

Abstract
Host protein synthesis is inhibited in cells infected with vesicular stomatitis virus (VSV). It has been proposed that viral mRNAs are subjected to the same inhibition but are predominantly translated because of their abundance. To compare translation efficiencies of viral and host mRNAs during infection, we used an enhanced green fluorescent protein (EGFP) reporter expressed from a recombinant virus or from the host nucleus in stably transfected cells. Translation efficiency of host-derived EGFP mRNA was reduced more than threefold at eight hours postinfection, while viral-derived mRNA was translated around sevenfold more efficiently than host-derived EGFP mRNA in VSV-infected cells. To test whether mRNAs transcribed in the cytoplasm are resistant to shutoff of translation during VSV infection, HeLa cells were infected with a recombinant simian virus 5 (rSV5) that expressed GFP. Cells were then superinfected with VSV or mock superinfected. GFP mRNA transcribed by rSV5 was not resistant to translation inhibition during superinfection with VSV, indicating that transcription in the cytoplasm is not sufficient for preventing translation inhibition. To determine if cis-acting sequences in untranslated regions (UTRs) were involved in preferential translation of VSV mRNAs, we constructed EGFP reporters with VSV or control UTRs and measured the translation efficiency in mock-infected and VSV-infected cells. The presence of VSV UTRs did not affect mRNA translation efficiency in mock- or VSV-infected cells, indicating that VSV mRNAs do not contain cis-acting sequences that influence translation. However, we found that when EGFP mRNAs transcribed by VSV or by the host were translated in vitro, VSV-derived EGFP mRNA was translated 22 times more efficiently than host-derived EGFP mRNA. This indicated that VSV mRNAs do contain cis-acting structural elements (that are not sequence based), which enhance translation efficiency of viral mRNAs.
All viruses rely on host translation machinery for synthesis of viral proteins. In addition, many viruses inhibit translation of host mRNA to suppress host antiviral responses (21). These viruses have developed a variety of mechanisms to inhibit host protein synthesis while viral mRNAs are preferentially translated. Understanding how viral mRNAs are translated at the same time that host mRNA translation is inhibited is critical for understanding viral replication. Furthermore, studying these viral mechanisms of translation has also increased our knowledge of cellular mechanisms for translational control. The goal of the experiments presented here was to address these questions for vesicular stomatitis virus (VSV).
VSV is widely studied as a model for other negative-sense, single-stranded RNA viruses. Following virus penetration and uncoating, viral mRNAs are synthesized by the viral RNA-dependent RNA polymerase. When viral proteins begin to accumulate, progeny viral genomes are replicated and are used for secondary transcription. mRNAs from both primary and secondary transcription are similar in structure to host mRNAs. They have a 5′ end containing 2′-O-methylated adenosine capped by 7mG linked by 5′-5′ triphosphate (18, 24, 25, 32, 33, 38). VSV mRNAs also have a 3′ poly(A) tail that is similar in length to that of cellular mRNAs (11, 13, 14). The synthesis of VSV mRNAs, including the 5′ and 3′ end modifications, is accomplished entirely in the cytoplasm of the infected cell (9).
During VSV infection, host translation is rapidly inhibited. This is likely a result of modification to the eukaryotic translation initiation factor 4F (eIF4F) (7, 8). However, it seems paradoxical that this modification would affect translation of host mRNAs but not of VSV mRNAs, since VSV mRNAs are structurally similar to host mRNAs. Yet, in cells infected with VSV, viral protein synthesis becomes predominant as host protein synthesis is inhibited (7, 8, 22, 26, 37, 40). The issue addressed here is whether VSV mRNAs are subject to the same inhibition of translation as host mRNAs. It has been suggested that the inhibition of translation in VSV-infected cells affects viral mRNAs as much as host mRNAs during infection but that the abundance of viral mRNAs leads to the predominance of viral protein synthesis (20). In the experiments described here, we compared the translation efficiencies of a viral-derived and host-derived reporter mRNA. We have determined that in VSV-infected cells, mRNAs derived from the viral genome are translated seven times more efficiently than host-derived mRNAs.
A common way for viral mRNAs to resist the inhibition of translation imposed on host mRNAs is through cis-acting sequences that recruit cellular or viral factors which promote translation. For example, picornavirus positive-strand genomes contain internal ribosome entry sites, in their 5′ untranslated regions (UTRs), that direct initiation of translation efficiently when cap-dependent translation is inhibited (16, 29, 30). Similarly, rotavirus mRNAs contain a cis-acting sequence in their 3′ UTRs that recruits a viral protein NSP3 which binds to eIF4F, competing with polyadenosine binding protein for a common binding site (6, 23, 28, 31, 36, 39). Another example is adenovirus late mRNAs, which contain cis-acting sequences in their 5′ UTRs that, along with adenovirus 100k protein, direct ribosome shunting (42-45). However, we have found that cis-acting sequences are not involved in preferential translation of VSV mRNAs. In contrast to these examples where cis-acting elements that function in resisting translation shutoff are embedded in mRNA nucleotide sequences, here we present a case where a negative-strand RNA virus produces mRNA that contains a cis-acting element that is not a nucleotide sequence. The cis-acting element acquired by VSV mRNAs allows VSV protein synthesis to predominate, in infected cells, by conferring high translation efficiencies to overcome translation inhibition.
MATERIALS AND METHODS
Cells and Viruses.
HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) containing 7.5% fetal bovine serum (FBS). Recombinant wild-type VSV (rwt virus) and recombinant VSV-expressing enhanced green fluorescent protein (EGFP) as a foreign gene (rVSV-EGFP) were grown in BHK cells as described previously (19). Recombinant simian virus 5 expressing GFP (rSV5-GFP) was isolated and grown in MDBK cells as described previously (12). The following cloning steps were required to produce the infectious cDNA clone corresponding to the rVSV-EGFP virus. The cassette vector pGEM.XK3.1 (19) was modified to include a VSV transcription control site closely followed by NotI and PacI restriction sites. Oligonucleotides of positive sense (5′ CGCGCTATGAAAAAAACTAACAGGCGGCCGCTTAATTAAGG TAC 3′) and negative sense (5′ CTTAATTCCGC GGCCGCCTGTTAGTTTTTTTCATAG 3′) were annealed and ligated into pGEM.XK3.1 prepared by digestion with BssHII and KpnI restriction endonucleases. The resulting plasmid was called pGEM-BNPK3.1. The DNA segment containing the EGFP coding region and 5′ multiple cloning site (MCS) was removed from the EGFP-N1 plasmid (Clontech) by digestion using BssHII and PacI and was ligated into pGEM-BNPK3.1. A DNA fragment containing the VSV-M gene, the new transcription stop-start site, and the EGFP gene was cleaved with SpeI and PacI and cloned into the recombinant VSV cDNA infectious clone pVSV.XK4.1 (41) to make the new infectious clone used to recover rVSV-EGFP, as described previously (19). Virus stocks were prepared as described previously (19). Infections with VSV were done at a multiplicity of infection of 10 PFU/cell in DMEM with 2% FBS. To infect HeLa cells with SV5-GFP, SV5 was added in DMEM without serum at 10 PFU/cell. After adding virus, cells were incubated for 1 h before replacing infection media with DMEM containing 2% FBS. The rwt virus was then added 11 h later in DMEM with 2% FBS at a multiplicity of infection of 10 PFU/cell.
EGFP plasmids.
The VSV-P gene 5′ UTR was cloned into pEGFP-N1 (Clontech) using complementary oligonucleotides of positive sense (5′ TTAAGCATAGGGATAGAAAAGACAGGATATTAGTTGTTCTTTATTCGCGCCTTAATTAATTAACTT 3′) and negative sense (5′ GTACAAGTAATTAATTAAGGCGCGAATAAAGAACAACTAATATCCTGTCTTTTCTATCCCTATGC 3′) that were annealed and ligated into pEGFP-N1 cleaved with AflII and BsrGI. The EGFP-N1 plasmid 3′ UTR was then replaced with a sequence of the VSV G gene 3′ UTR modified to include a eukaryotic poly(A) signal. Oligonucleotides of positive sense (5′ GTACAAGTAACCAACCAAGGCGCGAATAAAGAACAACTAATATCCTGTCTTTTCTATCCCTATGCTTAATAC 3′) and negative sense (5′ TTAAGCATAGGGATAGAAAAGACAGGATATTAGTTGTTCTTTATTCGCGCCTTAATTAATTACTT 3′) were annealed and ligated into the modified EGFP-N1 plasmid that was prepared by digestion with BsrGI and DraIII.
