

Abstract
Our previous studies using trans-complementation analysis of Kunjin virus (KUN) full-length cDNA clones harboring in-frame deletions in the NS3 gene demonstrated the inability of these defective complemented RNAs to be packaged into virus particles (W. J. Liu, P. L. Sedlak, N. Kondratieva, and A. A. Khromykh, J. Virol. 76:10766-10775). In this study we aimed to establish whether this requirement for NS3 in RNA packaging is determined by the secondary RNA structure of the NS3 gene or by the essential role of the translated NS3 gene product. Multiple silent mutations of three computer-predicted stable RNA structures in the NS3 coding region of KUN replicon RNA aimed at disrupting RNA secondary structure without affecting amino acid sequence did not affect RNA replication and packaging into virus-like particles in the packaging cell line, thus demonstrating that the predicted conserved RNA structures in the NS3 gene do not play a role in RNA replication and/or packaging. In contrast, double frameshift mutations in the NS3 coding region of full-length KUN RNA, producing scrambled NS3 protein but retaining secondary RNA structure, resulted in the loss of ability of these defective RNAs to be packaged into virus particles in complementation experiments in KUN replicon-expressing cells. Furthermore, the more robust complementation-packaging system based on established stable cell lines producing large amounts of complemented replicating NS3-deficient replicon RNAs and infection with KUN virus to provide structural proteins also failed to detect any secreted virus-like particles containing packaged NS3-deficient replicon RNAs. These results have now firmly established the requirement of KUN NS3 protein translated in cis for genome packaging into virus particles.
West Nile (WN) virus subtype Kunjin (KUN) belongs to the family Flaviviridae and is a useful model to study flavivirus biology (51). KUN is substantially less pathogenic than the New York 99 strain of WN virus and is being used to develop a candidate WN virus vaccine (13). The 11-kb positive-stranded KUN RNA genome consists of one long open reading frame encoding three structural proteins (C, prM, and E) that constitute the virus particle and seven nonstructural proteins (NS1, -2A, -2B, -3, -4A, -4B, and -5) that are mainly involved in virus replication (51) but also in virus assembly (29, 31) and in suppression of the host antiviral response (30, 32, 33). RNA replication in the cytoplasm is believed to take place within the replication complexes (RCs) located in virus-induced cellular membranous structures termed vesicle packets. The RC is composed of viral proteins NS1, NS2A, NS3, NS4A, and NS5 associated with the viral double-stranded RNA replication intermediate, replicative form, and some host proteins (36, 50).
Studies on flavivirus RNA replication were initially performed using a full-length infectious clone of KUN (24), but the subsequent development of subgenomic KUN replicons lacking the structural genes has enabled the uncoupling between replication and packaging (25). In particular, the extensive complementation studies in helper replicon cells of full-length and replicon RNAs with systematic deletions throughout the nonstructural coding region have identified and further specified the roles of nonstructural proteins in flavivirus replication (16, 19-21, 31). These and other studies led to the discovery that two NS proteins that are part of the RC, NS2A and NS3, were not only involved in RNA replication but, quite unexpectedly, were also essential for virus assembly in KUN and Yellow Fever (YF) viruses (27, 29, 31). NS2A is a small, hydrophobic, integral membrane protein shown to be essential for RNA replication (36, 52), assembly/secretion of virus particles (29), and in modulating the host antiviral interferon response (30, 32, 33). NS3 is a multifunctional protein with enzymatic activities required for polyprotein processing, viral RNA replication, and RNA capping (51). The NS3 gene encodes a serine protease at its N terminus, which together with cofactor NS2B cleaves the viral polyprotein at the junctions C-prM, NS2A-2B, NS2B-3, NS3-4A, NS4A-4B, and NS4B-5 (4, 52). Furthermore, NS3 encodes the viral helicase/nucleoside 5′-triphosphatase for unwinding of the double-stranded RNA template (12, 49), as well as an RNA 5′-triphosphatase at its C terminus (3), which together with a methyltransferase located in the N terminus of NS5 (26) caps the 5′ terminus of the displaced positive-stranded RNA.
The packaging defect caused by a single amino acid mutation in KUN NS2A at position 59 can be rescued in trans by a helper replicon expressing wild-type NS2A (29). We also showed that any deletions in the NS3 coding region allowing complementation of replication (amino acids 178 to 611) resulted in a defect in packaging of complemented replicon RNA (20, 31), suggesting a role for the NS3 gene product in cis in virus assembly. Similar experiments with complementation of YF virus replicons, however, did not confirm the requirement for NS3 protein in cis in RNA packaging (15), suggesting that some differences in the packaging requirements between these two viruses may exist.
One of the possible explanations for the observations of the packaging inability of NS3-deleted KUN RNAs could be that the functional, full-length NS3 protein must be translated in cis for packaging of the RNA molecule (20, 31). However, an alternative explanation could also be that the presence of a specific RNA sequence or RNA secondary structure within the NS3 coding sequence is required for genome encapsidation, similar, for example, to the packaging signal(s) found in alphaviruses (9, 53). Mutations/deletions of these RNA structures from the KUN genome would then prevent RNA packaging. This study aims to determine the reason for the previously demonstrated packaging inability of complemented KUN RNA molecules with in-frame deletions in the NS3 coding region (20). In the first approach the RNA structure of NS3 was mutated without changing the amino acid sequence, and the effect on replication and packaging was examined. In the second approach the amino acid sequence of NS3 was altered with minimal impact on the RNA structure. Complementation experiments were performed to answer the question of whether the functional NS3 protein or its RNA structure determines specific encapsidation of KUN RNA.
MATERIALS AND METHODS
RNA structure prediction and plasmid construction.
RNA structure modeling of the NS3 coding region was performed using the Mfold program (57). Based on the previously published RNA structure of KUN replicon C20DXrep (16), three regions in NS3 with low P-num values (41) were selected and used for site-specific mutagenesis (slA, KUN nucleotides [nt] 5042 to 5092; slB, KUN nt 5439 to 5471; and slC, KUN nt 6039 to 6074) (Fig. 1A). Silent mutations were designed to disrupt suspected stable RNA stem-loops without affecting amino acid sequence (Fig. 1A, slAm, slBm, and slCm) and were generated by quick-change PCR mutagenesis using primer pairs (Table 1). PCR was performed with Pfu DNA polymerase (Promega) followed by DpnI (New England BioLabs [NEB]) digestion and transformation into Escherichia coli DH5α cells. PCR was performed on an intermediate plasmid containing a 3.2-kb AvrII fragment encoding partial NS1, NS2A, NS2B, and almost the entire NS3 gene. After mutagenesis, the entire plasmid insert was sequenced to confirm the presence of the designed mutations and that no unwanted mutations were introduced during the PCR. The mutated AvrII fragments were then cloned back into replicon repPAC-βGal (31), generating repPAC-βgal-slAm, -slBm, and -slCm, respectively. Double frameshifts in the NS3 gene were introduced by two rounds of quick-change PCR mutagenesis on the same intermediate plasmid as described above. DfsI was constructed using primer pairs fs188-F with fs188-R and fs212-F with fs212-R. DfsII was constructed using primer pairs fs465-F with fs465-R and fs501-F with fs501-R. The mutated AvrII fragments were subsequently inserted into the full-length infectious clone FLSDXpro_HDVr (29) and replicon repPAC-βGal.
