order to determine whether infectious virions could be produced in nonhepatic cells, we transfected 10-3 cells and CaCo-2 cells (human intestinal cells) in one case or 293T cells (human embryonal kidney) in the other case with wild-type viral genomes, stained a sample for ORF2 protein, and prepared cell lysates in the same way as had been done for the hepatoma cells. Duplicate 100-μl aliquots of each lysate were inoculated onto naïve 10-3 cells and prepared for immunofluorescence microscopy 6 days later. Infectious virions were recovered from all lysates. Immunofluorescence staining of transfected cells indicated that five times more 10-3 cells were transfected than were CaCo-2 cells, which correlated well with the number of infectious virions recovered (65 and 68 compared to 19 and 11) for the 10-3 and CaCo-2 cells, respectively. Similarly, in the second experiment, twice as many 10-3 cells as 293T cells were transfected, and the number of infectious virions detected in 10-3 lysates was about double that in the 293T lysates (83 and 103 compared to 40 and 39). Therefore, it appeared that infectious virions could be produced in these nonhepatic cells as efficiently as in hepatic cells.
Neutralization of infectivity by antibody to HEV.
If infectious virions present in the cell lysate of transfected cells were identical to those produced in vivo, they should be neutralized by antibodies that were known to neutralize authentic wild-type HEV isolated from clinical samples. Duplicate samples of transfected cell lysate were incubated with PBS, rhesus monkey pre-HEV infection serum, or dilutions of the same rhesus monkey hepatitis E convalescent-phase serum. Samples were coded and then assayed for infectivity as described above (Fig. 2). The numbers of infected cells were similar in the cultures inoculated with the lysate mixed with either PBS or preinfection serum but were dramatically decreased in the cultures inoculated with lysate mixed with convalescent-phase serum diluted 10- or 100-fold, suggesting that authentic virions capable of being neutralized by anti-HEV were present in the cell lysate and accounted for most, if not all, infectivity.
FIG. 2.

Rhesus monkey anti-HEV convalescent-phase serum neutralizes wild-type HEV produced in vitro. A lysate of cells transfected with HEV genomes was incubated overnight with PBS or rhesus monkey serum collected prior to (Pre) or after (Post) infection with HEV, and the number of infectious virions was quantified on 10-3 cells. The preimmune serum was not diluted; dilutions of postinfection serum are indicated. Pairs of columns represent duplicate samples read before the code was broken. Undil, undiluted.
Previously, we utilized a panel of 13 monoclonal antibodies (MAbs) specific for the ORF2 capsid protein to study neutralization of infectivity of human isolates of HEV for HepG2/3CA cells (4). Of the 12 MAbs still available to us, three (HEV4, EBL56, and EBL9) had neutralized HEV in a human stool suspension, whereas the other nine MAbs did not neutralize the virus for HepG2/3CA cells. However, one of the nine MAbs (HEV31), which had not neutralized in the cell culture assay, had neutralized HEV when tested in rhesus monkeys (16). We tested these same preparations of MAbs for their ability to neutralize the HEV in the transfected 10-3 cell lysates when inoculated onto naïve 10-3 cells. As expected, HEV4, EBL56, and EBL9 all neutralized the virus and prevented infection of 10-3 cells, while eight of the nine remaining MAbs did not neutralize (Fig. 3). Thus, with one exception, the results with the virus produced in vitro were in total agreement with the results obtained previously with the virus in a clinical sample. The exception was HEV31 which, at the highest concentration tested, demonstrated minimal neutralization for the HepG2/3CA cells but effectively neutralized the virus for 10-3 cells; however, even in the 10-3 assay, HEV31 was not as potent as HEV4, and it did not neutralize when diluted only fivefold, whereas HEV4 provided almost total neutralization at either concentration tested (data not shown). The excellent agreement between these results and those obtained previously provides strong evidence that infectivity of the cell lysate represents infectious virions that are antigenically identical to those produced in vivo. In addition, they suggested that the 10-3 cells might be a better model for infection of macaque liver than HepG2/3CA cells were.
FIG. 3.

MAbs that neutralize clinical isolates of HEV neutralize HEV produced in vitro. The number of infectious virions remaining after overnight incubation with PBS or with a panel of MAbs specific for ORF2 protein was quantified on 10-3 cells. Pairs of columns represent duplicate samples. MAbs mapping to antigenic site 2 were the only MAbs to neutralize clinical isolates (17). The epitope for EBL1 bridged antigenic sites 1 and 2.
Infectious virus production in the absence of ORF3 protein.