Metabolic labeling.
Approximately 7 × 105 cells grown in six-well dishes were washed twice with DMEM without methionine and then incubated in DMEM without methionine for 0.5 h. Cells were pulse-labeled for one hour in DMEM containing 200 μCi/ml [35S]methionine. Cells were then harvested for immunoprecipitation using 500 μl radioimmunoprecipitation assay (RIPA) buffer (0.15 M NaCl, 1% deoxycholic acid, 1% Triton X-100, 10 mM Tris-Cl, pH 7.4, and 0.1% sodium dodecyl sulfate [SDS]) with 1 mg/ml bovine serum albumin (BSA), 10 mM benzamidine, and 10 mM phenylmethylsulfonyl fluoride. Plates containing RIPA buffer were rocked gently until cells were visibly lifted from the dish. Lysates were then centrifuged at 20,000 × g for 15 min at 4°C. For analysis of total protein synthesis, cells were harvested following pulse-labeling using 500 μl RIPA buffer without BSA, and 360 μl of cell extract was added to 40 μl of 10× SDS-polyacrylamide gel electrophoresis (PAGE) sample loading buffer.
Immunoprecipitation.
Immunoprecipitation of EGFP was performed by adding 3.8 μg goat anti-GFP (RDI; code RDI-GRNFP3abg) to 100 μl of cell lysate. Samples were incubated overnight at 4°C. Protein G-Sepharose (Sigma; 20 μl) in NETN buffer (20 mM Tris-Cl, pH 8.0, 1 mM EDTA, 150 mM NaCl, 0.5% NP-40, and 4% BSA) was added and incubated for 1 h. Samples were centrifuged at 500 × g at 4°C, and pellets were washed five times with 400 μl of RIPA buffer with high SDS (1% SDS). SDS loading buffer (5 μl) was added to final pellets, and samples were heated to ∼95°C and run on 12% SDS-PAGE gels. Gels were dried and analyzed by phosphor imaging (Molecular Dynamics). Quantitation was performed using ImageQuant 5.2 (Molecular Dynamics).
Northern blotting.
RNA was harvested from 6 × 106 HeLa cells using 3 ml of TRIzol (Invitrogen), according to the manufacturer's specifications. RNA (5 μg) harvested from stably transfected cells or 0.125 μg of RNA harvested from HeLa cells infected with rVSV-EGFP was run on a 1.2% glyoxal agarose gel. The gel was then incubated in 800 ml of transfer buffer (0.01 M NaOH, 3 M NaCl) for 20 min and then transferred to a GeneScreen Plus (PerkinElmer) hybridization transfer membrane by upward capillary transfer (35). [α-32P]dCTP-labeled EGFP probe was prepared using a prime-a-gene kit (Promega). Membranes were probed using ExpressHyb hybridization solution (BD Biosciences Clontech) according to the manufacturer's specifications. Membranes were analyzed by phosphorimaging.
Polysome profiles.
Cells were treated with puromycin (puromycin added to media to 360 μM) or mock treated 1 h prior to harvesting. Several minutes prior to harvesting cells, cycloheximide (CHX) was added to the media to a concentration of 0.1 mg/ml. HeLa cells (1.5 × 107 to 1.8 × 107) were prepared by scraping off the culture dish in ice-cold PBS containing 0.1 mg/ml CHX. Cells were pelleted and resuspended in ice-cold PBS containing 0.1 mg/ml CHX. Cells were pelleted again and resuspended in 0.2 ml RSB buffer (10 mM NaCl, 3 mM MgCl2, and 10 mM Tris-Cl, pH 7.4) containing 20% vanadyl adenosine ribonucleoside complex and 0.1 mg/ml CHX. Cells were incubated on ice for 5 min followed by addition of 0.2 ml RSB buffer containing 1% deoxycholic acid and 2% Tween 40; Cells were briefly vortexed before and after addition of RSB containing detergents. Cells were again incubated on ice for 5 min followed by brief vortexing and centrifugation at 2000 × g for 15 min at 4°C to pellet nuclei. Cytoplasmic fractions were transferred to a new tube, and 0.1 ml 5× HSB (5× HSB contains 2.5 M NaCl, 250 mM MgCl2, and 50 mM Tris-Cl, pH 7.4) was added and solutions were quickly mixed. Solutions were then carefully overlaid onto ∼10% to 50% sucrose gradients in 1× HSB. Gradients were centrifuged at 37,000 rpm for 1.75 h at 4°C. Polysome profiles were analyzed by pumping off the top of the gradient using an AUTO DENSI-FLOW (LABCONCO) gradient pump, through an EM-1 Econo UV monitor (Bio-Rad). Absorbance at 254 nm was recorded using a Rec-111 (GE Healthcare) recorder. Sixteen fractions were collected from each gradient using a Frac-100 (Pharmacia) fraction collector. RNA was precipitated by adding 20 μl glycogen and 0.5 ml isopropanol followed by overnight incubation at −20°C. RNA was pelleted by centrifugation for 20 min at 12,000 × g at 4°C. Pellets were washed with 70% ethanol and briefly centrifuged. Pellets were suspended in 300 μl 1% N-leuroyl sarcosine (N-LS) 10 μl proteinase K (20 mg/ml) was added, followed by incubation for 30 min at 37°C. GTC (300 μl; 4 M guanidine thiocyanate, 25 mM sodium citrate, pH 7.0, and 0.5% N-LS, with β-mercaptoethanol added to 0.7% immediately prior to use) and 600 μl isopropanol were added and mixed, and solutions were incubated at −20°C for longer than 30 min. RNA was pelleted by centrifugation at 12,000 × g for 15 min at 4°C, washed with 70% ethanol, and resuspended in 10 μl distilled water and 10 μl NorthernMax-Gly sample loading dye (Ambion). EGFP mRNA was analyzed by Northern blotting as described above.
In vitro translation.
RNA harvested eight hours postinfection with TRIzol (Invitrogen) was used to direct translation in rabbit reticulocyte lysates (Promega). Reactions were 0.1 ml in total volume with 27 μg total RNA or 1.6 μg of poly(A) RNA added. Poly(A) RNA was isolated from total RNA using oligo(dT)-cellulose columns (Amersham Biosciences) with two rounds of purification. Translation reactions were carried out in a water bath at 30°C for 2 h and stopped by incubating on ice. Three microliters of each reaction was removed for analysis of the all products of protein synthesis. Remaining volumes were diluted in 1.2 ml RIPA buffer plus 1 mg/ml BSA, 10 mM benzamidine, and 10 mM phenylmethylsulfonyl fluoride, and EGFP was immunoprecipitated as described above.
RESULTS
Lodish and Porter suggested in 1980 that VSV mRNA is subject to the same inhibition of translation as host mRNA during infection, based on their observation that viral and host mRNAs are associated with similar numbers of ribosomes in VSV-infected cells (20). They proposed that an abundance of viral mRNAs, rather than preferential translation, leads to the predominance of viral protein synthesis in VSV-infected cells. If this hypothesis is correct, then the rates of viral and host protein synthesis should be the same after normalizing for (dividing by) mRNA levels. To compare the translation efficiencies of viral and host mRNAs during infection, we compared the translation efficiencies of an EGFP reporter expressed from a recombinant virus, rVSV-EGFP (genome depicted in Fig. 1A), or from the nucleus of HeLa cells stably transfected with a plasmid that expresses EGFP driven by a cytomegalovirus immediate early promoter. HeLa cells stably expressing EGFP are referred to as HeLa-EGFP cells (a representation of incorporated plasmid DNA is shown in Fig. 1B). Using reporters with identical coding regions to measure viral and host translation efficiencies reduces complications from normalization for different proteins or from various effects of the coding region secondary structure or codon usage, etc., on translation. Relative translation efficiencies were determined by the ratios of the rates of protein synthesis to mRNA levels.
FIG. 1.