FIG. 1.

Predicted RNA structures in NS3 do not play a role in replication and packaging. (A) Mfold predictions of RNA structures with low P-num values. Three RNA stem-loop structures (slA, slB, and slC) were selected from an overall Mfold prediction of KUN replicon RNA C20Dxrep (17). Base-pairing in the stems of slA, slB, and slC was disrupted by silent mutations (arrows), giving mutants slAm, slBm, and slCm. Free energy values (dG, in kcal/mol) calculated by Mfold are indicated below the predicted RNA structures. Nucleotide numbers according to the full-length KUN genome sequence FLSDX (GenBank accession number AY274504) are indicated, and mutated nucleotides are encircled. The mutations were introduced in KUN replicon repPAC-βgal encoding a β-galactosidase marker gene to quantitate RNA replication levels in BHK cells electroporated with in vitro-transcribed replicon RNA. (B) RNA replication levels in cell culture. Capped in vitro RNA was transcribed and electroporated in BHK-21 cells. Cell lysates were prepared in duplicate at 48 h posttransfection, and β-galactosidase activity was measured. (C) Release of virus-like particles containing (mutant) repPAC-βgal RNAs upon electroporation into tetKUNCprME-packaging BHK-21 cells (14). Culture fluids were harvested every second day and titrated on VERO cells. Infected VERO cells were fixed and stained with X-Gal, and positive cells were counted.
TABLE 1.
Oligonucleotides for construction of plasmids, mutant full-length clones and replicons, PCR amplification, and cDNA synthesis
| Primer name | Sequence (5′ to 3′)a | Comment |
|---|---|---|
| slAm-F (GP9) | GAAACGGAGTAATCATGCCAAACGGTTCATACATAAGCGCGATAG | |
| slAm-R (GP10) | TGATTACTCCGTTTCCGTATAATCCTATCACATCACCGTTTTTG | |
| slBm-F (nGP11) | CTACAACCTATTCGTCATGGACGAGGCCCACTTCAC | |
| slBm-R (nGP12) | GACGAATAGGTTGTAGTTAGGCACCCTATGAGGAG | |
| slCm-F (GP13) | ATGAGGATGATAGCAACTGCGCACACTGGACTGAGGCACGAATC | |
| slCm-R (GP14) | TGCTATCATCCTCATTCGTATGACCTCCATAGCAGTACTC | |
| fs188-F (GP1) | AAACGGATC*CCGTTTTGGATCTTCACCCTG | BamHI site underlined |
| fs188-R (GP3) | CCAAAACGG*GATCCGTTTTTTCCTCAACATC | BamHI site underlined |
| fs212-F (GP2) | ATCAAAGAGGCCTATAAACAGGAGGCTGAG | StuI site underlined |
| fs212-R (GP4) | ATAGGCCTCTTTGATGATCTGTGGCAGAATC | StuI site underlined |
| fs465-F (GP5) | ATTGGTAGGA*TCCATCACAAGTTGGAGATG | BamHI site underlined |
| fs465-R (GP7) | TGATGGA*TCCTACCAATGCGTCCTCGTCTC | BamHI site underlined |
| fs501-F (GP6) | ACATGGCCAAACGGATTGATTGCTCAATTC | MscI site underlined |
| fs501-R (GP8) | CAATCCGTTTGGCCATGTTGATGTTATCGAG | MscI site underlined |
| NS2B-ApaI-F | ATGGGCCCACCATGGGGTGGCCTGCA | ApaI site underlined |
| NS3-XbaI-R | AATTTCTAGACTAGCGCTTCCCTGA | XbaI site underlined |
| 3'UTRDX-R | CACACTAAACACTATTATAAAGCTAAA | |
| NS3-F | GGAGGTGTGCTGTGGGACAC | |
| NS3-R | GCGCTTCCCTGAGGCGAAG |
Introduced nucleotide mutations are shown in boldface, and stop and start codons are in italics. Inserted nucleotides are shown in boldface italics, and positions with nucleotide deletions are indicated with an asterisk.
Cells, in vitro transcription, electroporation, and cell line selection.
BHK-21, repBHK (16), VERO (ATCC), and repVERO (C20SDrepVero) (37) cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen) supplied with 10% fetal bovine serum (Invitrogen) at 37°C at 5% CO2 in tissue culture flasks. The repBHK and repVERO cell lines were grown under selection with G418 (Sigma) at a concentration of 500 μg/ml. Replicon DNA templates were linearized with XhoI (NEB) and purified. In vitro RNA transcription and electroporation were performed as described previously (25). Cell lines were selected using various concentrations (1 to 5 μg/ml) of puromycin (Sigma), and cell clones were isolated after 2 weeks with cloning cylinders (Corning). After initial selection, the cell lines were grown under a puromycin selection pressure of 3 μg/ml.
RNA isolation and RT-PCR.
Cellular RNA was isolated from puromycin-selected cell lines (grown in six-well plates) using TRIzol reagent (Invitrogen) following the manufacturer's recommendations. RNA was quantified by UV spectroscopy. Reverse transcription (RT) was performed using Moloney murine leukemia virus reverse transcriptase (Promega) with the primer 3′UTRDX-R (Table 1) specifically annealing to the 3′-untranslated region (UTR) of repPAC-βgal but not to the 3′-UTR of C20Dxrepneo (21). PCR on cDNA was carried out with primers NS3-F (Table 1) and NS3-R (Table 1) using Taq polymerase (NEB). Restriction analysis of RT-PCR products was performed with BamHI (NEB), and the sizes of the digested bands were estimated using the Generuler DNA size marker (Fermentas).
Virus infection, plaque assay, and VLP titration.
Puromycin-selected cell lines in six-well plates were infected with Kunjin virus strain MRM61C at a multiplicity of infection (MOI) of 5. Culture fluids were harvested at 72 hours postinfection and stored in small aliquots at −80°C. A plaque assay was performed on 80 to 90% confluent monolayers of VERO cells in 24-well plates. Briefly, VERO cells were infected with dilutions of virus-containing culture fluids for 2 h and washed twice with Dulbecco's modified Eagle's medium. The cells were overlaid with 1 ml of 199 medium (Gibco) containing 1% carboxymethyl cellulose (ICN Biomedicals) and incubated for 4 days at 37°C in an incubator. Cells were fixed by adding 100 μl of 20% formaldehyde directly onto the overlay and incubation for 30 min at room temperature. Wells were washed under running tap water and stained with 0.2% crystal violet for 20 min. The plates were dried, and plaques were counted. Virus-like particles (VLPs) harboring defective repPAC-βgal RNA were titrated onto repVERO cells using in situ 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-Gal) staining.
Immunofluorescence, X-Gal staining, and β-Gal assay.