On the basis of the demonstration that the ORF3 protein is dispensable for genome replication in vitro but is absolutely required for infection of macaques, we had presumed ORF3 protein might be needed for virion production. However, to our surprise, the cell lysate from cells transfected with the ORF3-null mutant infected a comparable number of cells as did the wild-type virus produced in a parallel transfection (Fig. 4). In some experiments, more wild-type positive cells were detected than ORF3-null positive cells, but in other experiments, the opposite was true; however, each of the many times this experiment has been repeated, the numbers of infectious virions encoded by the wild-type virus and ORF3-null mutant have been of similar magnitude (see Fig. 6 for example). Immunofluorescence microscopy confirmed that cells transfected with the ORF3-null mutant made ORF2 protein but not ORF3 protein (data not shown). The lack of ORF3 protein in lysate containing the null mutant was confirmed by Western blotting (Fig. 5). Therefore, ORF3 protein was not needed for the production of virions infectious for Huh-7 cells.
FIG. 4.

An ORF3-null mutant produces a virus that is infectious in vitro. Lysates from cells transfected with wild-type viral genomes or genomes unable to encode ORF2 protein (ΔORF2) or ORF3 protein (ΔORF3) were inoculated onto 10-3 cells, and the number of cells expressing ORF2 protein was quantified. Pairs of columns represent duplicate samples.
FIG. 6.

Sucrose velocity sedimentation gradients of lysates from cells transfected with the wild-type, ORF3-null, or ORF2-null viral genomes or of a rhesus stool suspension containing wild-type HEV. Note the differences in the abscissa scales. All samples except the sample in panel A were treated with RNase, but the sample in panel A received less RNase than the samples in panels C to F. Samples in panels A and B were spiked with HAV. Samples in panels C, D, and E were prepared in parallel starting at the transfection step, and the sample in panel F was included at the RNase step. The numbers of viral RNA genomes loaded onto gradients in panels C, D, and E were 3.1 × 106, 3.8 × 106, and 2.3 × 106, respectively.
FIG. 5.

Western blot confirming that the ORF3-null viral mutant does not produce ORF3 protein. Equal aliquots of lysate from cells transfected with the ORF3-null mutant or wild-type viral genomes were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis and probed with anti-ORF3.
Comparison of virions made in the presence or absence of ORF3 protein.
Sucrose velocity sedimentation gradients and reverse transcription-PCR were performed in order to separate and detect encapsidated and free viral RNA. Initial attempts to detect mature virus particles were complicated by the overwhelming excess amount of free viral RNA (Fig. 6A). Therefore, in preliminary experiments, lysates were treated with various amounts of RNase A prior to loading onto the gradients; hepatitis A virions were added as an internal marker, since HAV has a reported sedimentation coefficient of 156 to 160S, which is close to the 183S value reported for HEV virions (1). As Fig. 6 demonstrates, even 5 Kunitz units of RNase reduced the amount of viral RNA at the top of the gradient, while a peak of RNA presumed to represent virions was not reduced, as expected for encapsidated RNA. Therefore, the RNase concentration was increased to 250 Kunitz units in subsequent experiments. In confirmatory experiments, authentic HEV in a clinical stool specimen also sedimented just ahead of the HAV marker (data not shown).
In a parallel comparison of a clinical isolate of HEV and lysates from cells transfected with wild-type, ORF3-null, and ORF2-null virus mutants, the virus particles produced by the wild-type virus and ORF3-null mutant displayed a rate of sedimentation that was similar to that of the clinical isolate. On the other hand, virtually all of the RNA in the ORF2-null lysate was eliminated by RNase treatment, and an RNase-resistant peak corresponding to those in the other two lysates was not observed. Reverse transcription-PCR of samples prior to RNase treatment indicated that the amount of viral RNA in any one of the three transfected cell lysates varied less than twofold from the amounts in the other two.
We next performed a neutralization test to determine whether the ORF3-null mutant could be neutralized by a hepatitis E immune serum that neutralized wild-type virus. Transfected cell lysates mixed with prevaccination serum were compared to those mixed with immune serum (Fig. 7). In each case, incubation with the immune serum neutralized the virus and decreased the number of cells infected by wild-type virus or the ORF3-null mutant by greater than 95% compared to the prevaccination control serum, suggesting that the ORF3-null mutant was antigenically similar to wild-type virus.
FIG. 7.