(A) Genome of rVSV-EGFP recombinant virus that expresses EGFP as a foreign gene. Viral genes and leader (le) and trailer (tr) sequences are indicated. (B) EGFP-N1 plasmid DNA. EGFP mRNA synthesis is directed by a cytomegalovirus (CMV) promoter in stably transfected HeLa-EGFP cells. (C) Analysis of EGFP synthesis. HeLa cells were infected with rVSV-EGFP, and HeLa-EGFP cells were mock infected or infected with rwt virus. Cells were pulse-labeled with [35S]methionine at 7.5 h postinfection and harvested at 8.5 h postinfection. EGFP was immunoprecipitated and analyzed by SDS-PAGE and phosphorimaging. Duplicate immunoprecipitates are shown. (D) Analysis of EGFP mRNA levels. HeLa cells were infected with rVSV-EGFP, and HeLa-EGFP cells were mock infected or infected with rwt virus. RNA was harvested at 8 h postinfection, and EGFP mRNA levels were analyzed by Northern blotting using a [32P]dCTP-labeled EGFP probe and phosphorimaging. Samples were run in duplicate. (E) Translation efficiencies (rates of protein synthesis divided by mRNA levels) of EGFP mRNAs shown relative to HeLa-EGFP cells that were mock infected. Data are shown as means ± SEs for four or five experiments.
HeLa cells (not transfected) were infected with rVSV-EGFP, and HeLa-EGFP cells were either mock infected or infected with rwt virus that does not express EGFP. Cells were pulse-labeled with [35S]methionine to measure the rate of EGFP synthesis at eight hours postinfection, a time at which host protein synthesis is effectively inhibited and viral protein synthesis is near maximal (7). EGFP was analyzed by immunoprecipitation, SDS-PAGE, and phosphorimaging. Figure 1C shows a representative phosphorimage used to determine EGFP synthesis rates. EGFP was synthesized at high levels in HeLa cells infected with rVSV-EGFP. Therefore, these lysates were diluted one to forty prior to immunoprecipitation, so that EGFP intensities in the phosphorimage would be comparable to those from the stably transfected HeLa-EGFP cells. In the phosphorimage (Fig. 1C), EGFP labeling intensity from the lysates (diluted 40-fold) of cells infected with rVSV-EGFP was similar to that in stably transfected HeLa-EGFP cells that were mock infected. Immunoprecipitates of labeled EGFP from HeLa-EGFP cells infected with rwt virus contained two bands. The upper band was a background band of VSV M protein, which was observed in VSV-infected cell lysates following immunoprecipitation using both specific and nonspecific antibodies. The lower band was labeled EGFP, which was synthesized at a much lower rate in rwt virus-infected cells than in uninfected HeLa-EGFP cells.
Quantitation of multiple experiments showed that the rates of synthesis of EGFP in HeLa-EGFP cells was reduced to 18% of the rates for the mock-infected control at eight hours postinfection with rwt virus (Table 1). While synthesis of EGFP in these cells was greatly reduced by VSV infection, EGFP mRNA levels, measured by Northern blotting, were similar (Fig. 1D), indicating that inhibition was at the level of translation, as expected (20). In addition, the signal of EGFP mRNA from rVSV-EGFP-infected cells (Fig. 1D), which was also diluted one to forty, was lower than EGFP signal intensity from HeLa-EGFP cells. This result, combined with the result shown in Fig. 1C, indicates that EGFP mRNA expressed from rVSV-EGFP was translated more efficiently than EGFP mRNA expressed from stably transfected plasmid DNA in both mock-infected and rwt virus-infected HeLa-EGFP cells.
TABLE 1.
EGFP translation efficiencya
| Cell type | Relative rate of EGFP synthesis | Relative level of EGFP mRNA | Translation efficiencyb |
|---|---|---|---|
| HeLa-EGFP mock | 1.0 | 1.0 | 1.0 |
| HeLa-EGFP rwt | 0.18 ± 0.03 | 0.62 ± 0.09 | 0.29 ± 0.21 |
| HeLa VSV-MCS | 22 ± 4.5 | 9.4 ± 2.0 | 2.3 ± 0.30 |
All values shown ± SE.
Translation efficiencies of EGFP were determined by dividing the relative rates of EGFP synthesis by the relative levels of mRNA.
The relative translation efficiencies of EGFP mRNAs were determined by quantitating data from multiple experiments; similar to the quantitations in Fig. 1C and 1D. These data are shown relative to HeLa-EGFP mock-infected cells in Table 1, after correction for the dilution factors in Fig. 1. To determine the relative translation efficiencies of EGFP mRNA, relative rates of EGFP protein synthesis, determined by pulse-labeling, were divided by the relative levels of EGFP mRNA from the Northern blots (Fig. 1E and Table 1). Comparison of relative translation efficiencies shows that infection with rwt virus reduced EGFP translation efficiency in HeLa-EGFP cells more than threefold. In contrast, EGFP mRNA encoded by rVSV-EGFP was translated over twofold more efficiently than EGFP mRNA in mock-infected HeLa-EGFP cells and around sevenfold more efficiently than EGFP mRNA in rwt virus-infected cells (Fig. 1E and Table 1). These results show that EGFP mRNA expressed from the viral genome is translated much more efficiently than host-derived EGFP mRNA during VSV infection.
Analysis of polysome profiles by Lodish and Porter suggested that VSV mRNAs are associated with a similar number of ribosomes as host mRNAs of similar size, following infection (20). This result led them to conclude that viral mRNAs are subject to the same inhibition of translation initiation as host mRNAs in infected cells. Therefore, we analyzed the distribution of EGFP mRNA among polysomes, to compare the number of ribosomes associated with EGFP mRNAs derived from the rVSV-EGFP genome or from the nucleus in HeLa-EGFP cells. HeLa cells were infected with the rVSV-EGFP virus, and HeLa-EGFP cells were mock infected or infected with rwt virus. Eight hours postinfection, cells were harvested and lysates were separated in sucrose density gradients. Polysome profiles were analyzed by monitoring the absorbance at 254 nm (Fig. 2A). Sixteen fractions were collected from each sucrose gradient, RNA was extracted, and EGFP mRNA was analyzed by Northern blotting and phosphorimaging (Fig. 2B). To determine the distribution of EGFP mRNA within the polysome profile, intensities of EGFP mRNA in Northern blots were quantitated and are shown graphically in Fig. 2C.
FIG. 2.

(A) Polysome profiles of mock-infected HeLa-EGFP cells, HeLa-EGFP cells infected with rwt virus, or HeLa cells infected with rVSV-EGFP. The bracket in the polysome profile from mock-infected cells shows the region of the gradient, containing monosomes and polysomes, that we are focused on. Abs, absorbance. (B) Distributions of EGFP mRNAs within sucrose gradients. Eight hours postinfection, cells were harvested for analysis of polysome profiles. Sucrose gradients were collected in 16 fractions, and EGFP mRNA levels in each fraction were analyzed by Northern blotting using a [32P]dCTP-labeled EGFP probe and phosphorimaging. Fractions from the tops of the gradients are on the left. (C) Quantitations of EGFP mRNA distributions from multiple experiments were analyzed to show the distributions, as a percentage of the total EGFP mRNA in each fraction (± SE), for HeLa cells infected with rVSV-EGFP (○) or for HeLa-EGFP cells that were mock infected (▪) or infected with rwt virus (▴).
The polysome profile of mock-infected cells indicated that most RNA is found in large polysomes, estimated to contain eight or nine ribosomes (Fig. 2A). In cells infected with rVSV-EGFP or rwt viruses, the polysome profiles were almost identical. In each case, the size of the polysomes was reduced while free ribosomal subunits were more abundant, as expected (20, 34). EGFP mRNA from mock-infected cells followed a distribution similar to the bulk of RNA and was distributed mainly in large polysomes (Fig. 2B). When these cells were infected with rwt virus, EGFP mRNA shifted to the lighter region of the sucrose gradient, suggesting that fewer ribosomes were associated with each mRNA. In HeLa cells infected with the rVSV-EGFP, EGFP mRNA transcribed from the viral genome appeared intermediate in its distribution, in that more mRNA was distributed throughout the gradient, although it was mainly distributed in the light region of the gradient, indicating that in infected cells, viral-derived and host-derived EGFP mRNAs were all found in complexes with similar sedimentation velocities. The similarity in distribution of EGFP mRNAs in infected cells is apparent in Fig. 2C, which shows quantitation of Northern blots from multiple experiments.
