

Abstract
Human CD4+ T cells are major targets for human immunodeficiency virus (HIV) infection. Resting T cells are resistant to HIV infection unless activated through the T-cell receptor (TCR) or by cytokine signals. How T-cell signaling promotes susceptibility of T cells to HIV infection remains poorly understood. Here we demonstrate that the VacA toxin produced by Helicobacter pylori can inhibit HIV infection of primary T cells, stimulated through the TCR or by cytokines alone. This activity of VacA was dependent on its ability to form membrane channels. VacA suppressed HIV infection of T cells at a stage after viral entry, post-reverse transcription and pre-two-long-terminal-repeat circle formation, similar to the cytokine signaling inhibitor rapamycin. Mechanistically, neither VacA nor rapamycin inhibited the activation of cytokine signal transduction components (STAT5, p42/44 mitogen-activated protein kinase, or p38), but both blocked activation of key regulatory proteins required for G1 cell cycle transition. In contrast to rapamycin, VacA did not suppress phosphorylation of p70 S6 kinase but caused mitochondrial depolarization and ATP depletion within primary T cells. These results suggest that VacA inhibits T-cell activation and HIV infection via a novel mechanism. Identifying the host cell targets of VacA could be useful for elucidating the HIV life cycle within primary T cells.
Human CD4+ T cells serve as the primary targets for human immunodeficiency virus (HIV) replication in vivo (40). Resting T cells are resistant to HIV infection unless activated via the T-cell receptor (TCR) or cytokines (11, 18, 32, 39, 42, 47, 52, 53). The stimulation of human T cells through TCR signals in combination with costimulatory signals results in changes in gene expression, increased metabolic activity, and entry into the cell cycle. Following TCR activation, the proliferation and differentiation of T cells are subsequently driven by sustained cytokine signals, such as those from interleukin-2 (IL-2). Activation of T cells through cytokine pathways in the absence of TCR signals is thought to mediate homeostatic proliferation and survival of T cells in vivo (21, 36, 46, 48, 49). Both TCR and cytokine signals can render resting T cells highly susceptible to HIV infection (9, 11, 32, 33, 41, 47). How T-cell activation promotes susceptibility of T cells to HIV infection remains poorly understood.
Several immunosuppressive drugs have been shown to interfere with HIV infection of primary human T cells (11, 13, 29). These agents include cyclosporine (CsA) and FK506, which block activation of calcineurin and thereby prevent activation of nuclear factor of activated T cells (NFAT), a key transcription factor required for the expression of IL-2 and the IL-2 high-affinity receptor (CD25) (37, 45), and rapamycin, which inhibits cytokine-induced activation of p70 S6 kinase and causes a G1-stage cell cycle arrest (1, 2, 20). These inhibitors block HIV infection of TCR- or cytokine-stimulated primary human T cells but do not block HIV infection of transformed T-cell lines, which are susceptible to infection in the absence of any external stimuli (29). Both cyclosporine and FK506 are effective in reducing HIV infection of primary T cells at the time of TCR activation, but these agents have little or no inhibitory effect on infection of cytokine-stimulated primary T cells (29). In contrast, rapamycin suppresses HIV infection of both TCR- and cytokine-stimulated primary T cells (29).
Helicobacter pylori is a gram-negative bacterium that persistently colonizes the human stomach and contributes to the development of peptic ulcer disease and gastric cancer (43). A secreted cytotoxin (VacA) produced by H. pylori facilitates colonization of the human stomach by this bacterium and has an important role in the pathogenesis of peptic ulcer disease and gastric cancer (7). VacA was recently shown to inhibit human T-cell activation and proliferation (3, 7, 15, 44). Inhibition of T-cell activation and proliferation by VacA may contribute to the ability of H. pylori to resist immune clearance and establish a lifelong persistent infection (3, 7, 15, 44). Studies of transformed Jurkat T cells indicate that VacA blocks the activation of NFAT (3, 15). The process by which VacA inhibits NFAT activation in Jurkat T cells (3, 15) is reportedly similar to the actions of the immunosuppressive drugs cyclosporine and FK506 (37, 45). However, VacA inhibits the activation-induced proliferation of primary human T cells by a mechanism independent of its effects on NFAT activation and IL-2 secretion (44). Inhibition of primary human T-cell activation by VacA may be due to disruption of cytokine signaling pathways, which results in cell cycle arrest (44).
Because TCR or cytokine signals are critical for rendering human T cells susceptible to infection by HIV, we hypothesized that VacA might inhibit HIV infection of primary human CD4+ T cells. We report here that VacA blocks HIV infection of activated T cells at a stage after viral entry, post-reverse transcription and pre-two-long-terminal-repeat (2-LTR) circle formation. This same stage of HIV infection is blocked by the immunosuppressive drug rapamycin, but we show that there are multiple distinguishable differences between the effects of VacA and rapamycin on T cells. These findings provide important new insights into the signaling pathways and host factors required for HIV infection of primary T cells and also help to elucidate the mechanisms by which VacA may alter the function of human T cells during H. pylori infection.
MATERIALS AND METHODS
Purification of VacA.
H. pylori strain 60190 is a wild-type strain, and AV452 is an isogenic mutant H. pylori strain that produces a dominant-negative form of VacA, VacAΔ6-27 (50). H. pylori strains were cultured in broth, and oligomeric forms of VacA were purified from broth culture supernatants of H. pylori as described previously (8). All experiments were performed by using acid-activated preparations of VacA (24) or acidified buffer control (phosphate-buffered saline [PBS]), unless stated otherwise. VacA was acid activated by addition of 250 mM HCl, thereby lowering the pH of VacA preparations to pH 3 (10, 24). The final VacA concentration was 10 μg/ml for all experiments, unless stated otherwise. For the dominant-negative assays, wild-type VacA was mixed with an equimolar concentration of VacAΔ6-27, and the mixture was acid activated before addition to cells (50).
Cell isolation and culture.