A chimpanzee anti-HEV immune serum that neutralizes wild-type virus also neutralizes virus encoded by the ORF3-null mutant genome. Lysates from cells transfected with wild-type or ORF3-null mutant genomes were incubated overnight with PBS or with undiluted preinoculation (Pre) or HEV postvaccination (Post) chimpanzee serum, and infectious virus was quantified in 10-3 cells. Pairs of columns represent duplicate samples.
Thermostability of the two viruses produced in vitro was determined also. Previously, we had demonstrated that infectivity of clinical isolates of HEV in stool suspensions was almost totally abolished by heating the virus to 56°C (3). Both the wild-type virus and ORF3-null mutant virus produced in vitro were similarly inactivated by greater than 99% by heating the cell lysate at 56°C for either 15 or 30 min (data not shown). Furthermore, heating at 50°C, a temperature that had in 30 min inactivated only a small portion of HEV in the clinical isolate, inactivated only 65% of the cell culture-produced viruses in 135 min, and the kinetics of inactivation of the wild-type and ORF3-null mutant viruses were indistinguishable in this experiment (Fig. 8). In two other experiments, wild-type virus was inactivated by incubation at 50°C for approximately 2 h on average 58% compared to 54% for the ORF3-null mutant.
FIG. 8.

Thermostability of virions produced in 10-3 cells. Duplicate 100-μl aliquots of lysates from cells transfected with wild-type (squares) or ORF3-null (diamonds) viral genomes were incubated at 50°C for the indicated times, and residual infectivity was quantified in 10-3 cells. Each point is the average of duplicate samples.
In summary, comparisons by different tests failed to reveal any differences between virions produced in the presence or absence of ORF3 protein. Furthermore, the high-titered anti-ORF3 antibody used in the immunofluorescence assays did not neutralize wild-type virions (data not shown) in agreement with a previous report that anti-ORF3 failed to neutralize HEV in a cynomolgus primary hepatocyte culture system (18). These data strongly suggest that the protein encoded by ORF3 is either not present in or is not an essential component of virions.
DISCUSSION
Identification of a subclone of human hepatoma cells that could be relatively efficiently transfected or infected with HEV will permit a more thorough investigation of the molecular biology of HEV. We do not know what portion of the large excess nonencapsidated viral RNA in the cytoplasm (Fig. 6A) represented residual transcripts introduced during transfection compared to de novo-synthesized RNA. However, although virion production was not robust enough to permit structural or biochemical analysis of virions, clearly the system can provide answers to many relevant questions. The level of HEV replication and morphogenesis following transfection of one T25 flask of 10-3 cells was sufficient to provide enough virus for 10 to 20 assays. This system enabled us to perform cell culture-based infectivity assays, neutralization studies, and mutant analyses that previously had to be performed in monkeys.
The characteristics of the infectious virions produced in cell culture appeared to be identical to those of virions produced in vivo and recovered from experimentally infected primates. Virions produced in vitro were neutralized by the same antibodies to the capsid protein, had similar thermostability, and similar sedimentation properties to virions produced in vivo.
It is interesting to note that HAV, like HEV, may also remain strongly cell associated in vitro; recovery of infectious HAV virions is often accomplished by lysing the cells with cycles of freezing and thawing. In the case of HAV, much of the virus released from the cells is associated with lipids, which prevent its neutralization by antibodies (12). In contrast, the lysis of cells containing HEV was accomplished by a combination of hypotonicity and mechanical disruption (vortexing). It should be noted that HEV extracted from cells by this method was not protected from neutralizing antibodies and the vast majority of wild-type virus or ORF3-null mutant virus were neutralized without additional treatments to remove lipids (Fig. 7). It is also noteworthy that ORF3 protein was recovered in the cell lysate generated by the water extraction procedure. Since overexpressed recombinant ORF3 protein has been reported by others to bind to and partition with the cytoskeletal fraction of cells (23), we expected most of it to copellet with cellular components during the clarification step. However, the Western blot analysis detected approximately 50% of the ORF3 protein in the clarified cytosol following the water extraction procedure (data not shown).