Similar distributions of viral-derived and host-derived EGFP mRNAs in infected cells (Fig. 2C) are consistent with Lodish and Porter's data. However, interpretation of these sedimentation data are further complicated by the data of Rosen et al. (34), who showed that in VSV-infected cells, a large portion of mRNA was found in nontranslating messenger ribonucleoprotein particles (mRNPs). This raises the possibility that the distribution of EGFP mRNA shown in Fig. 2C may largely represent translationally inactive mRNPs rather than active polysome complexes. In an effort to distinguish mRNPs from active polysome complexes and estimate the proportion of mRNA being translated, we treated cells with the translation inhibitor puromycin to disrupt polysomes and obtain distributions of EGFP mRNPs. Puromycin causes dissociation of elongating ribosomes from mRNA. HeLa cells were infected with rVSV-EGFP, and HeLa-EGFP cells were mock infected or infected with rwt virus. At seven hours postinfection, the cells were treated with puromycin or mock treated for one hour before harvesting cytoplasmic extracts, at eight hours postinfection, for analysis by sucrose density gradient centrifugation. Distributions of total RNA (A254) and of EGFP mRNA (from Northern blots) from cells treated with puromycin and from mock-treated cells are shown in Fig. 3, panels A through D.
FIG. 3.
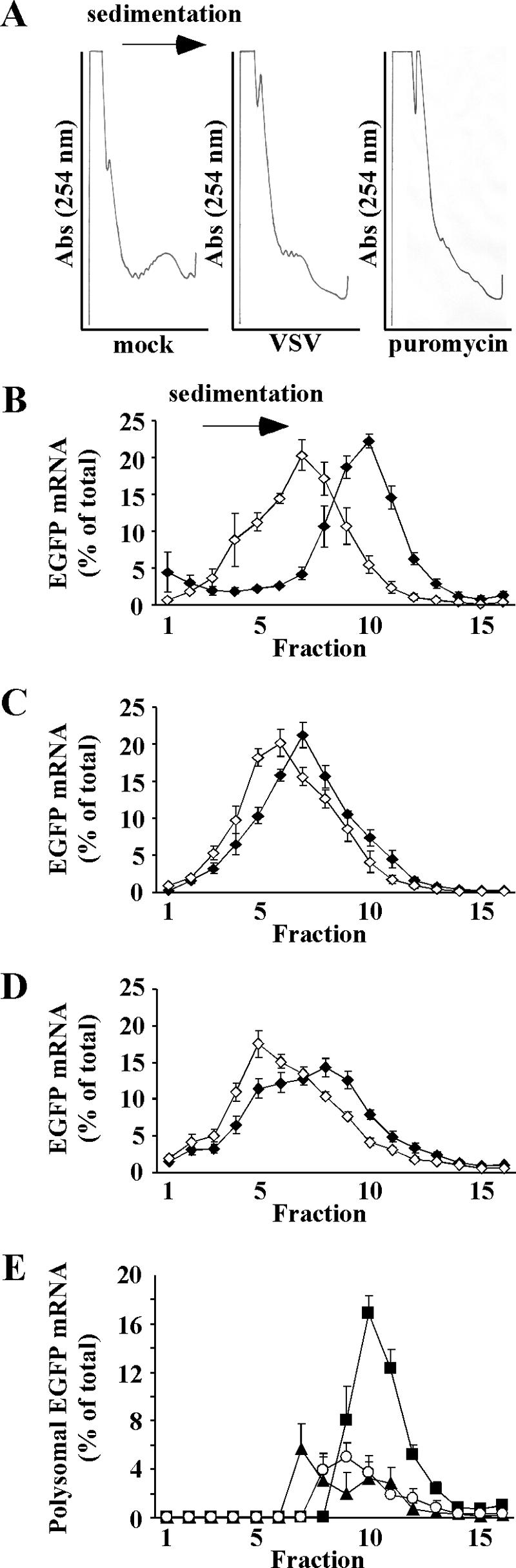
(A) Polysome profiles of HeLa-EGFP cells that were mock infected, infected with rwt virus, or mock infected and treated with puromycin. Abs, absorbance. (B to D) EGFP mRNA distributions in HeLa-EGFP cells that were mock infected, HeLa-EGFP cells infected with rwt virus, or HeLa cells infected with rVSV-EGFP, respectively, that were untreated (⧫) or treated with puromycin (⋄). Sixteen fractions were collected from sucrose gradients, and EGFP mRNA levels were analyzed by Northern blotting. EGFP mRNA in each fraction is shown as an average of multiple experiments ± SE. (E) Distributions of EGFP mRNAs in large polysomes (± SE); determined by subtracting EGFP mRNA signals in each fraction in puromycin-treated cells from the corresponding fraction in untreated cells, for HeLa-EGFP cells that were mock infected (▪), HeLa-EGFP cells infected with rwt virus (▴), or HeLa cells infected with rVSV-EGFP (○).
As expected, puromycin treatment caused a shift in the total RNA distributions towards the region of the sucrose gradient containing individual ribosome subunits (Fig. 3A). In mock-infected HeLa-EGFP cells, puromycin treatment caused a large redistribution of EGFP mRNA from the region of the gradient containing large polysomes to the lighter region of the gradient where mRNPs and small polysomes sediment (Fig. 3B). In contrast, in HeLa-EGFP cells infected with rwt virus, there was only a small difference in the distributions of EGFP mRNAs from puromycin-treated and mock-treated cells (Fig. 3C). This indicates that a large portion of EGFP mRNA, in infected cells, was associated with mRNP complexes that sediment with velocities similar to those for nontranslating mRNPs. These complexes could be small polysomal complexes or nontranslating mRNPs. Similarly, there was little difference in sedimentations of EGFP mRNAs from cells infected with rVSV-EGFP that were either treated with puromycin or mock treated (Fig. 3D). To estimate the percentage of mRNA in polysomes, EGFP mRNP distributions from puromycin-treated cells were subtracted from EGFP mRNA distributions from untreated cells to eliminate the contribution from inactive mRNPs (Fig. 3E). Negative values where EGFP mRNPs were more abundant than polysomes were plotted as zero to reflect that most EGFP mRNA was not in polysomes in these fractions. The resulting distributions represent an estimate of the minimum proportion of EGFP mRNA in active polysomes (Fig. 3E). Since small polysomes and mRNPs sediment with similar velocities, small polysomes may be underrepresented by this method. In mock-infected HeLa-EGFP cells, large polysomes (polysomes that sediment faster than nontranslating mRNPs) containing EGFP mRNA were more abundant than in rwt virus-infected cells. The total amount of EGFP mRNA in large polysomes was calculated as the sum of EGFP mRNA in all fractions in Fig. 3E. In mock-infected cells, 48% of EGFP mRNA was found in large polysomes while in infected cells, approximately 18% of EGFP mRNA was associated with large polysomal complexes, regardless of the mRNA source. These data indicate that viral mRNAs are no more likely to be in large polysomes than host mRNAs, which is consistent with Lodish and Porter's findings (20). However, the higher translation efficiency of EGFP mRNA expressed from rVSV-EGFP (Fig. 1) suggests that viral mRNAs in these polysomes are more efficient in directing protein synthesis than host polysomal mRNAs.
Since VSV mRNAs are synthesized by the viral RNA polymerase in the cytoplasm, the lack of association of viral mRNAs with host factors in the nucleus may allow them to be preferentially translated during infection. To determine if transcription in the cytoplasm confers resistance to translation inhibition during VSV infection, HeLa cells were infected with another virus, negative-strand recombinant simian virus 5, that expressed GFP, (rSV5-GFP). Eleven hours postinfection with rSV5-GFP, cells were mock superinfected or superinfected with VSV. Approximately six hours postsuperinfection, cells were pulse-labeled with [35S]methionine to measure the rate of GFP synthesis, or RNA was harvested for Northern blotting to measure GFP mRNA levels (Fig. 4A). Figure 4B shows a representative phosphorimage. In this image, it is apparent that GFP was synthesized in cells that were mock superinfected, but in cells superinfected with VSV, GFP synthesis was reduced dramatically. Superinfection with VSV had little if any effect on GFP mRNA levels (Fig. 4C), as expected. Data from several experiments were quantitated to determine the relative translation efficiencies of GFP in cells infected with rSV5-GFP and superinfected with VSV relative to cells that were mock superinfected. Superinfection with VSV reduced GFP translation efficiency to below 20% of mock-superinfected levels (Fig. 4D). These results indicate that mRNAs transcribed in the cytoplasm by another viral RNA polymerase are not resistant to inhibition of translation during VSV infection.