Resting CD4+ T cells were purified from peripheral blood mononuclear cells obtained from healthy adult donors by using magnetic bead purification as described previously (26, 47). Purified CD4+ T cells were typically 99% pure, as determined by postpurification flow cytometry analysis. The culture medium used in all experiments was as described previously (26, 47). Purified resting T cells were activated by cross-linking with plate-bound anti-CD3 antibody (OKT-3; American Type Culture Collection) and soluble anti-CD28 antibody (BD Biosciences, Franklin Lakes, NJ) (hereafter termed TCR/CD28 stimulation). Cells were removed from the activation signals after 48 h and expanded and maintained in culture medium supplemented with 200 U/ml recombinant human IL-2 (Chiron Corporation). When indicated, cells were cultured in the presence of 10 μM zidovudine (AZT; Sigma) to inhibit HIV reverse transcription. To inhibit TCR/CD28 stimulation or IL-2-driven stimulation, T cells were treated with cyclosporine (50 nM; Alexis Biochemicals, Lausen, Switzerland) or rapamycin (200 ng/ml; Alexis Biochemicals), respectively. To inhibit ATP production, T cells were treated with 2,4-dinitrophenol (100 μM; Aldrich) or sodium azide (2 mM; Sigma).
Virus production and infections.
Replication-incompetent HIV pseudotyped with vesicular stomatitis virus G protein (VSV-G) and replication-competent R5-tropic viruses, each containing the green fluorescent protein (GFP) gene as a marker gene, were generated as previously described (26, 47). Supernatants were either used immediately for infections or frozen in aliquots at −80°C. The viral titers were determined by infection of a Hut 78 T-cell line that expresses CCR5 (provided by V. KewalRamani) with serially diluted virus supernatant and typically ranged from 2 × 106 to 5 × 106 infectious units per ml for replication-competent viruses and 10 × 106 to 30 × 106 infectious units/ml for VSV-G-pseudotyped HIV. T cells were infected at a multiplicity of infection of 5 to 10 in 96-well plates. In some experiments, cells inoculated with virus were centrifuged for 1 h at 2,000 rpm to enhance infectivity, as described previously (28). Infection was assessed by GFP expression using flow cytometry at 72 h postinfection.
Quantitative analysis of HIV reverse transcription and 2-LTR circle formation in target cells.
Viral DNA was quantified by real-time PCR using an ABI 7700 instrument (PE Biosystems) with SYBR green chemistry (PE Biosystems). The reaction was amplified and analyzed as previously described (29). The sequences of primers for amplification of the late reverse transcription product (R and 5NC) were 5′-TGTGTGCCCGTCTGTTGTGT (forward) and 5′-GAGTCCTGCGTCGAGAGAGC (reverse), as previously described (5). For the quantification of 2-LTR circles, the primer sequences were 5′-GTGCCCGTCTGTTGTGTGACT-3′ (forward) and 5′-CTTGTCTTCTTTGGGAGAGAATTAGC-3′ (reverse), and the probe sequence was 5′-(6-carboxyfluorescein)-TCCACACTGACTAAAAGGGTCTGAGGGATCTCT-(6-carboxytetramethylrhodamine)-3′. The 2-LTR circle reaction was performed exactly as described previously (5).
Cell cycle, mitochondrial membrane depolarization, ATP, and FACS analyses.
For these assays, primary human T cells were activated via TCR/CD28 stimulation and then expanded in the presence of IL-2 for a total of 7 days. At this point, cells are totally dependent on IL-2 for their survival and proliferation. These activated T cells were removed from IL-2 for 17 h and then pretreated with the different additives (i.e., VacA, buffer, or rapamycin) for 8 h. Cells were then restimulated with IL-2, cultured for the indicated time intervals, harvested, and stained with propidium iodide (PI) to assess cell cycle progression and cell viability. Mito Flow reagent (Cell Technology) and Cell Glow reagent (Promega) were used to analyze mitochondrial membrane polarization and cellular ATP levels, respectively. PI staining was performed as described previously (44), and Mito Flow and Cell Glow staining were performed according to the manufacturers' instructions. For the PI and Mito Flow assays 1 × 105 activated T cells were used and for assays of ATP 5 × 104 cells. The PI and Mito Tracker data were collected by fluorescence-activated cell sorting (FACS) and analyzed using flow cytometry (FACSCalibur; BD Biosciences). Live cells were gated based on forward and side scatter profiles and analyzed using the FlowJo (Treestar) program. Measurements of ATP levels in the Cell Glow assay were done using a microplate fluorescence reader.
Immunoblot analysis.
For immunoblotting experiments to investigate the expression of proteins involved in cell cycle progression, resting T cells were pretreated with PBS, rapamycin, or VacA for 1 h, and then were TCR/CD28-stimulated and incubated for the indicated times. Alternatively, TCR/CD28-activated T cells were expanded in the presence of IL-2 for 7 days and washed and maintained in IL-2-free medium for 24 h. During this period, cells were treated with PBS, rapamycin, or VacA. Cells were then stimulated with IL-2 for the indicated times and were lysed with 4× lysis buffer (0.25 M Trizma base, pH 6.8, 8% sodium dodecyl sulfate [SDS], and 0.1% β-mercaptoethanol). Protein extracts were mixed with 6× SDS loading buffer, electrophoresed on a 4 to 20% gradient precast-acrylamide gel (Bio-Rad), and transferred to polyvinylidene difluoride membranes. Membranes were immunoblotted with antibodies against Rb, cyclin D3, and p19 (BD Transduction Laboratories), followed by horseradish peroxidase-conjugated secondary antibodies (Bio-Rad). Immune complexes were revealed by the SuperSignal West Pico chemiluminescent substrate (Pierce).
For immunoblotting experiments to investigate the activation of IL-2/IL-2 receptor signaling pathways, primary human T cells were activated via TCR/CD28 stimulation and then expanded in the presence of IL-2 for a total of 7 days. At this point, these cells are totally dependent on IL-2 for their survival and proliferation. These IL-2-stimulated primary T cells were then removed from IL-2 for 17 h. The cells (1 × 106 activated T cells per condition) were then pretreated with the different additives (i.e., VacA, buffer, or rapamycin) for 8 h, followed by restimulation of the cells with IL-2 for 5 or 20 min. Cells were then spun down, washed, and lysed in 4% SDS in 0.25 M Trizma base in the presence of protease inhibitors, NaF (10 mM) and NaOV3 (2 mM). Samples were boiled for 10 min and stored at −70°C. Protein extracts were mixed with 6× SDS loading buffer, electrophoresed on a 4 to 20% gradient precast-acrylamide gel (Bio-Rad), and transferred to polyvinylidene difluoride membranes. Membranes were then blotted according to the manufacturer's instructions using specific antibodies to phosphorylated Stat5 and Stat3 (P-Stat5 and P-Stat3, respectively; Cell Signaling), total Stat5 and Stat3 (T-Stat5 and T-Stat3, respectively; Cell Signaling), phosphorylated p38 (Thr180/Tyr182; Cell Signaling), phosphorylated p44/42 (Thr202/Tyr204; Cell Signaling), phosphorylated AKT (Ser 473; Cell Signaling), and actin (Santa Cruz Biotechnology). Following incubation with the primary antibodies, membranes were then immunoblotted with horseradish peroxidase-conjugated secondary antibodies (Bio-Rad). The immune complexes were revealed by the SuperSignal West Pico chemiluminescent substrate (Pierce) and X-ray film.