In spite of the advantages afforded by this cell culture system, it is clear that it cannot substitute totally for in vivo models. The receptor used for infection of the 10-3 cells may well differ somewhat from that in rhesus monkeys, since MAb HEV31 was a potent neutralizer of HEV inoculated into rhesus monkeys but provided relatively inefficient neutralization of HEV inoculated onto 10-3 cells. Additionally, the ORF3-null mutant, which in the present study made infectious virions in vitro, was the exact same mutant that was unable to infect rhesus monkeys. The rhesus monkey model of HEV infection requires that virus is spread within the liver to generate the titers of virus needed to produce serum liver enzyme elevations and seroconversion to HEV that are used to document infection with wild-type virus. Since, in cell culture, apparently authentic virions were produced even when ORF3 protein was not synthesized, ORF3 protein most likely does not participate in virion production in vivo either. This leaves the interesting possibility that one critical function of the ORF3 protein is to promote cell-to-cell spread. Cell-to-cell spread of the ORF3 mutant was not observed in vitro, but neither was it observed with the wild-type virus. This inability of the virus to spread in vitro may be the key to the difference between the in vitro and in vivo systems. Since ORF3 is absolutely required for detectable infection in vivo but is not necessary for viral RNA replication, viral protein production, virion morphogenesis, or virion infectivity in vitro, one function may be to promote viral egress, perhaps by interaction with cellular proteins or pathways that are not functional in vitro. ORF3 protein thus appears to be a viral regulatory protein rather than a structural component of virions. As others have reported, ORF3 protein contains sequence motifs which may interact with proteins involved in cell signaling pathways (11). Studies with overexpressed recombinant ORF3 protein have provided evidence for its interaction with mitogen-activated protein kinase, and this will be a very interesting possibility to explore further. Also, the adaptive immune system is absent in vitro and could conceivably play a role in vivo in viral release mediated by ORF3 protein. It will most likely require new approaches to determine exactly what essential functions ORF3 protein is performing in vivo. All indications are that it will be an interesting story.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute of Allergy and Infectious Diseases.
We thank Sandra Chang for excellent secretarial help.
Footnotes
Published ahead of print on 23 August 2006.
REFERENCES
- 1.Bradley, D. W., A. G. Andjaparidze, E. H. Cook, Jr., K. McCausland, M. Balayan, M. Stetler, O. Velazquez, B. Robertson, C. Humphrey, M. Kane, and I. Weisfuse. 1988. Etiologic agent of enterically transmitted non-A, non-B hepatitis. J. Gen. Virol. 69:731-738. [DOI] [PubMed] [Google Scholar]
- 2.Emerson, S. U., D. Anderson, A. Arankalle, X.-J. Meng, M. Purdy, G. G. Schlauder, and S. A. Tsarev. 2004. Hepevirus, p. 851-855. In C. M. Fauquet, M. A. Mayo, J. Maniloff, U. Desselberger, and L. A. Ball (ed.), Virus taxonomy. Eighth report of the International Committee on Taxonomy of Viruses. Elsevier/Academic Press, London, United Kingdom.
- 3.Emerson, S. U., V. A. Arankalle, and R. H. Purcell. 2005. Thermal stability of hepatitis E virus. J. Infect. Dis. 192:930-933. [DOI] [PubMed] [Google Scholar]
- 4.Emerson, S. U., P. Clemente-Casares, N. Moiduddin, V. A. Arankalle, U. Torian, and R. H. Purcell. 2006. Putative neutralization epitopes and broad cross-genotype neutralization of hepatitis E virus confirmed by a quantitative cell-culture assay. J. Gen. Virol. 87:697-704. [DOI] [PubMed] [Google Scholar]
- 5.Emerson, S. U., H. Nguyen, J. Graff, D. A. Stephany, A. Brockington, and R. H. Purcell. 2004. In vitro replication of hepatitis E virus (HEV) genomes and of an HEV replicon expressing green fluorescent protein. J. Virol. 78:4838-4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emerson, S. U., M. Zhang, X. J. Meng, H. Nguyen, M. St. Claire, S. Govindarajan, Y. K. Huang, and R. H. Purcell. 2001. Recombinant hepatitis E virus genomes infectious for primates: importance of capping and discovery of a cis-reactive element. Proc. Natl. Acad. Sci. USA 98:15270-15275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engle, R. E., C. Yu, S. U. Emerson, X.-J. Meng, and R. H. Purcell. 