Immunofluorescence was performed on cells grown on coverslips, which were fixed with 100% ice-cold acetone for 30 s. Detection of complementation and expression of the mutated full-length KUN RNAs were performed by indirect immunofluorescence analysis with a mix of three monoclonal antibodies against the KUN E protein (2B2, 3.91D, and 3.67G; gifts from Roy Hall) or polyclonal antibodies against the KUN E protein (gift from Roy Hall). Detection of replication and expression of mutated replicon RNAs was performed by in situ staining with X-Gal or immunofluorescence using anti-β-galactosidase (β-Gal) monoclonal antibody (Promega). Secondary antibodies (AF488 anti-mouse, AF488 anti-rabbit, and AF594 anti-mouse) were purchased from Molecular Probes/Invitrogen. Cells in duplicate 24-well plates were lysed in 200 μl reporter lysis buffer (Promega), and the β-Gal assay was performed using the β-galactosidase enzyme assay system (Promega) following the manufacturer's instructions. Absorbance was measured using a Lucy2 plate reader/luminometer (Anthos, Austria).
In vitro translation.
For in vitro translation of the NS2B3 precursor, the NS2B3 gene cassettes (wild type, with deletion and with frameshift mutations) were amplified using Pfu DNA polymerase with primers NS2B-ApaI-F and NS3-XbaI-R (Table 1), digested with XbaI, and cloned into pcDNA3 vector (Invitrogen) digested with EcoRV and XbaI. Resulting plasmid DNAs were linearized with XbaI, and capped mRNA was prepared using T7 RNA polymerase (Promega) and 1 to 2 μg of linearized plasmid DNA essentially as described above. RNA was extracted with phenol-chloroform, precipitated with ethanol, and quantified by UV spectroscopy. In vitro translation reaction mixtures (25 μl) were prepared with rabbit reticulocyte lysate (Promega) and were supplied with 1 μl trans-[35S]methionine label (MPI Biomedicals) and 3 μl canine pancreatic microsomal membranes (Promega) to facilitate translation and proteolytic cleavage of the NS2B3 gene product. Membranes were pelleted in 20 min at 14,000 rpm in a microcentrifuge and washed with phosphate-buffered saline. Samples were loaded onto a 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel followed by exposing the dried gel to X-ray film (Kodak).
RESULTS
Computer-predicted conserved RNA structures in the KUN NS3 gene do not play a role in RNA replication and packaging.
The first set of experiments aimed to answer the question of whether Mfold-predicted RNA structures in the NS3 coding region located in the deleted regions used in our previous packaging-complementation experiments (20, 31) are involved in the specific packaging of genomic RNA. Three predicted conserved stem-loop structures with low P-num values (indicated by degrees of coloration in the overall RNA structure, as shown in reference 17) were selected in the NS3 coding region and named slA (nt 5042 to 5092), slB (nt 5439 to 5471), and slC (nt 6039 to 6074) (Fig. 1A). Multiple silent mutations were subsequently introduced by PCR-directed mutagenesis as described in Materials and Methods, generating repPAC-βgal-slAm, -slBm and -slCm, respectively (Fig. 1A). In vitro-transcribed replicon RNAs were electroporated into BHK cells, and the levels of RNA replication were compared at 48 h posttransfection by quantifying the amount of β-galactosidase production from the replicon RNA. The β-Gal assay has previously been shown to be a reliable method to indirectly measure RNA replication levels (30, 31). The results from the β-Gal assay demonstrated that there was no significant difference in RNA replication levels between the parental replicon repPAC-βgal and the replicons with mutated stem-loop structures in NS3 at 48 h after transfection (Fig. 1B). Next, these replicon RNAs were introduced into a BHK packaging cell line expressing KUN structural proteins C, prM, and E, capable of packaging replicon RNA into secreted VLPs (14). Titration of secreted VLPs at 2-day intervals showed, again, no significant difference between the parental replicon and the mutants (Fig. 1C). In summary, these experiments showed that multiple mutations disrupting secondary structure in three separate, conserved Mfold-predicted RNA structures with low P-num values in the KUN NS3 gene did not have any effect on either replication or packaging.
Complementation of full-length KUN RNAs with double frameshift mutations in the NS3 helicase region.
We next examined whether the previously observed defect in packaging of complemented KUN full-length or replicon RNA containing deletions in NS3 (20, 31) could alternatively be explained by the absence in the NS3 region of a longer RNA sequence or overall secondary RNA structure essential for packaging. In order to preserve the secondary RNA structure in the NS3 gene but to make the NS3 gene product functionally inactive, we introduced single nucleotide insertions and deletions resulting in double frameshifts (dfs) in the NS3 coding sequence with minimal impact on its RNA structure. Two mutants were designed and constructed that had double frameshifts in the NS3 helicase coding region from codons 188 to 212 and from codons 465 to 501, respectively (Fig. 2A and B). Mfold predictions of the mutated NS3 RNA sequence showed that no major structural changes were generated as a result of the nucleotide insertions/deletions. Frameshift mutation at position 188 only changes the ranking of predicted RNA structures provided by Mfold, whereas frameshift mutations at positions 212 and 465 are located in predicted loops that do not change upon mutation. Frameshift mutation at position 501 is located in a predicted stem with high P-num values, which decreases the relevance of this particular prediction. As a control for a defect in packaging upon successful complementation, a previously generated construct with an in-frame deletion within the NS3 gene (dNS3. 8) (31) was included in the assay.
FIG. 2.

Double frameshift mutations in NS3 do not affect NS2B-3 protease activity. (A) NS3 constructs with double frameshifts or an in-frame deletion in the NS3 coding sequence. PRO, serine protease; HEL/NTP, helicase/nucleoside 5′-triphosphatase; RTP, RNA 5′-triphosphatase. Two different sets of nucleotide insertion/deletions were introduced to generate a partial frameshift in the NS3 coding sequence from amino acids 188 to 212 (NS3dfsI) and from amino acids 465 to 501 (NS3dfsII). The deletion in NS3 was published previously and denoted dNS3.8 (31). (B) Amino acid sequence resulting from double frameshift mutations (dfsI and dfsII) in the NS3 coding sequence. Conserved helicase motifs Walker A (motif I) and motif VI are indicated in boldface. Changed amino acid residues are underlined, and amino acid numbers of the NS3 protein are indicated. (C) In vitro translation (IVT) and autolytic cleavage of KUN NS2B-NS3 serine protease. Wild-type and mutated/deleted NS2B3 precursors were cloned into the pcDNA3 vector, and in vitro-transcribed capped mRNA was used as a template for in vitro translation reactions using [35S]methionine. IVT reaction mixtures were supplied with canine microsomal membranes to facilitate translation and allow self-cleavage of the membrane-bound 2B3 serine protease. Membranes were pelleted by centrifugation for concentration of labeled membrane-associated NS2B3 proteins and run in a sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel, and the dried gel was exposed by autoradiography. NS2B3 precursor and cleaved NS3 gene products are indicated by arrows. Note the smaller gene products for dNS3.8, harboring an in-frame deletion of the C terminus of NS3.