RESULTS
VacA inhibits HIV infection of TCR/CD28-activated primary human T cells.
Based on our previous findings that VacA can interfere with proliferation of primary human T cells (44), we hypothesized that VacA might also block HIV infection of activated T cells. To test this hypothesis, CD4+ T cells were purified from healthy adult donor blood and activated using anti-CD3 and anti-CD28 antibodies (TCR/CD28 stimulation). The T cells were treated at the time of activation with PBS, AZT, CsA, or wild-type VacA and were concurrently infected with VSV-G-pseudotyped HIV (HIV.VSVG; Fig. 1A). As expected, AZT, an inhibitor of HIV reverse transcription, and CsA inhibited HIV infection of TCR-activated T cells (Fig. 1A). VacA also inhibited HIV infection (Fig. 1A) in a dose-dependent manner (Fig. 1B). Consistent with previous results (44), VacA treatment did not have any detrimental effects on the viability of T cells, as determined by flow cytometric analyses (data not shown). Furthermore, purified oligomeric VacA required acid activation to inhibit HIV infection (Fig. 1A), similar to the requirement of acid activation in order for VacA to interfere with activation of primary T cells (44). These results demonstrate that VacA inhibits HIV infection of TCR/CD28-activated primary human T cells.
FIG. 1.
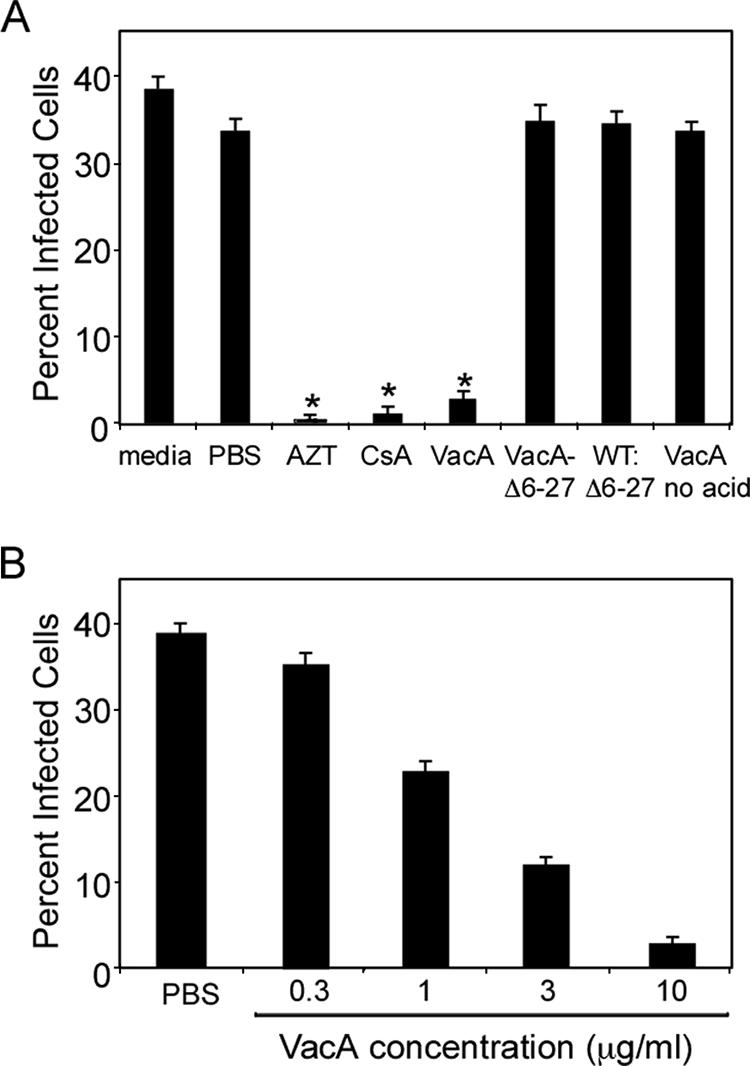
VacA inhibits HIV infection of primary human T cells. (A) Resting primary human CD4+ T cells were activated using antibodies against CD3 and CD28 and concurrently infected with HIV.VSVG in the presence of AZT, CsA, acid-activated wild-type VacA, acid-activated mutant VacA protein (VacAΔ6-27), acid-treated PBS, or nonactivated wild-type VacA (VacA no acid). Concentrations of these agents are described in Materials and Methods. Cells were also incubated with an equimolar mixture of acid-activated wild-type VacA and VacAΔ6-27 (WT: Δ6-27). After 72 h, the percentage of infected cells was determined by flow cytometry, based on detection of GFP expression. (B) Cells treated in similar fashion were incubated with various concentrations of acid-activated wild-type VacA. Results are representative of three experiments with T cells isolated from different donors. The asterisks indicate P < 0.05 compared to PBS-treated cells at the same time point. Statistical significance between groups was determined by a two-tailed t test.
In contrast to acid-activated wild-type VacA, an acid-activated VacA mutant toxin (VacAΔ6-27) did not inhibit HIV infection (Fig. 1A). VacAΔ6-27 has previously been shown to lack detectable activity when tested in epithelial cell assays and is defective in the capacity to form membrane channels (50). VacAΔ6-27 is also able to block several activities of wild-type VacA in a dominant-negative fashion (50), via a process dependent on the formation of inactive mixed oligomeric complexes (23). Therefore, we hypothesized that VacAΔ6-27 would neutralize the ability of wild-type VacA to inhibit HIV infection. To test this hypothesis, VacA and VacAΔ6-27 were mixed in an equimolar ratio and added to CD4+ T cells at the time of activation and infection. Indeed, VacAΔ6-27 blocked the ability of wild-type VacA to inhibit HIV infection, suggesting that functional oligomeric forms of VacA are required for this activity (Fig. 1A).