FIG. 4.
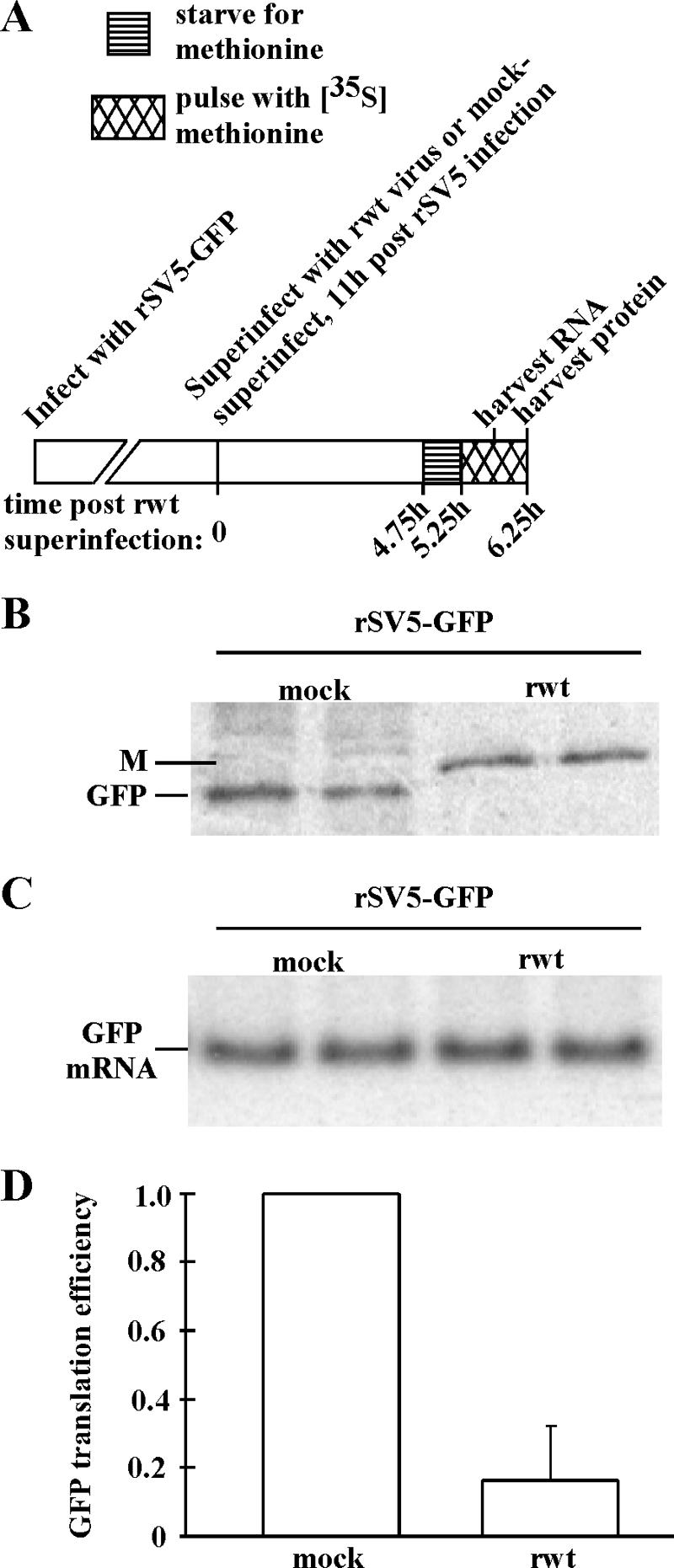
(A) Experimental design for infecting HeLa cells with rSV5-GFP followed by superinfection with rwt virus or mock superinfection. Cells were starved for methionine for 30 min, pulse-labeled with [35S]methionine for 1 h, and harvested at 6.25 h postsuperinfection—or RNA was harvested at 6 h postsuperinfection. (B) Labeled GFP analyzed by immunoprecipitation, SDS-PAGE, and phosphorimaging. In rwt virus-superinfected cells, VSV M protein is the prominent band. Duplicate samples are shown. (C) Phosphorimage of Northern blot for GFP mRNA; samples were run in duplicate. (D) Translation efficiencies of GFP in rwt virus-superinfected cells relative to mock-superinfected cells; efficiencies were determined by dividing translation rates by mRNA levels (± SEs for three experiments).
For many viruses, cis-acting sequences in viral mRNAs recruit translation machinery and allow preferential translation over host mRNAs. VSV mRNAs have short 5′ UTRs, all of which share a conserved sequence at the 5′ end (AACAG) followed by five to forty-five nucleotides that are specific to each mRNA. To determine if the length of the VSV 5′ UTR is involved in preferential translation, a recombinant virus was generated that expressed EGFP from an mRNA with a twenty-nine-nucleotide 5′ UTR containing the sequence from the P gene 5′ UTR (rVSV-P; Fig. 5A); complete sequences of UTRs are shown in Table 2. The rate of translation of EGFP in cells infected with this virus was compared to the rate in cells infected with the rVSV-EGFP virus analyzed in Fig. 1 to 3. The 5′ UTR of the EGFP mRNA expressed by rVSV-EGFP contains the 5′ AACAG sequence required for viral transcription followed by a ninety-nine-nucleotide vector-derived sequence consisting primarily of a multiple cloning site (rVSV-MCS; Fig. 5A). The 3′ UTR for both viruses was derived from the VSV G gene. Cells were infected with either virus and pulse-labeled with [35S]methionine at approximately eight hours postinfection followed by immunoprecipitation of EGFP. As shown in Fig. 5B and C, EGFP was synthesized at similar rates in cells infected with either virus. This result indicates that the length of the 5′ UTR (in this size range) has little if any effect on the translation of mRNA transcribed from the viral genome.
FIG. 5.

(A) Recombinant viruses rVSV-P and rVSV-MCS that stably express EGFP from mRNA containing either the VSV P gene 5′ UTR or the 5′ UTR from the EGFP-N1 vector, respectively. (B) Analysis of EGFP translated from viral-derived mRNA. HeLa cells were infected with recombinant viruses and pulse-labeled at 7.5 h postinfection with [35S]methionine and harvested 8.5 h postinfection. EGFP was immunoprecipitated and analyzed by SDS-PAGE and phosphorimaging. (C) EGFP synthesis in HeLa cells infected with recombinant viruses. Synthesis is shown as an average of two experiments ± SE, relative to synthesis in HeLa cells infected with rVSV-P virus. (D) Plasmid DNA that directs synthesis of EGFP-N1 and EGFP-P-G mRNAs in stably transfected HeLa cells. EGFP-N1 mRNA contains negative control UTRs from the pEGFP-N1 plasmid. EGFP-P-G mRNA contains the VSV P gene 5′ UTR and the VSV G gene 3′ UTR. (E) EGFP synthesis in stably transfected cells mock-infected or infected with rwt virus. At 7.5 h postinfection the cells were pulse-labeled with [35S]methionine, and they were harvested at 8.5 h postinfection. EGFP was analyzed by immunoprecipitation, SDS-PAGE, and phosphorimaging. Duplicate immunoprecipitates are shown. (F) EGFP translation efficiencies in rwt virus-infected cells relative to mock-infected cells, determined by normalizing EGFP synthesis to mRNA levels (± SEs for three experiments).
TABLE 2.