Before the double frameshift mutations in NS3 were tested in the KUN full-length infectious clone, it was first verified that no adverse effects on NS2B3 protease processing were caused by scrambling parts of the amino acid sequence of the (downstream) helicase region of NS3. The respective NS2B3 gene cassettes were therefore cloned into pcDNA3 vectors, and in vitro-transcribed capped mRNA was subsequently used as a template for in vitro translation reactions (Fig. 2C). These in vitro translation reactions were supplemented with canine microsomal membranes for proper membrane translocation of the NS2B3 precursor. The results showed that translation and subsequent cleavage of the NS2B3 precursor protein into the individual NS2B and NS3 proteins were not affected by the introduced mutations.
Next, the double frameshift substitutions were introduced into the KUN full-length infectious clone (Fig. 3A), and in vitro-transcribed KUN RNAs were electroporated into two different helper KUN replicon-expressing cell lines, repBHK and repVERO cells. Immunofluorescence using anti-E antibodies (Fig. 3B) showed that complementation of replication (and thus expression of E protein) indeed occurred in both repBHK and repVERO, but this complementation was rather inefficient, as was previously observed for the complementation of KUN RNAs with deletions in NS3 (20). As in previous complementation experiments with KUN RNAs containing deletions in NS3, no secreted virus particles containing encapsidated KUN full-length RNAs with double frameshift mutations in NS3 were detected in the culture fluid of transfected repBHK and repVERO cells (data not shown).
FIG. 3.

Complementation of full-length RNAs with double frameshift mutations in NS3 in the helper replicon-expressing cell lines. (A) Full-length clones with deletions in NS1 and NS3 (FLdNS1. 1/3. 8) and double frameshift mutations in NS3 (FLNS3dfsI and FLNS3dfsII) used for complementation in repBHK and repVERO cells. (B) Immunofluorescence analysis of repBHK and repVERO cells electroporated with in vitro-transcribed (defective) full-length RNAs. Cells were grown on glass coverslips, fixed 5 days postelectroporation, and stained with anti-E monoclonal antibodies.
Generation and characterization of cell lines stably producing complemented replicon RNAs with double frameshift mutations in NS3.
To increase the sensitivity of the complementation-packaging assay, another approach was chosen to establish repBHK cell lines harboring both helper replicon (C20DXrepNeo) and the complemented puromycin-selectable KUN replicons with the deletions/frameshift mutations in NS3 and a β-Gal marker gene. These established cell lines should provide ample amounts of complemented, actively replicating defective replicon RNAs available for packaging by KUN structural proteins which could be supplied in trans. To generate such stable cell lines, the double frameshift mutations were cloned into the selectable replicon repPAC-βgal (Fig. 4A), and in vitro-transcribed replicon RNAs were electroporated into repBHK cells. Cell clones containing actively replicating defective replicon RNAs were selected with puromycin in six-well plates, and cells in parallel wells were fixed and stained in situ with X-Gal to demonstrate rescued replication of the complemented replicons (Fig. 4B). Although the complementation efficiencies for repNS3dfsI and repNS3dfsII RNAs were slightly lower than for repdNS1. 1/3. 8 RNA (Fig. 4B), puromycin-resistant cell lines for each construct were established. Since the level of marker gene expression (in this case β-Gal) is an accurate measure of the level of RNA replication (31), a β-Gal assay was performed on cell lysates of the different cell lines (Fig. 5A). The result showed that the level of β-Gal expression in these established cell lines was between 65 and 100% of that detected in the cell line containing replication-competent replicon RNA (repPAC-βgal), indicating that a sufficient amount of actively replicating defective replicon RNA was available for packaging. To confirm that the initially designed and introduced mutations were still present in the replicons and had not reverted back to generate replication-competent RNA, cDNA was made from cellular RNA using a primer that specifically binds to the 3′-UTR of repPAC-βgal RNA but not of C20DXrepNeo RNA (21). Next, a PCR amplifying the NS3 gene (using primers NS3-F and NS3-R) (Fig. 5B) was performed, followed by BamHI digestion (Fig. 5C). The restriction analysis confirmed that the introduced BamHI sites were retained in the complemented repNS3dfsI and repNS3dfsII RNA and that a major deletion was still present in complemented repdNS1.1/3.8 RNA.
FIG. 4.

Establishment of cell lines continuously producing complemented defective replicon RNAs using the dual replicon complementation system. (A) Schematic representation of puromycin-selectable and β-Gal-expressing replicon constructs containing deletions in the NS1 and NS3 genes (repdNS1. 1/3. 8) and double frameshift mutations in the NS3 gene (repNS3dfsI and repNS3dfsII), which were used to establish stable cell lines continuously producing complemented defective replicon RNAs. (B) repBHK cells stably producing replicating KUN replicon RNA C20DXrepNeo (21) were electroporated with defective replicon RNAs shown in panel A and incubated in the medium with 5 μg/ml of puromycin for 10 days, fixed, and stained with X-Gal.
FIG. 5.
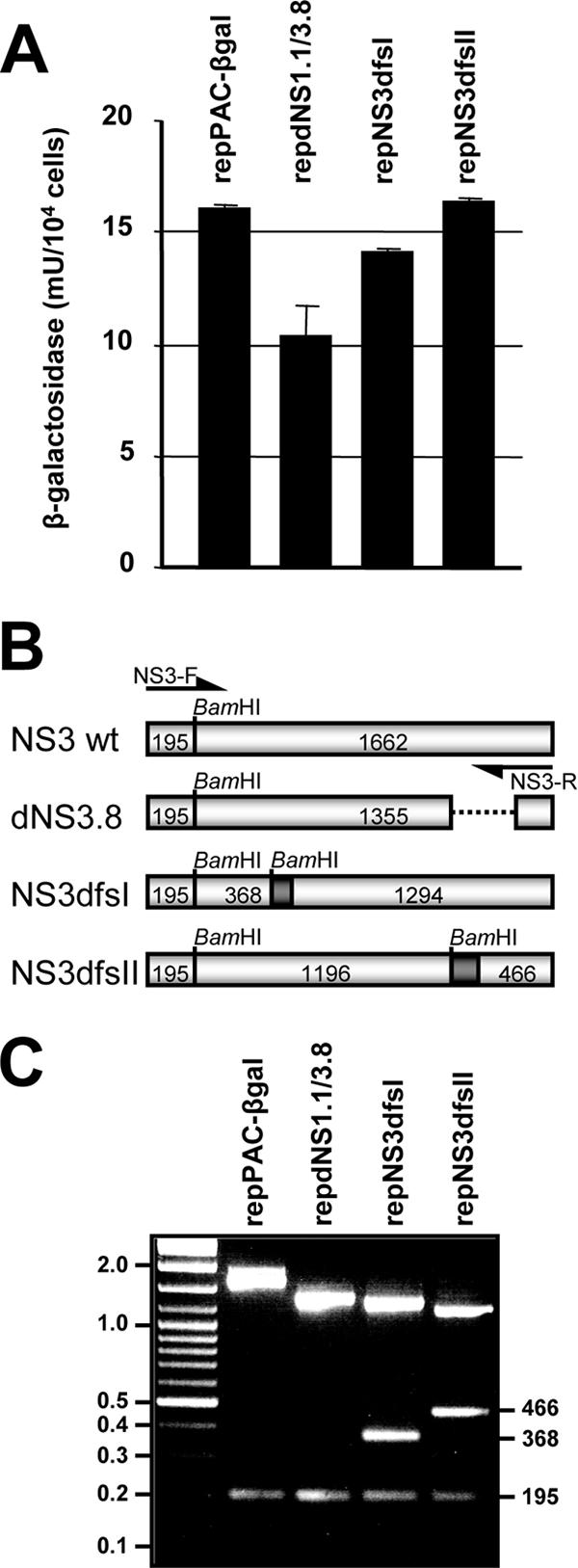
Characterization of stable cell lines producing complemented defective replicon RNAs. (A) β-Galactosidase expression in selected stable cell lines. Cell lysates of the stable cell lines were analyzed by β-Gal assay. (B) PCR amplification of NS3 genes from complemented defective replicon RNA. cDNA for PCR amplification with NS3-F and NS3-R primers was synthesized with a primer specific for the 3′-UTR of the defective replicon but not of the helper replicon (21). Expected sizes (in nucleotides) of PCR products after digestion with BamHI are indicated. (C) Retention of introduced mutations in complemented defective replicon RNA. NS3 PCR products were digested with BamHI. Sizes (in nucleotides) are indicated on the right.