VacA does not block HIV infection of a transformed T-cell line.
Our results (Fig. 1) demonstrated that VacA effectively inhibits HIV infection of activated human T cells. However, these data do not exclude the possibility that the inhibition of HIV infection might be due to VacA-mediated inactivation of the virus. To address this possibility, we determined whether VacA could inhibit HIV infection of Hut 78 cells, a transformed T-cell line that is highly susceptible to HIV infection and does not require any activation signals (such as TCR/CD28 or IL-2 receptor-mediated signaling). For this experiment, Hut 78 cells were treated with either VacA or other additives for 8 h and then washed and infected with HIV.VSVG or replication-competent CCR5-tropic HIV (HIV.R5) in the presence of VacA or other additives (data not shown). VacA, similarly to rapamycin, had no effect on HIV infection of Hut 78 cells, whereas HIV infection was inhibited by AZT, as expected (data not shown) (29). This finding strongly suggests that VacA does not have a direct effect on HIV.
VacA inhibits HIV infection of cytokine-stimulated primary human T cells.
We next evaluated whether VacA could inhibit HIV infection of T cells that are dependent on IL-2 for proliferation and survival. For this experiment, primary CD4+ T cells were TCR/CD28 stimulated and expanded for 5 days in IL-2 (Fig. 2A). At this stage, primary T cells are completely dependent on IL-2 signals for proliferation and survival. T cells were then removed from IL-2 and pretreated with VacA or other additives for 8 h, washed to remove additives, and infected with HIV.VSVG (Fig. 2A) or HIV.R5 (Fig. 2B) in the presence of IL-2 and the indicated additives. VacA, but not VacAΔ6-27, inhibited both HIV.VSVG and HIV.R5 infection of these cells (Fig. 2A and 2B). VacA-mediated inhibition was observed not only if T cells were treated with VacA prior to HIV infection but also if VacA was added to cells at the time of infection with HIV.VSVG or HIV.R5 (data not shown). HIV infection of IL-2-stimulated T cells was also suppressed by rapamycin but not CsA (Fig. 2A and 2B), as we have previously shown (29).
FIG. 2.

VacA inhibits HIV infection of IL-2-dependent TCR-activated and cytokine-stimulated primary T cells. (A) Resting primary human CD4+ T cells were TCR/CD28 activated, and cells were then expanded in the presence of IL-2 (yielding “preactivated” cells). After 7 days, cells were removed from IL-2 and treated with VacA, AZT, rapamycin (Rap), CsA, or VacAΔ6-27 for 8 h. Concentrations of these agents were as described in Materials and Methods. Cells were then washed, restimulated with IL-2, and infected with HIV.VSVG (A) or HIV.R5 (B) in the presence of the indicated additives. (C and D) Cytokine-stimulated CD4+ T cells (cultured in IL-2-containing medium for 7 days in the absence of TCR stimulation) were pretreated with the indicated additives for 8 h and then washed and infected with either HIV.VSVG (C) or HIV.R5 (D) in the presence of the indicated additives. Infection was assessed by analyzing GFP expression using flow cytometry at 72 h. Results are representative of three experiments performed in triplicate with T cells isolated from different donors. Asterisks indicate P < 0.05 compared to PBS-treated cells at the same time point. Statistical significance between groups was determined by a two-tailed t test.
We and others have shown that resting T cells can be rendered susceptible to productive HIV infection by stimulation with the IL-2 family of cytokines (IL-2, IL-4, IL-7, and IL-15) in the absence of any prior TCR signals (9, 41, 47). Therefore, we investigated whether VacA could inhibit HIV infection of human T cells stimulated only with cytokines. For these experiments, purified CD4+ T cells were stimulated with IL-2 for 7 days to render them susceptible to infection, as previously described (47). The IL-2-stimulated T cells were pretreated with VacA or other additives for 8 h, washed, and then infected with HIV.VSVG (Fig. 2C) or HIV.R5 (Fig. 2D) in the presence of the indicated additives. We found that VacA inhibited HIV infection of IL-2-stimulated T cells (Fig. 2C and 2D), similar to the effects of rapamycin. In contrast to VacA and rapamycin, CsA treatment of T cells had no detectable effect on HIV infection of these cytokine-stimulated T cells. Taken together, these results indicate that VacA can block HIV infection of T cells, either after TCR stimulation when the cells are dependent on cytokine signals for their proliferation and survival or when T cells are rendered susceptible to infection with cytokines alone.
VacA interferes with cell cycle machinery in primary T cells.
We have previously shown that VacA inhibits the proliferation of TCR/CD28-activated primary human T cells (44). The mechanism by which VacA inhibits T-cell proliferation may be relevant for understanding how VacA inhibits HIV infection of T cells (44). To investigate effects of VacA on cell cycle progression, primary human T cells were pretreated with VacA, rapamycin, or PBS for 1 h prior to TCR/CD28 stimulation. The activated T cells were cultured for 3 days and were then analyzed by propidium iodide staining and flow cytometry to assess cell cycle distribution or were lysed for immunoblot analyses. In comparison to treatment of cells with PBS, treatment of cells with either VacA or rapamycin resulted in an increased proportion of cells in G1 and a decreased proportion of cells in S and G2/M, indicative of a cell cycle arrest at the G1 stage (Fig. 3A). Expression and activation of two cell cycle regulators, retinoblastoma protein (Rb) and cyclin D3, were analyzed in VacA-treated and untreated cells. These proteins are activated in resting T cells upon TCR/CD28 stimulation and are required for cell cycle progression and proliferation (12, 34). Both VacA and rapamycin inhibited TCR/CD28-induced expression and hyperphosphorylation of Rb (the hyperphosphorylated form migrates as a higher-molecular-weight band) and inhibited the expression of cyclin D3 (Fig. 3B). Similar results were obtained in experiments using IL-2-dependent activated T cells (data not shown). These results indicate that VacA, like rapamycin, inhibits TCR/CD28- and IL-2-mediated cell cycle progression by blocking the activation of key regulatory proteins important in the G1 transition, resulting in the impairment of cell proliferation.
FIG. 3.