Untranslated regions of EGFP reporter mRNAs
| mRNA | 5′ UTR | 3′ UTR |
|---|---|---|
| EGFP-N1 | 5′-AGAUCCGCUAGCGCUACCGGACUCAGAUCUCGAGCUCAAGCUUCGAAUUCUGCAGUCGACGGUACCGCGGGCCCGGGAUCCACCGGUCGCCACC(AUG) | (UAA)AGCGGCCGCGACUCUAGAUCAUAAUCAGCCAUACCACAUUUGUAGAGGUUUUACUUGCUUUAAAAAACCUCCCACACCUCCCCCUGAACCUGAAACAUAAAAUGAAUGCAAUUGUUGUUGUUAACUUGUUUAUUGCAGCUUAUAAUGGUUACAAAUAAGCAAUAGCAUCACAAAUUUCACAAAUAAAGCAUUUUUUUCACUGC-poly(A)-3′ |
| EGFP-P-G | 5′-AAACAGAUAUCAGCUAGCCGGUCGCCACC(AUG) | (UAA)UUAAUUAAGGCGCGAAUAAAGAACAACUAAUAUCCUGUCUUUUCUAUCCCUAUGCUUAAUAC-poly(A)-3′ |
| rVSV-EGFP (rVSV-MCS) | 5′-AACAGAGAUCCGAUAGCGCUACCGGACUCAGAUCUCGAGCUCAAGCUUCGAAUUCUGCAGUCGACGGUACCGCGGGCCCGGGAUCCACCGGUCGCCACC(AUG) | (UAA)UUAAUUAAGGCGCGCCUCUCGAACAACUAAUAUCCUGUCUUUUCUAUCCCUAUGAAAAAAA-poly(A)-3′ |
| rVSV-P | 5′-AACAGAUAUCAGCUAGCCGGUCGCCACC(AUG) | (UAA)UUAAUUAAGGCGCGCCUCUCGAACAACUAAUAUCCUGUCUUUUCUAUCCCUAUGAAAAAAA-poly(A)-3′ |
To determine if VSV cis-acting sequences in mRNA prevent the inhibition of translation of host-derived mRNAs, the VSV P gene 5′ UTR and G gene 3′ UTR (Fig. 5A) were substituted for the vector-derived 5′ and 3′ UTRs in pEGFP-N1 (pEGFP-P-G; Fig. 5D). The translation efficiency of this mRNA was compared to that of mRNA derived from pEGFP-N1, which lacks viral sequences. Translation rates were measured by pulse-labeling with [35S]methionine and immunoprecipitation and were divided by relative mRNA levels from Northern blots to determine translation efficiencies (Fig. 5E and F). It is apparent in Fig. 5E that for each construct, the EGFP translation rate was greatly reduced in VSV-infected cells relative to mock-infected cells. The results shown in Fig. 5F indicate that translation efficiency of each construct was reduced during VSV infection and that mRNA flanked by viral UTRs was not translated any better than control mRNA during VSV infection.
If VSV UTRs contained cis-acting sequences involved in translation, then we would expect a reporter flanked by viral UTRs to be more resistant to inhibition of translation during VSV infection than a control mRNA. However, translation of mRNA with VSV UTRs was inhibited as much as translation of control mRNA (Fig. 5F). Therefore, we concluded that viral UTRs do not contain cis-acting sequences involved in preferential translation.
To determine if another property of VSV mRNAs, besides their sequence, allows them to be translated more efficiently than host-transcribed mRNAs, RNA extracted from cells was translated in vitro. RNA was extracted from HeLa-EGFP cells that were mock infected or infected with rwt virus and from HeLa cells infected with rVSV-EGFP. Total RNA (27 μg) was used to direct translation in rabbit reticulocyte lysates in the presence of [35S]methionine. Translation of EGFP was analyzed by immunoprecipitation, SDS-PAGE, and phosphorimaging (Fig. 6, lanes 1 to 5). Total protein synthesis was also analyzed (Fig. 6, lanes 6 to 10) by running 3% of the total in vitro translation reaction along with the immunoprecipitated products. Translation of RNA from mock-infected HeLa-EGFP cells (Fig. 6, lane 1) resulted in two immunoprecipitated bands: EGFP and an unidentified host protein that migrated slightly slower than EGFP (indicated with an asterisk). The immunoprecipitated sample from HeLa-EGFP cells infected with rwt virus (Fig. 6, lane 2) also contained an intensely labeled M protein band that was nonspecifically precipitated. Translations of EGFP from mRNAs from mock- and rwt virus-infected HeLa-EGFP cells were similar (Fig. 6, lanes 1 and 2), consistent with previous reports that host RNA is not degraded during VSV infection (17, 20). Lane 3 shows translation of EGFP from RNA from HeLa cells infected with rVSV-EGFP. This RNA was mixed with RNA from mock-infected HeLa cells in a 1:20 ratio so that the amount of EGFP mRNA would be similar to the samples in lanes 1 and 2, while keeping the total amount of RNA constant at 27 μg. The striking result is the intense labeling of EGFP in this sample, indicating a much greater translation of EGFP from viral-derived mRNA than from host-derived mRNA in lanes 1 and 2. Lanes 4 and 5 show additional specificity controls: RNA from rwt virus-infected HeLa cells (1:20) and RNA from mock-infected HeLa cells alone.
FIG. 6.

Analysis of in vitro translation of RNA from infected cells. RNA isolated from infected cells at 8 h postinfection was translated in vitro in reticulocyte lysates in the presence of [35S]methionine. EGFP synthesis was analyzed by immunoprecipitation, SDS-PAGE, and phosphorimaging (lanes 1 to 5). All translation products (3% of total) were also analyzed (lanes 6 to 10). VSV M protein and an unknown host protein (*) were nonspecifically immunoprecipitated along with EGFP. Translation was directed by 27 μg of total RNA from the following: HeLa-EGFP cells that were mock infected (lanes 1 and 6), HeLa-EGFP cells infected with rwt virus for 8 h (lanes 2 and 7), HeLa cells infected with rVSV-EGFP (1.4 μg) and mock-infected HeLa cells (25.6 μg) (lanes 3 and 8), HeLa cells infected with rVSV-EGFP (1.4 μg) and HeLa cells infected with rwt virus for 8 h (25.6 μg) (lanes 4 and 9), or mock-infected HeLa cells (lanes 5 and 10).
Translation efficiencies of EGFP mRNA in the in vitro reactions were calculated from data similar to those in Fig. 6 and Northern blot analysis of EGFP mRNA in these samples (data not shown). EGFP mRNA from rVSV-EGFP was translated 22 ± 3 (standard error [SE]) times more efficiently than EGFP mRNA from HeLa-EGFP cells. These results were from five in vitro translation experiments using four different total RNA samples. Results for two experiments using poly(A) RNA purified by oligo(dT) chromatography were similar; VSV-transcribed RNA was translated 24 ± 9 (SE) times more efficiently than host-transcribed EGFP mRNA (data not shown). These results indicate that EGFP mRNA transcribed from the viral genome contains a structural element that enhances translation efficiency over that of mRNAs derived from the host. However, this element must be an mRNA feature other than sequence, since EGFP with VSV UTRs is translated with an efficiency similar to that of EGFP mRNA with control UTRs.
DISCUSSION
A striking feature of VSV infection is the rapid shutoff of host translation and the abundant synthesis of viral proteins. The inhibition of host translation is at the level of translation initiation (20) and is likely due to virus-induced modification of the cap-binding complex eIF4F (7, 8). The conclusion that host translation is inhibited at the level of translation initiation was based on analysis of polysome profiles, by Lodish and Porter, in which host mRNAs were found to be in smaller polysomes in VSV-infected cells compared to mock-infected cells (20). They also observed that viral mRNAs were present in the same size polysomes as host mRNAs of similar size, suggesting that viral mRNAs are subject to the same inhibition of translation as host mRNAs (20). In this model, the predominant synthesis of viral proteins is due to the high abundance of viral mRNAs. The question of how VSV protein synthesis predominates in infected cells, in the face of inhibition of host protein synthesis, has remained unresolved (3, 10).