Complemented replicon RNAs with defective NS3 protein cannot be packaged into secreted virus-like particles by the structural proteins provided in trans by KUN virus infection.
To provide KUN structural proteins for packaging, the established cell lines producing defective replicon RNA were infected with KUN virus at an MOI of 5, and culture fluids were harvested 3 days postinfection. All the cells were positive for both β-Gal and KUN E expression at 24 h postinfection (Fig. 6A), demonstrating that both complemented RNA and the KUN structural genes were expressed in the vast majority of infected cells. Dual immunofluorescence using anti-β-Gal monoclonal and anti-E polyclonal antibodies was also carried out to confirm that individual cells were simultaneously expressing actively replicating defective replicon RNAs and KUN structural proteins (Fig. 6B). The presence of VLPs containing complemented replicon RNAs in the culture fluid of infected cells was tested by infection of repVERO cells followed by X-Gal staining. If the complemented replicon RNA had become packaged by the structural proteins provided by KUN infection, repVERO cells infected with these VLPs would complement the defective RNA and, thus, expression of β-Gal should be detected. To estimate the levels of secretable structural proteins produced by KUN virus infection in cell lines with NS3-mutated replicons, culture fluids from infected cells were examined for the presence of infectious KUN virus by plaque assay on VERO cells. Clearly, sufficient amounts of structural proteins for packaging were provided by KUN virus infection as demonstrated by the release of 2. 2 × 108 to 3. 3 × 108 infectious virus particles per ml in all tested culture fluids by 72 h postinfection (data not shown). The culture fluid of cells stably expressing replication-competent repPAC-βgal RNA and infected with KUN virus contained 1. 8 × 105 virus-like particles with encapsidated repPAC-βgal RNA per ml, as judged by the X-Gal staining of repVERO cells infected with this culture fluid (Fig. 6C). As an additional control for packaging of complemented replicon RNA, we used infection of stable cells continuously producing complemented replicon RNA with a large deletion in the NS5 gene and expressing β-Gal, repdNS5AB (Fig. 6A). We showed previously that full-length RNA with this deletion could be efficiently complemented in repBHK cells and packaged into defective viral particles (20). A total of 1.1 × 103/ml of VLPs containing encapsidated complemented repdNS5AB replicon RNA were detected in the culture fluid of repdNS5AB cells infected with KUN virus (Fig. 6C). Although packaging of NS5-deleted replicon RNA was somewhat less efficient than that of the wild-type replicon RNA, the results show that both wild-type and the NS5-deleted complemented defective replicon RNAs could be packaged by the structural proteins provided in trans by KUN virus infection. However, despite the presence of abundant amounts of complemented replicon RNA available for packaging and the structural proteins provided by the virus infection (Fig. 6A and B), no VLPs containing complemented replicon RNAs with double frameshift mutations in NS3 were detected (Fig. 6C). A barely detectable number of VLPs (two positive cells in a well infected with 100 μl of undiluted culture fluid) containing complemented replicon RNA with a deletion in NS3 could be detected. Such a low number of detected VLPs clearly showed that packaging, if it occurred, was extremely inefficient. Overall, the results showed that the defect in NS3 protein caused by the double frameshift mutations blocks packaging of the RNA template from which it is translated and that this packaging defect cannot be complemented in trans by supplying functional NS3 protein from the helper replicon or full-length genomic KUN RNAs.
FIG. 6.

trans-packaging of the defective replicon RNAs using KUN virus infection. (A) Stable cell lines were fixed and stained with X-Gal prior to KUN virus infection to confirm that all the cells were producing actively replicating defective replicon RNAs. Next, the cell lines were infected with KUN virus at an MOI of 5 to provide KUN structural proteins, and the infection was confirmed by immunofluorescence with anti-E monoclonal antibodies. (B) Dual immunofluorescence using anti-β-Gal monoclonal and anti-E polyclonal antibodies to confirm that individual cells were simultaneously expressing actively replicating defective replicon RNAs and KUN structural proteins. (C) At 72 h after infection, culture fluids were harvested and either titrated on VERO cell monolayers by plaque assay to determine the titers of KUN virus or titrated on repVero cells to determine the titers of virus-like particles harboring defective β-Gal-expressing replicon RNAs.
DISCUSSION
In view of the demonstrated inability of complemented KUN full-length and replicon RNAs with in-frame deletions in NS3 to be packaged into virus/virus-like particles (20, 31), we hypothesized that a structural RNA element required for packaging may be present in the KUN NS3 gene. Examples of RNA structures in coding regions of positive-strand viruses with important roles in viral RNA translation, replication, and packaging include the cyclization sequence (18) and a hairpin structure (7) in the flavivirus core gene, the cis-acting replication elements in the poliovirus 2C gene (10, 11) and in the hepatitis C virus NS5B gene (28, 55), the alphavirus translation enhancer sequence in the capsid gene (8), and packaging signals in the Sindbis virus nsP1 (methyltransferase) (9) and in the Semliki Forest virus nsP2 (nucleoside triphosphatase/helicase) genes (53). Surprisingly, mutations disrupting the secondary structure of three computer-predicted RNA structures with low P-num values (17) in the KUN NS3 coding region did not have any effect on KUN RNA packaging or replication, although the presence of other cis structural elements in the NS3 coding region that were not predicted by Mfold cannot be excluded. These findings raise some doubts about the biological significance of computer-predicted, low-P-num-value RNA structures in viral coding regions. Covariance analyses of various RNA virus genomes using a parsimony-based algorithm (46) indicated that flaviviruses may not have genome-scaled ordered RNA structures in their coding regions like those found, for example, in hepaciviruses (43). Instead, flaviviruses appear to have cis-requirements for virus replication localized in their 5′- and 3′-UTRs and in the core coding sequence; these RNA structures are clearly vitally important for initiation of translation and of synthesis of minus- and plus-strand RNAs (17, 18, 34, 39, 56), and perhaps for packaging as well.