VacA inhibits G1-to-S cell cycle progression. (A) Primary human CD4+ T cells were activated with CD3/CD28 antibodies in the presence of the indicated additives. Cells were then harvested 24, 48, and 72 h postactivation, and cell cycle analysis was performed using propidium iodide staining as described in Materials and Methods. The combined percentages of cells in S and G2/M are indicated. (B) Cells from panel A were harvested at 48 h postactivation for immunoblot analysis as described in Materials and Methods. Protein lysates were generated and immunoblotted with the indicated antibodies. Immunoblotting with antibody against total p19 was used as a loading control. Results are representative of three independent experiments.
Effects of VacA on IL-2-induced signaling pathways.
VacA can inhibit IL-2-induced proliferation of T cells (44) and can render IL-2-stimulated T cells resistant to HIV infection. Therefore, we assessed the effects of VacA on activation of several molecules that are important in the IL-2 signaling pathway. We first investigated whether VacA blocks membrane-proximal signaling events mediated by IL-2-dependent activation of signal transducer and activator of transcription 3 and 5 (Stat3 and Stat5, respectively). In control cells, removal of IL-2 from activated CD4+ T resulted in the dephosphorylation of both Stat3 and Stat5, and readdition of IL-2 was associated with rapid Stat activation (Fig. 4A). Neither VacA nor rapamycin treatment inhibited IL-2-dependent phosphorylation of Stat3 or Stat5 (Fig. 4A). A second signaling pathway that can be activated by binding of IL-2 to the IL-2R is the extracellular signal-regulated protein kinase 1/2 mitogen-activated protein kinase pathway (p42/44 MAPK), which plays a role in T-cell proliferation (12). We investigated whether VacA had any effect on IL-2-induced activation of p42/p44 MAPK. As expected, p42/44MAPK was not activated in control cells that were removed from IL-2 but was rapidly activated when cells were restimulated with IL-2 (Fig. 4B). The activation of p42/44MAPK was not perturbed by VacA intoxication (Fig. 4B). VacA stimulated phosphorylation of p38, a result consistent with a previous study (3). These data indicate that VacA inhibits IL-2-mediated proliferation and IL-2-dependent HIV infection of human T cells without affecting the activation of Stat5 or p42/44 MAPK pathways.
FIG. 4.

Analysis of VacA effects on IL-2 signaling. Preactivated CD4+ T cells (7 days post-CD3/CD28 activation) were removed from IL-2 and treated with the indicated additives for 8 h. Cells were then stimulated with IL-2 for 5 min (A and right panel of D) or for 20 min (B and C). Alternatively (left panel of D), cells were pretreated with the indicated additives and then TCR/CD28 activated and cultured for 2 days prior to harvesting. Cells were then lysed, and samples were immunoblotted with indicated antibodies as described in Materials and Methods. Reactivity with phosphorylated (P) or total (T) proteins is indicated. Results are representative of three independent experiments.
IL-2 signals also activate the phosphatidylinositol 3-kinase signal transduction pathway. Phosphatidylinositol 3-kinase activation, in turn, leads to the activation of AKT and p70 S6 kinase. The activity of p70 S6 kinase is controlled by phosphorylation at multiple sites throughout the protein, but phosphorylation of Thr389 is tightly linked to p70 S6 kinase activity in vivo (12, 29, 38). Phosphorylation of this site is induced by growth factors including IL-2 and can be blocked by rapamycin (6). Rapamycin-dependent inhibition of p70 S6 kinase activation is mediated by the interaction of rapamycin with FRAP/mTOR, which operates upstream of p70 S6 kinase (29). Since there are several similarities in the effects of VacA and rapamycin on T cells, we investigated whether VacA inhibits IL-2-induced phosphorylation of Akt or IL-2-induced phosphorylation of Thr389 in p70 S6 kinase. Neither VacA nor rapamycin treatment blocked IL-2-induced activation of AKT (Fig. 4C). Rapamycin inhibited the phosphorylation of p70 S6 kinase, whereas VacA had no effect (Fig. 4D). Neither rapamycin nor VacA had any effect on the activation of p85 S6 kinase, which was used as a loading control. These data indicate that there are distinguishable differences between the effects of rapamycin and VacA on T cells.
Effects of VacA on mitochondria and ATP levels in primary human T cells.
Since the results presented above did not identify a specific stage in the IL-2 signaling pathway that was blocked by VacA, we hypothesized that VacA might affect IL-2/IL-2 receptor-induced activation of energy production, which is required for cell cycle progression, cell proliferation, and HIV infection of activated T cells (12, 29, 38). Therefore, we analyzed ATP levels in VacA-treated human T cells. In control cells, removal of IL-2 from activated CD4+ T cells resulted in a decrease in ATP levels (Fig. 5A), indicating a role of IL-2/IL-2 receptor signals for the production of energy. Additionally, treatment of cells with inhibitors of ATP synthesis (dinitrophenol and sodium azide) resulted in a decrease in ATP levels (Fig. 5A). Treatment of T cells with wild-type VacA resulted in a statistically significant reduction of ATP levels (Fig. 5A). In contrast, treatment with VacAΔ6-27 did not result in reduced ATP levels (data not shown).
FIG. 5.

Effects of VacA on ATP levels and mitochondrial membrane polarization. Primary human CD4+ T cells were activated with CD3/CD28 and cultured in the presence of IL-2 for 7 days as described in Materials and Methods. Cells were then incubated for 16 h without IL-2 and then treated with the indicated additives for 8 h without IL-2. (A) After treatment, cells were stimulated with IL-2 for 24 or 48 h and then the total ATP levels were analyzed using Cell Glow reagent as described in Materials and Methods. (B) Cell viability analysis for panel A. Cells were treated as stated for panel A, and then at 24 and 48 h cells were analyzed for viability based on forward and side scatter analysis with flow cytometry. (C) For analysis of mitochondrial membrane potential and cell viability, cells were stimulated with IL-2 for 24 h and stained using Mito Flow reagent as described in Materials and Methods. Cell viability was determined based on forward and side scatter analysis and PI staining (data not shown), and mitochondrial membrane potential was determined as described in Materials and Methods. Asterisks indicate P < 0.05 compared to PBS-treated cells at the same time point. Statistical significance between groups was determined by a two-tailed t test. Results are representative of three experiments performed in triplicate with T cells isolated from different donors. DNP, dinitrophenol; NaN3, sodium azide; Rap, rapamycin.