2002. Hepatitis E virus (HEV) capsid antigens derived from viruses of human and swine origin are equally efficient for detecting anti-HEV by enzyme immunoassay. J. Clin. Microbiol. 40:4576-4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Graff, J., H. Nguyen, C. Yu, W. R. Elkins, M. St. Claire, R. H. Purcell, and S. U. Emerson. 2005. The open reading frame 3 gene of hepatitis E virus contains a cis-reactive element and encodes a protein required for infection of macaques. J. Virol. 79:6680-6689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Graff, J., U. Torian, H. Nguyen, and S. U. Emerson. 2006. A bicistronic subgenomic mRNA encodes both the ORF2 and ORF3 proteins of hepatitis E virus. J. Virol. 80:5915-5926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang, Y. W., G. Haqshenas, C. Kasorndorkbua, P. G. Halbur, S. U. Emerson, and X. J. Meng. 2005. Capped RNA transcripts of full-length cDNA clones of swine hepatitis E virus are replication competent when transfected into Huh-7 cells and infectious when intrahepatically inoculated into pigs. J. Virol. 79:1552-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Korkaya, H., S. Jameel, D. Gupta, S. Tyagi, R. Kumar, M. Zafrullah, M. Mazumdar, S. K. Lal, L. Xiaofang, D. Sehgal, S. R. Das, and D. Sahal. 2001. The ORF3 protein of hepatitis E virus binds to Src homology 3 domains and activates MAPK. J. Biol. Chem. 276:42389-42400. [DOI] [PubMed] [Google Scholar]
- 12.Lemon, S. M., and L. N. Binn. 1985. Incomplete neutralization of hepatitis A virus in vitro due to lipid-associated virions. J. Gen. Virol. 66:2501-2505. [DOI] [PubMed] [Google Scholar]
- 13.Magden, J., N. Takeda, T. Li, P. Auvinen, T. Ahola, T. Miyamura, A. Merits, and L. Kaariainen. 2001. Virus-specific mRNA capping enzyme encoded by hepatitis E virus. J. Virol. 75:6249-6255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakabayashi, H., K. Taketa, K. Miyano, T. Yamane, and J. Sato. 1982. Growth of human hepatoma cell lines with differentiated functions in chemically defined medium. Cancer Res. 42:3858-3863. [PubMed] [Google Scholar]
- 15.Purcell, R. H., and S. U. Emerson. 2001. Hepatitis E virus, p. 3051-3061. In D. Knipe, P. Howley, D. Griffin, R. Lamb, M. Martin, B. Roizman, and S. Straus (ed.), Fields virology, 4th ed. Lippincott, Williams and Wilkins, Philadelphia, Pa.
- 16.Schofield, D. J., J. Glamann, S. U. Emerson, and R. H. Purcell. 2000. Identification by phage display and characterization of two neutralizing chimpanzee monoclonal antibodies to the hepatitis E virus capsid protein. J. Virol. 74:5548-5555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schofield, D. J., R. H. Purcell, H. T. Nguyen, and S. U. Emerson. 2003. Monoclonal antibodies that neutralize HEV recognize an antigenic site at the carboxy terminus of an ORF2 protein vaccine. Vaccine 22:257-267. [DOI] [PubMed] [Google Scholar]
- 18.Tam, A. W., R. White, P. O. Yarbough, B. J. Murphy, C. P. McAtee, R. E. Lanford, and T. R. Fuerst. 1997. In vitro infection and replication of hepatitis E virus in primary cynomolgus macaque hepatocytes. Virology 238:94-102. [DOI] [PubMed] [Google Scholar]
- 19.Torresi, J., F. Li, S. A. Locarnini, and D. A. Anderson. 1999. Only the non-glycosylated fraction of hepatitis E virus capsid (open reading frame 2) protein is stable in mammalian cells. J. Gen. Virol. 80:1185-1188. [DOI] [PubMed] [Google Scholar]
- 20.Tyagi, S., H. Korkaya, M. Zafrullah, S. Jameel, and S. K. Lal. 2002. The phosphorylated form of the ORF3 protein of hepatitis E virus interacts with its non-glycosylated form of the major capsid protein, ORF2. J. Biol. Chem. 277:22759-22767. [DOI] [PubMed] [Google Scholar]
- 21.Tyagi, S., M. Surjit, A. K. Roy, S. Jameel, and S. K. Lal. 2004. The ORF3 protein of hepatitis E virus interacts with liver-specific α1-microglobulin and its precursor α1-microglobulin/bikunin precursor (AMBP) and expedites their export from the hepatocyte. J. Biol. Chem. 279:29308-29319. [DOI] [PubMed] [Google Scholar]
- 22.Zafrullah, M., M. H. Ozdener, R. Kumar, S. K. Panda, and S. Jameel. 1999. Mutational analysis of glycosylation, membrane translocation, and cell surface expression of the hepatitis E virus ORF2 protein. J. Virol. 73:4074-4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zafrullah, M., M. H. Ozdener, S. K. Panda, and S. Jameel. 1997. The ORF3 protein of hepatitis E virus is a phosphoprotein that associates with the cytoskeleton. J. Virol. 71:9045-9053. [DOI] [PMC free article] [PubMed] [Google Scholar]