In the subsequent set of experiments we tested and confirmed the hypothesis that translation of NS3 protein in cis is essential for RNA packaging. For this purpose, KUN full-length and replicon RNAs containing double frameshift mutations in the NS3 gene were analyzed for packaging following complementation by helper replicon. During complementation in repBHK cells, the RNAs encoding defective NS3 become translated and the defective RC is presumably assembled on the 3′-UTR (21, 51). The defective RNA templates may then be released from the defective RC and reattach to a functional RC produced from the helper replicon or, alternatively, components of the defective and helper RCs may exchange (21). Whichever is the mechanism of complementation, the result is that a functional RC is copying defective RNA templates which may subsequently be released and become available for packaging. In the first set of complementation experiments with the NS3-mutated full-length RNAs in the helper replicon-expressing cells, the structural proteins were provided in cis from the same (full-length) RNA. It is possible that the low efficiency of complementation (and thus low expression of structural proteins) observed in the first set of experiments with NS3-mutated full-length RNAs precluded us from detecting low levels of released virus particles containing defective RNA.
Our second set of experiments with the NS3-mutated replicon RNAs was therefore designed to really force the packaging by producing large amounts of complemented replicon RNA and of the structural proteins in the majority of cells. This was achieved by establishing stable cell lines continuously producing complemented NS3-defective replicon RNA and by infecting these cells with KUN virus. However, despite the presence of abundant amounts of NS3-mutated, actively replicating, complemented replicon RNAs and of the structural proteins provided by virus infection in the great majority of cells, we failed to detect any secreted virus-like particles containing packaged replicon RNAs with double frameshift mutations in the NS3 helicase. This experiment firmly established that not the KUN NS3 RNA sequence or secondary structure but the NS3 gene product itself is required in cis for the packaging of KUN RNA. These results are in contradiction with the recent studies on complementation experiments with YF virus replicons containing deletions in the NS3 gene and encoding a luciferase marker gene (15). In YF virus experiments, replication of replicons with deletions in the NS3 gene was first complemented in helper BHK-REP cells, and then their packaging into secreted virus-like particles was achieved (albeit inefficiently) by the YF virus structural proteins provided in trans from the Sindbis virus replicon vector (15). Although it is difficult to explain the differences between our results and the results with YF virus replicons, it should be noted that KUN and YF viruses belong to two distantly related subgroups of flaviviruses, and it is possible that they may have different requirements for complementation of packaging.
Viruses have evolved different strategies for specific packaging of viral RNA genomes in virus particles. A common mechanism to specifically encapsidate viral RNAs is the use of packaging signals in the RNA genome, which are recognized by a viral structural protein(s) that can be expressed in trans from a separate RNA molecule (6, 42, 45). A strict cis requirement for a viral structural protein in packaging is found in Flock House virus, where translation of capsid protein in cis directs packaging of replicating Flock House virus genomic RNA by direct association with the RNA from which it is translated (47). However, the mechanism by which NS3 protein in cis may be involved in assembly of KUN viral RNA from which it is translated must be clearly different, given that no NS3 protein is found in the virus particles and that replicon RNA can be efficiently encapsidated by C protein supplied in trans from a separate mRNA (14, 23). More relevant examples of nonstructural “replication” proteins involved in the packaging process are the bovine viral diarrhea virus NS5B (RdRp) (2), the uncleaved bovine viral diarrhea virus NS2-3 precursor (1), and the Venezuelan equine encephalitis virus nsP1-3 precursor (48). These studies and our results with NS3 further support the coupling between replication and packaging demonstrated previously for flaviviruses (22) and poliovirus (40).
It is logical to assume that for the viral RNA to be encapsidated, the NS3 helicase and RNA 5′-triphosphatase activities are required to release the nascent positive-stranded RNA from the RC and cap the RNA at its 5′ terminus, respectively (3, 12, 49). It is rather unlikely, however, that in our complementation-packaging experiments the expression of defective NS3 from complemented RNA would block these activities and thereby prevent RNA packaging, since the fully functional NS3 is present in the helper RC which replicates the defective RNA. To explain then how translation in cis of functional KUN NS3 may be required for packaging of genomic RNA, we tend to favor a model where the newly synthesized NS3 protein is directly associated with the progeny RNA template and at the same time also interacts with other viral and/or host proteins necessary for the translocation of this RNA-protein complex to the site of RNA encapsidation/virus assembly. Given that only newly replicated KUN RNA can become packaged (22), NS3 may need to be translated in cis from the progeny (+) RNA strand released from the RC to allow specific packaging of replication-competent RNA. This would mean that providing NS3 in trans from a helper replicon will not result in rescue of packaging of the viral RNA, since the helper NS3 will not be able to associate with the defective RNA. There is a remote possibility that interactions between NS3 and a viral/host factor essential for packaging are disrupted by the two double frameshift mutations (dfsI and dfsII) in NS3. A way to resolve this would involve substitution of individual residues in conserved helicase motifs and analysis of packaging of complemented replicon RNA. However, constructs FLdNS1.1/3.8, FLNS3dfsI, FLNS3dfsII, and full-length clones with a range of other in-frame deletions in NS3 (20) all displayed the same phenotype, i.e., complemented RNA was packaging deficient. Therefore, it is highly unlikely that the same putative interaction between NS3 and a virus/host factor would be affected in all of these mutants.
In brome mosaic virus replication, nascent 2A peptide (polymerase) associated with both the viral RNA and the ribosome is sequestered by 1A (capping enzyme and helicase) to membrane-bound RNA replication spherules formed by multiple 1A proteins (5). The necks of the spherules are thought to serve as channels to export newly synthesized progeny RNA to the cytoplasm for translation and packaging (42). In analogy with this system, the KUN NS3-RNA complex may be sequestered by another NS or host protein to the site of RNA packaging/virus assembly, where multiple copies of core protein (C) are present to encapsidate the RNA. A role for NS3 at the site of RNA packaging/virus assembly can be envisaged, as it has been shown that cytoplasmic cleavage between C and prM by the viral NS2B-3 protease is required to release mature C protein for encapsidation of viral RNA and subsequent virion formation (35, 44, 54). Some evidence from immunofluorescence analysis suggests that NS3 and E colocalize (J. M. Mackenzie, unpublished results) with similar intracellular NS3 and E staining patterns observed in the perinuclear region (38).
A putative candidate viral protein to sequester the presumed NS3-RNA complex to the sites of virus assembly could be NS2A, given its demonstrated role in packaging (27, 29), its interaction with 3′-UTR of genomic RNA (36), and its proposed interaction with NS3 (27). In this context, it is also noteworthy that the YF virus NS2A mutation blocking virus assembly was rescued by a compensatory mutation in the helicase domain of NS3 at amino acid position 343 (27). This site was predicted to be exposed on the NS3 protein surface and may thus be available for direct interaction with other proteins, such as NS2A. In our NS3 mutants with double frameshifts and/or in-frame deletions, this interaction with NS2A may be prohibited, resulting in a block in packaging. In future experiments, identification of putative binding domains between NS2A and NS3 may shed more light on the mechanism of their involvement in RNA packaging/virus assembly.