To elucidate the potential mechanism by which VacA causes ATP depletion, we analyzed the mitochondrial membrane potential of VacA-treated cells. T cells treated with control buffer, VacAΔ6-27, or rapamycin all exhibited similar levels of mitochondrial membrane polarization. In contrast, treatment with wild-type VacA markedly decreased the mitochondrial membrane potential of T cells (Fig. 5C). The ability of VacA to decrease the mitochondrial membrane potential of primary human T cells was not due to toxin-induced cell death, since the viability of VacA-intoxicated cells (measured by both forward and side scatter properties of the cells as well as PI staining) was similar to the viability of cells treated with the other additives (Fig. 5B and data not shown). Together these data suggest that VacA inhibits the proliferation of primary human T cells by interfering with energy production, without altering cell viability.
Dissecting the stage of HIV infection that is inhibited by VacA.
We next sought to determine the stage in the HIV life cycle that is affected by VacA. We first analyzed the production of HIV late reverse transcription products formed in TCR-stimulated T cells by real-time PCR analyses. We found that VacA, similarly to rapamycin, did not block the production of second-strand HIV reverse transcripts (Fig. 6A). Inhibitors of ATP synthesis (dinitrophenol and sodium azide) also did not significantly block the production of second-strand HIV transcripts (Fig. 6A). Thus, VacA does not have a direct effect on HIV virion integrity or early steps in the HIV life cycle, such as HIV binding to cells or the synthesis of viral cDNA.
FIG. 6.

VacA blocks HIV infection post-reverse transcription. Preactivated CD4+ T cells (7 days post-CD3/CD28 activation) were infected with HIV.VSVG in the presence of VacA, AZT, rapamycin, dinitrophenol, sodium azide, or PBS. Cells were then lysed at the indicated time points postinfection and processed for real-time PCR using primers for late HIV reverse transcript products (A) or primers that detect 2-LTR formation of reverse-transcribed viral DNA (B). Results are representative of three experiments performed in triplicate with T cells isolated from different donors.
We next investigated whether VacA inhibits the formation of circularized HIV DNA between 2-LTR circles, which are associated with the completion of reverse transcription and nuclear translocation of the HIV preintegration complex (19). We found that 2-LTR circle formation was greatly diminished in both VacA- and rapamycin-treated cells (Fig. 6B). Because it has been suggested that import of viral preintegration complexes into the nucleus is energy dependent (42), we investigated the effects of ATP synthesis inhibitors on this stage of the viral life cycle. Treatment of activated primary T cells with ATP synthesis inhibitors resulted in a highly significant reduction of 2-LTR circle formation, similar to that which was observed in VacA- or rapamycin-treated cells. Finally, to determine whether VacA was able to inhibit transcription of the integrated HIV provirus, we used destabilized GFP expressing HIV to monitor HIV-LTR-mediated transcription in infected T cells (30). VacA did not have any effect on GFP expression, which indicated that the inhibition of infection by VacA is likely to be at a step before provirus integration and transcription of HIV genes (data not shown). Collectively, these data indicate that VacA interferes with HIV infection of primary human T cells at a stage following viral reverse transcription but prior to viral integration into host DNA.
DISCUSSION
In this study we demonstrate that the H. pylori VacA toxin can inhibit HIV infection of primary human T cells. A previous study reported that another channel-forming bacterial toxin, aerolysin produced by Aeromonas hydrophila, can inhibit HIV infection by disrupting the viral envelope (27). In contrast, our results show that VacA does not directly affect virion integrity, since it did not inhibit the infection of a transformed T-cell line and did not block HIV reverse transcription in primary T cells.
After HIV enters a susceptible target cell, reverse transcriptase converts the viral genome RNA into linear double-stranded cDNA, which is imported into the nucleus, where it either integrates into the host cell genome or is circularized by host cell factors. To achieve a productive infection, the reverse-transcribed cDNA of HIV must integrate into the host cell genome. It has also been shown that T cells must be in the G1b phase of the cell cycle for optimal completion of HIV reverse transcription (19). The circularization of retroviral cDNA containing 1- or 2-LTR circles is presumed to represent an abortive pathway of infection (6). Because the machinery required for the creation of circular retroviral cDNA is nuclear, the circles provide an important surrogate to detect nuclear migration of the viral genome (6). We found that VacA does not appear to interfere with reverse transcription but does block 2-LTR circle formation. Moreover, VacA does not block transcription of HIV once the provirus is integrated into the chromosome. These results suggest that VacA blocks HIV at a post-reverse transcription and preintegration stage of the viral life cycle.
Several possible mechanisms can be suggested to explain how VacA inhibits HIV infection of primary human T cells. We provide experimental evidence indicating that VacA causes mitochondrial membrane depolarization and a reduction in cellular ATP levels in T cells. Previously, VacA has been reported to localize to the mitochondria in HeLa and HEp-2 cells and causes mitochondrial depolarization and a reduction in ATP levels in these cells (14, 17, 51). A reduction of ATP levels in T cells is expected to affect the translocation of HIV preintegration complexes into the nucleus, since this has been shown to be an energy-dependent process (4, 25). Other mechanisms also may be operative. For example, if VacA disrupts intracellular trafficking in T cells, similarly to what is observed in epithelial cels (22, 35), this might in turn affect the localization of host factors required for the nuclear translocation of HIV DNA (16). Studies using HeLa cells indicate that VacA can insert into the plasma membrane of cells to form anion-selective channels, can be internalized by cells, and can cause alterations in endocytic compartments and mitochondria (14, 22, 31, 51). It is possible that VacA might have effects on other cell compartments, including the nuclear membrane, which could result in alteration of the nuclear pore complex-mediated transport of DNAs, RNAs, transcription factors, and other large molecules. Therefore, if VacA perturbs nuclear pore complex formation, this would be expected to cause cell cycle arrest and inhibit translocation of HIV preintegration complexes to the nucleus.