It has been proposed that VSV has developed mechanisms to avoid the inhibition of translation (that limits host protein synthesis) of viral mRNAs (3). However, the idea that predominant synthesis of viral proteins is due simply to the abundance of viral mRNAs has not been ruled out previously. We addressed this question by comparing translation efficiencies of EGFP reporter mRNAs expressed from the host nucleus or from the viral genome as a foreign gene. We found that the translation efficiency of host-derived EGFP mRNA was decreased dramatically in VSV-infected cells compared to mock-infected cells, as expected. We also found that VSV-derived EGFP mRNA was translated seven times more efficiently than host-derived EGFP mRNA in infected cells. This indicates that the predominance of VSV protein synthesis is not a simple result of viral mRNA abundance.
Even though EGFP mRNAs derived from the VSV genome were translated more efficiently than those transcribed in the nucleus, we found that in infected cells, VSV-derived mRNAs followed a similar pattern of polysomal association as EGFP mRNAs from the nucleus (Fig. 2). These results were consistent with Lodish and Porter's observation that viral mRNAs were associated with similar numbers of ribosomes as host mRNAs of similar length, in infected cells (20). Although Lodish and Porter found that during infection, host and viral mRNAs were in polysomes of similar sizes, Rosen et al. found later, by treating cells with puromycin, that most mRNAs from VSV-infected cells were in nontranslating mRNPs (34). mRNPs containing VSV mRNAs were isolated by Rosen et al. and found to contain VSV N protein as a well as a host protein with an apparent molecular weight of 90,000 (34). It is likely that these mRNPs also contain other RNA binding proteins that were not labeled by [35S]methionine in VSV-infected cells. This suggested that the small polysomes described by Lodish and Porter may instead be nontranslating mRNPs (34). Our experiments with puromycin (Fig. 3) were consistent with those of Rosen et al. and showed that a large portion of mRNAs transcribed either from the viral genome or in the host nucleus were in nontranslating mRNPs. Therefore, we have concluded that only a small portion, around eighteen percent, of viral mRNAs is translated efficiently and that those mRNAs are in large polysomes.
Many viruses have been shown to use cis-acting sequences to enhance translation of viral mRNAs under conditions in which translation of normal host mRNAs is compromised. However, we have found that when EGFP reporter mRNAs contain viral UTRs, they are no more resistant to the shutoff of translation during VSV infection than an EGFP reporter mRNA with control UTRs (Fig. 5). We have also found that VSV UTRs do not enhance translation efficiency of EGFP mRNAs over control UTRs in uninfected cells (data not shown). Therefore, we have concluded that VSV UTRs do not contain cis-acting sequences that affect translation. We have also concluded, based on the levels of expression of EGFP as a foreign gene, that the coding regions of VSV mRNAs do not contain cis-acting sequences that affect translation. After normalizing for thirty percent transcription attenuation (15) and normalizing for methionine content, EGFP protein was found to be translated at levels similar to those for VSV proteins in cells infected with rVSV-EGFP (data not shown).
Many viruses have developed mechanisms to evade the inhibition of translation that often occurs during infections. These mechanisms involve recruitment of translation initiation factors such as eIF4F to viral cis-acting sequences in mRNAs, often through intermediate trans-acting factors. For example, the rotavirus NSP3 protein binds to a cis-acting sequence in rotavirus 3′ UTRs and to the eIF4G scaffolding subunit of eIF4F. Similarly, adenovirus 100k protein binds to the 5′ UTR of adenovirus late mRNAs and to eIF4G, enhancing translation of adenovirus mRNAs that contain the cis-acting sequence recognized by 100k protein (42-45). However, VSV mRNAs do not contain cis-acting sequences that promote translation, and the translation advantage of VSV mRNAs over host mRNAs, in infected cells, is not a result of VSV mRNAs being resistant to inhibition of translation. This is evident by the failure of rVSV-EGFP-derived mRNAs to exceed or even maintain, in infected cells, their 22-fold advantage in translation efficiency over host-derived EGFP mRNAs that is observed in vitro. Instead of using cis-acting sequences to evade translation inhibition, VSV achieves predominance in gene expression by transcribing mRNAs containing a cis-acting element (not sequence) that enhances translation efficiency over normal host mRNAs. While translation of VSV mRNAs appears to be inhibited in cells, the cis-acting element functions so well as to allow high levels of translation even when inhibited.
Several groups have studied features of VSV mRNA structure that are known to influence translation efficiency. These features are the 5′ guanosine cap, methylation at the 5′ end, and the 3′ poly(A) tail. So far, these features have been found to be similar for VSV mRNAs and host mRNAs. Efficient translation of mRNAs in eukaryotic cells requires a 5′ guanosine nucleotide cap methylated at position seven of the guanine base linked to the 5′ end of the mRNA by a 5′-5′ pyrophosphate bond (1, 4, 5, 27, 38, 46). In addition to the methyl group of the cap, eukaryotic mRNAs are also methylated to various extents at the first two nucleotides on the 2′-O of the ribose ring and at various internal positions. Some mRNAs are methylated at the first two coded nucleotides (cap 2), some at only the first (cap 1), while for other mRNAs the only methyl group is that of the 7-methylguanosine cap (cap 0) (1). VSV polymerase catalyzes capping and methylation of viral mRNAs to form 7mGppp2′OmApA (cap 1) (24, 25, 33). In some cell types, mRNAs may contain additional methyl groups. However, in these situations, it appears that host mRNAs and viral mRNAs are methylated similarly (1, 24). VSV mRNAs have also been shown to have 3′ poly(A) tails similar in length to those of host mRNAs (2, 13). Based on our data that VSV mRNAs do not contain cis-acting sequences and on previous reports of VSV mRNA structure, we believe this cis-acting element is a structural element other than methylation or poly(A) tail. Additional studies on the chemical structure of VSV mRNAs will address the specific nature of the translation-stimulating element.
Acknowledgments
We thank Maryam Ahmed and Elizabeth Pettit-Kneller for helpful advice and comments on the manuscript. We thank David Ornelles for help and advice regarding polysome profile experiments. We thank David Ornelles and Felicia Goodrum for their work developing the method used for extracting RNA from sucrose gradient fractions. We also thank the reviewers for their insightful comments and suggestions that helped in developing this story.
This work was supported by NIH grant RO1AI052304.
Footnotes
Published ahead of print on 27 September 2006.
REFERENCES
- 1.Banerjee, A. K. 1980. 5′-Terminal cap structure in eucaryotic messenger ribonucleic acids. Microbiol. Rev. 44:175-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banerjee, A. K., S. A. Moyer, and D. P. Rhodes. 1974. Studies on the in vitro adenylation of RNA by vesicular stomatitis virus. Virology 61:547-558. [DOI] [PubMed] [Google Scholar]
- 3.Barber, G. N. 2005. VSV-tumor selective replication and protein translation. Oncogene 24:7710-7719. [DOI] [PubMed] [Google Scholar]
- 4.Both, G. W., A. K. Banerjee, and A. J. Shatkin. 1975. Methylation-dependent translation of viral messenger RNAs in vitro. Proc. Natl. Acad. Sci. USA 72:1189-1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Both, G. W., Y. Furuichi, S. Muthukrishnan, and A. J. Shatkin. 1975. Ribosome binding to reovirus mRNA in protein synthesis requires 5′ terminal 7-methylguanosine. Cell 6:185-195. [DOI] [PubMed] [Google Scholar]
- 6.Bushell, M., and P. Sarnow. 2002. Hijacking the translation apparatus by RNA viruses. J. Cell Biol. 158:395-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Connor, J. H., and D. S. Lyles. 2005. Inhibition of host and viral translation during vesicular stomatitis virus infection. eIF2 is responsible for the inhibition of viral but not host translation. J. Biol. Chem. 280:13512-13519. [DOI] [PubMed] [Google Scholar]
- 8.Connor, J. H., and D. S. Lyles. 2002. Vesicular stomatitis virus infection alters the eIF4F translation initiation complex and causes dephosphorylation of the eIF4E binding protein 4E-BP1. J. Virol. 76:10177-10187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fields, B. N., D. M. Knipe, and P. M. Howley. 1996. Fundamental virology, 3rd ed. Lippincott-Raven, Philadelphia, Pa.