Acknowledgments
We thank Jason Mackenzie for helpful discussions and providing repVERO cells and other reagents. We thank Roy Hall for providing anti-E monoclonal antibodies.
The work was supported by the grants to A.A.K. from the National Health and Medical Research Council of Australia.
Footnotes
Published ahead of print on 13 September 2006.
REFERENCES
- 1.Agapov, E. V., C. L. Murray, I. Frolov, L. Qu, T. M. Myers, and C. M. Rice. 2004. Uncleaved NS2-3 is required for production of infectious bovine viral diarrhea virus. J. Virol. 78:2414-2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ansari, I. H., L. M. Chen, D. Liang, L. H. Gil, W. Zhong, and R. O. Donis. 2004. Involvement of a bovine viral diarrhea virus NS5B locus in virion assembly. J. Virol. 78:9612-9623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bartelma, G., and R. Padmanabhan. 2002. Expression, purification, and characterization of the RNA 5′-triphosphatase activity of dengue virus type 2 nonstructural protein 3. Virology 299:122-132. [DOI] [PubMed] [Google Scholar]
- 4.Chambers, T. J., R. C. Weir, A. Grakoui, D. W. McCourt, J. F. Bazan, R. J. Fletterick, and C. M. Rice. 1990. Evidence that the N-terminal domain of nonstructural protein NS3 from yellow fever virus is a serine protease responsible for site-specific cleavages in the viral polyprotein. Proc. Natl. Acad. Sci. USA 87:8898-8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen, J., A. Noueiry, and P. Ahlquist. 2003. An alternate pathway for recruiting template RNA to the brome mosaic virus RNA replication complex. J. Virol. 77:2568-2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clever, J. L., D. Mirandar, Jr., and T. G. Parslow. 2002. RNA structure and packaging signals in the 5′ leader region of the human immunodeficiency virus type 1 genome. J. Virol. 76:12381-12387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clyde, K., and E. Harris. 2006. RNA secondary structure in the coding region of dengue virus type 2 directs translation start codon selection and is required for viral replication. J. Virol. 80:2170-2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frolov, I., and S. Schlesinger. 1996. Translation of Sindbis virus mRNA: analysis of sequences downstream of the initiating AUG codon that enhance translation. J. Virol. 70:1182-1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frolova, E., I. Frolov, and S. Schlesinger. 1997. Packaging signals in alphaviruses. J. Virol. 71:248-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goodfellow, I., Y. Chaudhry, A. Richardson, J. Meredith, J. W. Almond, W. Barclay, and D. J. Evans. 2000. Identification of a cis-acting replication element within the poliovirus coding region. J. Virol. 74:4590-4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goodfellow, I. G., D. Kerrigan, and D. J. Evans. 2003. Structure and function analysis of the poliovirus cis-acting replication element (CRE). RNA 9:124-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gorbalenya, A. E., A. P. Donchenko, E. V. Koonin, and V. M. Blinov. 1989. N-terminal domains of putative helicases of flavi- and pestiviruses may be serine proteases. Nucleic Acids Res. 17:3889-3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hall, R. A., D. J. Nisbet, K. B. Pham, A. T. Pyke, G. A. Smith, and A. A. Khromykh. 2003. DNA vaccine coding for the full-length infectious Kunjin virus RNA protects mice against the New York strain of West Nile virus. Proc. Natl. Acad. Sci. USA 100:10460-10464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harvey, T. J., W. J. Liu, X. J. Wang, R. Linedale, M. Jacobs, A. Davidson, T. T. Le, I. Anraku, A. Suhrbier, P. Y. Shi, and A. A. Khromykh. 2004. Tetracycline-inducible packaging cell line for production of flavivirus replicon particles. J. Virol. 78:531-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones, C. T., C. G. Patkar, and R. J. Kuhn. 2005. Construction and applications of yellow fever virus replicons. Virology 331:247-259. [DOI] [PubMed] [Google Scholar]
- 16.Khromykh, A. A., M. T. Kenney, and E. G. Westaway. 1998. trans-Complementation of flavivirus RNA polymerase gene NS5 by using Kunjin virus replicon-expressing BHK cells. J. Virol. 72:7270-7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khromykh, A. A., N. Kondratieva, J. Y. Sgro, A. Palmenberg, and E. G. Westaway. 2003. Significance in replication of the terminal nucleotides of the flavivirus genome. J. Virol. 77:10623-10629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khromykh, A. A., H. Meka, K. J. Guyatt, and E. G. Westaway. 2001. Essential role of cyclization sequences in flavivirus RNA replication. J. Virol. 75:6719-6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khromykh, A. A., P. L. Sedlak, K. J. Guyatt, R. A. Hall, and E. G. Westaway. 1999. Efficient trans-complementation of the flavivirus kunjin NS5 protein but not of the NS1 protein requires its coexpression with other components of the viral replicase. J. Virol. 73:10272-10280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khromykh, A. A., P. L. Sedlak, and E. G. Westaway. 2000. cis- and trans-acting elements in flavivirus RNA replication. J. Virol. 74:3253-3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khromykh, A. A., P. L. Sedlak, and E. G. Westaway. 1999. trans-Complementation analysis of the flavivirus Kunjin ns5 gene reveals an essential role for translation of its N-terminal half in RNA replication. J. Virol. 73:9247-9255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Khromykh, A. A., A. N. Varnavski, P. L. Sedlak, and E. G. Westaway. 2001. Coupling between replication and packaging of flavivirus RNA: evidence derived from the use of DNA-based full-length cDNA clones of Kunjin virus. J. Virol. 75:4633-4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khromykh, A. A., A. N. Varnavski, and E. G. Westaway. 1998. Encapsidation of the flavivirus kunjin replicon RNA by using a complementation system providing Kunjin virus structural proteins in trans. J. Virol. 72:5967-5977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khromykh, A. A., and E. G. Westaway. 1994. Completion of Kunjin virus RNA sequence and recovery of an infectious RNA transcribed from stably cloned full-length cDNA. J. Virol. 68:4580-4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khromykh, A. A., and E. G. Westaway. 1997. Subgenomic replicons of the flavivirus Kunjin: construction and applications. J. Virol. 71:1497-1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koonin, E. V. 1993. Computer-assisted identification of a putative methyltransferase domain in NS5 protein of flaviviruses and lambda 2 protein of reovirus. J. Gen. Virol. 74:733-740. [DOI] [PubMed] [Google Scholar]