The results of the current study indicate that there are several interesting similarities between the inhibitory effect of VacA on HIV infection and the inhibitory effect of the immunosuppressive drug rapamycin. Both agents (i) inhibit HIV infection and inhibit proliferation of either TCR/CD28- or IL-2-stimulated T cells, (ii) block a post-reverse transcription stage of HIV infection, and (iii) inhibit the activity of cell cycle regulatory proteins and result in a G1 cell cycle arrest. Both VacA and rapamycin blocked the expression of Rb and cyclin D3, resulting in the impairment of cell proliferation upon TCR/IL-2 stimulation of T cells. Rapamycin inhibits a mitogen-activated Ser/Thr protein kinase (p70 S6 kinase) that is required for G1-to-S cell cycle progression upon IL-2 signaling (1, 2, 20). However, VacA did not inhibit p70 S6 kinase phosphorylation. Our results also clearly indicate that VacA does not have inhibitory effects on several steps in the proximal IL-2 signaling pathway. As discussed above, VacA caused mitochondrial depolarization in T cells whereas rapamycin did not, and this could be one potential mechanism by which it slows cell progression in primary T cells. The results of this study highlight several important differences between primary human T cells and transformed T-cell lines in permissiveness to HIV infection and activation states. The inhibitory effects of VacA and rapamycin on HIV infection and T-cell proliferation were observed with primary human T cells but not with a transformed T-cell line (Hut 78). We speculate that VacA may modulate the activity of host factors that are turned on during activation of primary T cells.
In summary, our results indicate that VacA inhibits HIV infection of primary human T cells. This activity of VacA might serve as a very useful tool for further dissecting T-cell signaling pathways required for HIV infection and for identifying host factors that regulate HIV infection of T cells. This knowledge in turn could result in the development of novel therapeutic agents that inhibit HIV infection by targeting host factors, an approach that may circumvent the ability of HIV to develop escape mutations.
Acknowledgments
We thank Beverly Hosse for assistance with VacA preparation and Vineet KewalRamani, Amanda Johnson, and Karla Eger for helpful discussions and critical reading of the manuscript.
This work was supported by NIH grant R01-AI49131 (D. Unutmaz), by R01-AI39657 and DK53623 and the Medical Research Department of the Department of Veterans Affairs (T. L. Cover), and by a Vanderbilt University Medical Center Discovery Grant/CFAR development grant to T.L.C. and D.U. V. J. Torres was supported in part by the GM070061-02 NIH Ruth L. Kirschstein predoctoral fellowship. S. E. VanCompernolle is supported by a postdoctoral fellowship grant, NIH AI63975. Immunology core services and FACS analysis were supported by Vanderbilt-Meharry Center for AIDS Research (CFAR), NIH P30 AI 54999.
Footnotes
Published ahead of print on 27 September 2006.
REFERENCES
- 1.Abraham, R. T. 1998. Mammalian target of rapamycin: immunosuppressive drugs uncover a novel pathway of cytokine receptor signaling. Curr. Opin. Immunol. 10:330-336. [DOI] [PubMed] [Google Scholar]
- 2.Abraham, R. T., and G. J. Wiederrecht. 1996. Immunopharmacology of rapamycin. Annu. Rev. Immunol. 14:483-510. [DOI] [PubMed] [Google Scholar]
- 3.Boncristiano, M., S. R. Paccani, S. Barone, C. Ulivieri, L. Patrussi, D. Ilver, A. Amedei, M. M. D'Elios, J. L. Telford, and C. T. Baldari. 2003. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J. Exp. Med. 198:1887-1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bukrinsky, M. I., N. Sharova, M. P. Dempsey, T. L. Stanwick, A. G. Bukrinskaya, S. Haggerty, and M. Stevenson. 1992. Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc. Natl. Acad. Sci. USA 89:6580-6584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Butler, S. L., M. S. Hansen, and F. D. Bushman. 2001. A quantitative assay for HIV DNA integration in vivo. Nat. Med. 7:631-634. [DOI] [PubMed] [Google Scholar]
- 6.Butler, S. L., E. P. Johnson, and F. D. Bushman. 2002. Human immunodeficiency virus cDNA metabolism: notable stability of two-long terminal repeat circles. J. Virol. 76:3739-3747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cover, T. L., and S. R. Blanke. 2005. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat. Rev. Microbiol. 3:320-332. [DOI] [PubMed] [Google Scholar]
- 8.Cover, T. L., P. I. Hanson, and J. E. Heuser. 1997. Acid-induced dissociation of VacA, the Helicobacter pylori vacuolating cytotoxin, reveals its pattern of assembly. J. Cell Biol. 138:759-769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dardalhon, V., S. Jaleco, S. Kinet, B. Herpers, M. Steinberg, C. Ferrand, D. Froger, C. Leveau, P. Tiberghien, P. Charneau, N. Noraz, and N. Taylor. 2001. IL-7 differentially regulates cell cycle progression and HIV-1-based vector infection in neonatal and adult CD4+ T cells. Proc. Natl. Acad. Sci. USA 98:9277-9282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Bernard, M., E. Papini, V. de Filippis, E. Gottardi, J. Telford, R. Manetti, A. Fontana, R. Rappuoli, and C. Montecucco. 1995. Low pH activates the vacuolating toxin of Helicobacter pylori, which becomes acid and pepsin resistant. J. Biol. Chem. 270:23937-23940. [DOI] [PubMed] [Google Scholar]
- 11.Ducrey-Rundquist, O., M. Guyader, and D. Trono. 2002. Modalities of interleukin-7-induced human immunodeficiency virus permissiveness in quiescent T lymphocytes. J. Virol. 76:9103-9111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellery, J. M., and P. J. Nicholls. 2002. Alternate signalling pathways from the interleukin-2 receptor. Cytokine Growth Factor Rev. 13:27-40. [DOI] [PubMed] [Google Scholar]