- 10.Gale, M., Jr., S. L. Tan, and M. G. Katze. 2000. Translational control of viral gene expression in eukaryotes. Microbiol. Mol. Biol. Rev. 64:239-280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gupta, A. K., M. Mathur, and A. K. Banerjee. 2002. Unique capping activity of the recombinant RNA polymerase (L) of vesicular stomatitis virus: association of cellular capping enzyme with the L protein. Biochem. Biophys. Res. Commun. 293:264-268. [DOI] [PubMed] [Google Scholar]
- 12.He, B., R. G. Paterson, C. D. Ward, and R. A. Lamb. 1997. Recovery of infectious SV5 from cloned DNA and expression of a foreign gene. Virology 237:249-260. [DOI] [PubMed] [Google Scholar]
- 13.Hunt, D. M. 1983. Vesicular stomatitis virus mutant with altered polyadenylic acid polymerase activity in vitro. J. Virol. 46:788-799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hunt, D. M., E. F. Smith, and D. W. Buckley. 1984. Aberrant polyadenylation by a vesicular stomatitis virus mutant is due to an altered L protein. J. Virol. 52:515-521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iverson, L. E., and J. K. Rose. 1981. Localized attenuation and discontinuous synthesis during vesicular stomatitis virus transcription. Cell 23:477-484. [DOI] [PubMed] [Google Scholar]
- 16.Jang, S. K., H. G. Krausslich, M. J. Nicklin, G. M. Duke, A. C. Palmenberg, and E. Wimmer. 1988. A segment of the 5′ nontranslated region of encephalomyocarditis virus RNA directs internal entry of ribosomes during in vitro translation. J. Virol. 62:2636-2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jaye, M. C., W. Godchaux III, and J. Lucas-Lenard. 1982. Further studies on the inhibition of cellular protein synthesis by vesicular stomatitis virus. Virology 116:148-162. [DOI] [PubMed] [Google Scholar]
- 18.Keene, J. D., and R. A. Lazzarini. 1976. A comparison of the extents of methylation of vesicular stomatitis virus messenger RNA. Virology 69:364-367. [DOI] [PubMed] [Google Scholar]
- 19.Kopecky, S. A., M. C. Willingham, and D. S. Lyles. 2001. Matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis virus. J. Virol. 75:12169-12181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lodish, H. F., and M. Porter. 1980. Translational control of protein synthesis after infection by vesicular stomatitis virus. J. Virol. 36:719-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lyles, D. S. 2000. Cytopathogenesis and inhibition of host gene expression by RNA viruses. Microbiol. Mol. Biol. Rev. 64:709-724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McAllister, P. E., and R. R. Wagner. 1976. Differential inhibition of host protein synthesis in L cells infected with RNA− temperature-sensitive mutants of vesicular stomatitis virus. J. Virol. 18:550-558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Michel, Y. M., D. Poncet, M. Piron, K. M. Kean, and A. M. Borman. 2000. Cap-Poly(A) synergy in mammalian cell-free extracts. Investigation of the requirements for poly(A)-mediated stimulation of translation initiation. J. Biol. Chem. 275:32268-32276. [DOI] [PubMed] [Google Scholar]
- 24.Moyer, S. A., G. Abraham, R. Adler, and A. K. Banerjee. 1975. Methylated and blocked 5′ termini in vesicular stomatitis virus in vivo mRNAs. Cell 5:59-67. [DOI] [PubMed] [Google Scholar]
- 25.Moyer, S. A., and A. K. Banerjee. 1976. In vivo methylation of vesicular stomatitis virus and its host-cell messenger RNA species. Virology 70:339-351. [DOI] [PubMed] [Google Scholar]
- 26.Mudd, J. A., and D. F. Summers. 1970. Protein synthesis in vesicular stomatitis virus-infected HeLa cells. Virology 42:328-340. [DOI] [PubMed] [Google Scholar]
- 27.Muthukrishnan, S., G. W. Both, Y. Furuichi, and A. J. Shatkin. 1975. 5′-Terminal 7-methylguanosine in eukaryotic mRNA is required for translation. Nature 255:33-37. [DOI] [PubMed] [Google Scholar]
- 28.Patton, J. T., and E. Spencer. 2000. Genome replication and packaging of segmented double-stranded RNA viruses. Virology 277:217-225. [DOI] [PubMed] [Google Scholar]
- 29.Pelletier, J., G. Kaplan, V. R. Racaniello, and N. Sonenberg. 1988. Cap-independent translation of poliovirus mRNA is conferred by sequence elements within the 5′ noncoding region. Mol. Cell. Biol. 8:1103-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pelletier, J., and N. Sonenberg. 1988. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature 334:320-325. [DOI] [PubMed] [Google Scholar]
- 31.Piron, M., P. Vende, J. Cohen, and D. Poncet. 1998. Rotavirus RNA-binding protein NSP3 interacts with eIF4GI and evicts the poly(A) binding protein from eIF4F. EMBO J. 17:5811-5821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rhodes, D. P., and A. K. Banerjee. 1975. 5′-Terminal sequence of vesicular stomatitis virus mRNA's synthesized in vitro. J. Virol. 17:33-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rhodes, D. P., S. A. Moyer, and A. K. Banerjee. 1974. In vitro synthesis of methylated messenger RNA by the virion-associated RNA polymerase of vesicular stomatitis virus. Cell 3:327-333. [DOI] [PubMed] [Google Scholar]
- 34.Rosen, C. A., H. L. Ennis, and P. S. Cohen. 1982. Translational control of vesicular stomatitis virus protein synthesis: isolation of an mRNA-sequestering particle. J. Virol. 44:932-938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sambrook, J., and D. W. Russell. 2001. Molecular cloning: a laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 36.Sarnow, P., P. Hearing, C. W. Anderson, D. N. Halbert, T. Shenk, and A. J. Levine. 1984. Adenovirus early region 1B 58,000-dalton tumor antigen is physically associated with an early region 4 25,000-dalton protein in productively infected cells. J. Virol. 49:692-700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stanners, C. P., A. M. Francoeur, and T. Lam. 1977. Analysis of VSV mutant with attenuated cytopathogenicity: mutation in viral function, P, for inhibition of protein synthesis. Cell 11:273-281. [DOI] [PubMed] [Google Scholar]
- 38.Toneguzzo, F., and H. P. Ghosh. 1976. Characterization and translation of methylated and unmethylated vesicular stomatitis virus mRNA synthesized in vitro by ribonucleoprotein particles from vesicular stomatitis virus-infected L cells. J. Virol. 17:477-491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vende, P., M. Piron, N. Castagne, and D. Poncet. 2000. Efficient translation of rotavirus mRNA requires simultaneous interaction of NSP3 with the eukaryotic translation initiation factor eIF4G and the mRNA 3′ end. J. Virol. 74:7064-7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wertz, G. W., and J. S. Youngner. 1972. Inhibition of protein synthesis in L cells infected with vesicular stomatitis virus. J. Virol. 9:85-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Whelan, S. P., L. A. Ball, J. N. Barr, and G. T. Wertz. 1995. Efficient recovery of infectious vesicular stomatitis virus entirely from cDNA clones. Proc. Natl. Acad. Sci. USA 92:8388-8392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xi, Q., R. Cuesta, and R. J. Schneider. 2005. Regulation of translation by ribosome shunting through phosphotyrosine-dependent coupling of adenovirus protein 100k to viral mRNAs. J. Virol. 79:5676-5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xi, Q., R. Cuesta, and R. J. Schneider. 2004. Tethering of eIF4G to adenoviral mRNAs by viral 100k protein drives ribosome shunting. Genes Dev. 18:1997-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yueh, A., and R. J. Schneider. 1996. Selective translation initiation by ribosome jumping in adenovirus-infected and heat-shocked cells. Genes Dev. 10:1557-1567. [DOI] [PubMed] [Google Scholar]
- 45.Yueh, A., and R. J. Schneider. 2000. Translation by ribosome shunting on adenovirus and hsp70 mRNAs facilitated by complementarity to 18S rRNA. Genes Dev. 14:414-421. [PMC free article] [PubMed] [Google Scholar]
- 46.Zan-Kowalczewska, M., M. Bretner, H. Sierakowska, E. Szczesna, W. Filipowicz, and A. J. Shatkin. 1977. Removal of 5′-terminal m7G from eukaryotic mRNAs by potato nucleotide pyrophosphatase and its effect on translation. Nucleic Acids Res. 4:3065-3081. [DOI] [PMC free article] [PubMed] [Google Scholar]