- 27.Kummerer, B. M., and C. M. Rice. 2002. Mutations in the yellow fever virus nonstructural protein NS2A selectively block production of infectious particles. J. Virol. 76:4773-4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee, H., H. Shin, E. Wimmer, and A. V. Paul. 2004. cis-Acting RNA signals in the NS5B C-terminal coding sequence of the hepatitis C virus genome. J. Virol. 78:10865-10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu, W. J., H. B. Chen, and A. A. Khromykh. 2003. Molecular and functional analyses of Kunjin virus infectious cDNA clones demonstrate the essential roles for NS2A in virus assembly and for a nonconservative residue in NS3 in RNA replication. J. Virol. 77:7804-7813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu, W. J., H. B. Chen, X. J. Wang, H. Huang, and A. A. Khromykh. 2004. Analysis of adaptive mutations in Kunjin virus replicon RNA reveals a novel role for the flavivirus nonstructural protein NS2A in inhibition of beta interferon promoter-driven transcription. J. Virol. 78:12225-12235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu, W. J., P. L. Sedlak, N. Kondratieva, and A. A. Khromykh. 2002. Complementation analysis of the flavivirus Kunjin NS3 and NS5 proteins defines the minimal regions essential for formation of a replication complex and shows a requirement of NS3 in cis for virus assembly. J. Virol. 76:10766-10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu, W. J., X. J. Wang, D. C. Clark, M. Lobigs, R. A. Hall, and A. A. Khromykh. 2006. A single amino acid substitution in the West Nile virus nonstructural protein NS2A disables its ability to inhibit alpha/beta interferon induction and attenuates virus virulence in mice. J. Virol. 80:2396-2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu, W. J., X. J. Wang, V. V. Mokhonov, P. Y. Shi, R. Randall, and A. A. Khromykh. 2005. Inhibition of interferon signaling by the New York 99 strain and Kunjin subtype of West Nile virus involves blockage of STAT1 and STAT2 activation by nonstructural proteins. J. Virol. 79:1934-1942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lo, M. K., M. Tilgner, K. A. Bernard, and P. Y. Shi. 2003. Functional analysis of mosquito-borne flavivirus conserved sequence elements within 3′ untranslated region of West Nile virus by use of a reporting replicon that differentiates between viral translation and RNA replication. J. Virol. 77:10004-10014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lobigs, M. 1993. Flavivirus premembrane protein cleavage and spike heterodimer secretion require the function of the viral proteinase NS3. Proc. Natl. Acad. Sci. USA 90:6218-6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mackenzie, J. M., A. A. Khromykh, M. K. Jones, and E. G. Westaway. 1998. Subcellular localization and some biochemical properties of the flavivirus Kunjin nonstructural proteins NS2A and NS4A. Virology 245:203-215. [DOI] [PubMed] [Google Scholar]
- 37.Mackenzie, J. M., A. A. Khromykh, and E. G. Westaway. 2001. Stable expression of noncytopathic Kunjin replicons simulates both ultrastructural and biochemical characteristics observed during replication of Kunjin virus. Virology 279:161-172. [DOI] [PubMed] [Google Scholar]
- 38.Mackenzie, J. M., and E. G. Westaway. 2001. Assembly and maturation of the flavivirus Kunjin virus appear to occur in the rough endoplasmic reticulum and along the secretory pathway, respectively. J. Virol. 75:10787-10799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Markoff, L. 2003. 5′- and 3′-noncoding regions in flavivirus RNA. Adv. Virus Res. 59:177-228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nugent, C. I., K. L. Johnson, P. Sarnow, and K. Kirkegaard. 1999. Functional coupling between replication and packaging of poliovirus replicon RNA. J. Virol. 73:427-435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palmenberg, A. C., and J.-Y. Sgro. 1997. Topological organization of picornaviral genomes: statistical prediction of RNA structural signals. Semin. Virol. 8:231-241. [Google Scholar]
- 42.Rao, A. L. 2006. Genome packaging by spherical plant RNA viruses. Annu. Rev. Phytopathol 44:3.1-3.27. [DOI] [PubMed] [Google Scholar]
- 43.Simmonds, P., A. Tuplin, and D. J. Evans. 2004. Detection of genome-scale ordered RNA structure (GORS) in genomes of positive-stranded RNA viruses: implications for virus evolution and host persistence. RNA 10:1337-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stocks, C. E., and M. Lobigs. 1998. Signal peptidase cleavage at the flavivirus C-prM junction: dependence on the viral NS2B-3 protease for efficient processing requires determinants in C, the signal peptide, and prM. J. Virol. 72:2141-2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tchatalbachev, S., R. Flick, and G. Hobom. 2001. The packaging signal of influenza viral RNA molecules. RNA 7:979-989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tuplin, A., J. Wood, D. J. Evans, A. H. Patel, and P. Simmonds. 2002. Thermodynamic and phylogenetic prediction of RNA secondary structures in the coding region of hepatitis C virus. RNA 8:824-841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Venter, P. A., N. K. Krishna, and A. Schneemann. 2005. Capsid protein synthesis from replicating RNA directs specific packaging of the genome of a multipartite, positive-strand RNA virus. J. Virol. 79:6239-6248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Volkova, E., R. Gorchakov, and I. Frolov. 2006. The efficient packaging of Venezuelan equine encephalitis virus-specific RNAs into viral particles is determined by nsP1-3 synthesis. Virology 344:315-327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wengler, G. 1991. The carboxy-terminal part of the NS 3 protein of the West Nile flavivirus can be isolated as a soluble protein after proteolytic cleavage and represents an RNA-stimulated NTPase. Virology 184:707-715. [DOI] [PubMed] [Google Scholar]
- 50.Westaway, E. G., J. M. Mackenzie, M. T. Kenney, M. K. Jones, and A. A. Khromykh. 1997. Ultrastructure of Kunjin virus-infected cells: colocalization of NS1 and NS3 with double-stranded RNA, and of NS2B with NS3, in virus-induced membrane structures. J. Virol. 71:6650-6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Westaway, E. G., J. M. Mackenzie, and A. A. Khromykh. 2003. Kunjin RNA replication and applications of Kunjin replicons. Adv. Virus Res. 59:99-140. [DOI] [PubMed] [Google Scholar]
- 52.Westaway, E. G., J. M. Mackenzie, and A. A. Khromykh. 2002. Replication and gene function in Kunjin virus. Curr. Top. Microbiol. Immunol. 267:323-351. [DOI] [PubMed] [Google Scholar]
- 53.White, C. L., M. Thomson, and N. J. Dimmock. 1998. Deletion analysis of a defective interfering Semliki Forest virus RNA genome defines a region in the nsP2 sequence that is required for efficient packaging of the genome into virus particles. J. Virol. 72:4320-4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yamshchikov, V. F., and R. W. Compans. 1993. Regulation of the late events in flavivirus protein processing and maturation. Virology 192:38-51. [DOI] [PubMed] [Google Scholar]
- 55.You, S., D. D. Stump, A. D. Branch, and C. M. Rice. 2004. A cis-acting replication element in the sequence encoding the NS5B RNA-dependent RNA polymerase is required for hepatitis C virus RNA replication. J. Virol. 78:1352-1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu, L., and L. Markoff. 2005. The topology of bulges in the long stem of the flavivirus 3′ stem-loop is a major determinant of RNA replication competence. J. Virol. 79:2309-2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zuker, M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31:3406-3415. [DOI] [PMC free article] [PubMed] [Google Scholar]