- 13.Francois, F., and M. E. Klotman. 2003. Phosphatidylinositol 3-kinase regulates human immunodeficiency virus type 1 replication following viral entry in primary CD4+ T lymphocytes and macrophages. J. Virol. 77:2539-2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Galmiche, A., J. Rassow, A. Doye, S. Cagnol, J. C. Chambard, S. Contamin, V. de Thillot, I. Just, V. Ricci, E. Solcia, E. Van Obberghen, and P. Boquet. 2000. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J. 19:6361-6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gebert, B., W. Fischer, E. Weiss, R. Hoffmann, and R. Haas. 2003. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301:1099-1102. [DOI] [PubMed] [Google Scholar]
- 16.Kilzer, J. M., T. Stracker, B. Beitzel, K. Meek, M. Weitzman, and F. D. Bushman. 2003. Roles of host cell factors in circularization of retroviral dna. Virology 314:460-467. [DOI] [PubMed] [Google Scholar]
- 17.Kimura, M., S. Goto, A. Wada, K. Yahiro, T. Niidome, T. Hatakeyama, H. Aoyagi, T. Hirayama, and T. Kondo. 1999. Vacuolating cytotoxin purified from Helicobacter pylori causes mitochondrial damage in human gastric cells. Microb. Pathog. 26:45-52. [DOI] [PubMed] [Google Scholar]
- 18.Korin, Y. D., and J. A. Zack. 1999. Nonproductive human immunodeficiency virus type 1 infection in nucleoside-treated G0 lymphocytes. J. Virol. 73:6526-6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Korin, Y. D., and J. A. Zack. 1998. Progression to the G1b phase of the cell cycle is required for completion of human immunodeficiency virus type 1 reverse transcription in T cells. J. Virol. 72:3161-3168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kuo, C. J., J. Chung, D. F. Fiorentino, W. M. Flanagan, J. Blenis, and G. R. Crabtree. 1992. Rapamycin selectively inhibits interleukin-2 activation of p70 S6 kinase. Nature 358:70-73. [DOI] [PubMed] [Google Scholar]
- 21.Lantz, O., I. Grandjean, P. Matzinger, and J. P. Di Santo. 2000. Gamma chain required for naive CD4+ T cell survival but not for antigen proliferation. Nat. Immunol. 1:54-58. [DOI] [PubMed] [Google Scholar]
- 22.Li, Y., A. Wandinger-Ness, J. R. Goldenring, and T. L. Cover. 2004. Clustering and redistribution of late endocytic compartments in response to Helicobacter pylori vacuolating toxin. Mol. Biol. Cell 15:1946-1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McClain, M. S., P. Cao, H. Iwamoto, A. D. Vinion-Dubiel, G. Szabo, Z. Shao, and T. L. Cover. 2001. A 12-amino-acid segment, present in type s2 but not type s1 Helicobacter pylori VacA proteins, abolishes cytotoxin activity and alters membrane channel formation. J. Bacteriol. 183:6499-6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McClain, M. S., W. Schraw, V. Ricci, P. Boquet, and T. L. Cover. 2000. Acid activation of Helicobacter pylori vacuolating cytotoxin (VacA) results in toxin internalization by eukaryotic cells. Mol. Microbiol. 37:433-442. [DOI] [PubMed] [Google Scholar]
- 25.McDonald, D., M. A. Vodicka, G. Lucero, T. M. Svitkina, G. G. Borisy, M. Emerman, and T. J. Hope. 2002. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 159:441-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Motsinger, A., D. W. Haas, A. K. Stanic, L. Van Kaer, S. Joyce, and D. Unutmaz. 2002. CD1d-restricted human natural killer T cells are highly susceptible to human immunodeficiency virus 1 infection. J. Exp. Med. 195:869-879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nguyen, D. H., Z. Liao, J. T. Buckley, and J. E. Hildreth. 1999. The channel-forming toxin aerolysin neutralizes human immunodeficiency virus type 1. Mol. Microbiol. 33:659-666. [DOI] [PubMed] [Google Scholar]
- 28.O'Doherty, U., W. J. Swiggard, and M. H. Malim. 2000. Human immunodeficiency virus type 1 spinoculation enhances infection through virus binding. J. Virol. 74:10074-10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oswald-Richter, K., S. M. Grill, M. Leelawong, and D. Unutmaz. 2004. HIV infection of primary human T cells is determined by tunable thresholds of T cell activation. Eur. J. Immunol. 34:1705-1714. [DOI] [PubMed] [Google Scholar]
- 30.Oswald-Richter, K., S. M. Grill, N. Shariat, M. Leelawong, M. S. Sundrud, D. W. Haas, and D. Unutmaz. 2004. HIV infection of naturally occurring and genetically reprogrammed human regulatory T-cells. PLoS Biol. 2:E198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papini, E., M. de Bernard, E. Milia, M. Bugnoli, M. Zerial, R. Rappuoli, and C. Montecucco. 1994. Cellular vacuoles induced by Helicobacter pylori originate from late endosomal compartments. Proc. Natl. Acad. Sci. USA 91:9720-9724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Polacino, P. S., H. A. Liang, and E. A. Clark. 1995. Formation of simian immunodeficiency virus long terminal repeat circles in resting T cells requires both T cell receptor- and IL-2-dependent activation. J. Exp. Med. 182:617-621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Polacino, P. S., H. A. Liang, E. J. Firpo, and E. A. Clark. 1993. T-cell activation influences initial DNA synthesis of simian immunodeficiency virus in resting T lymphocytes from macaques. J. Virol. 67:7008-7016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reed, S. I. 1997. Control of the G1/S transition. Cancer Surv. 29:7-23. [PubMed] [Google Scholar]
- 35.Satin, B., N. Norais, J. Telford, R. Rappuoli, M. Murgia, C. Montecucco, and E. Papini. 1997. Effect of Helicobacter pylori vacuolating toxin on maturation and extracellular release of procathepsin D and on epidermal growth factor degradation. J. Biol. Chem. 272:25022-25028. [DOI] [PubMed] [Google Scholar]
- 36.Schluns, K. S., W. C. Kieper, S. C. Jameson, and L. Lefrancois. 2000. Interleukin-7 mediates the homeostasis of naive and memory CD8 T cells in vivo. Nat. Immunol. 1:426-432. [DOI] [PubMed] [Google Scholar]
- 37.Schreiber, S. L., and G. R. Crabtree. 1992. The mechanism of action of cyclosporin A and FK506. Immunol. Today 13:136-142. [DOI] [PubMed] [Google Scholar]
- 38.Smith, K. A. 1988. Interleukin-2: inception, impact, and implications. Science 240:1169-1176. [DOI] [PubMed] [Google Scholar]
- 39.Sonza, S., A. Maerz, N. Deacon, J. Meanger, J. Mills, and S. Crowe. 1996. Human immunodeficiency virus type 1 replication is blocked prior to reverse transcription and integration in freshly isolated peripheral blood monocytes. J. Virol. 70:3863-3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stebbing, J., B. Gazzard, and D. C. Douek. 2004. Where does HIV live? N. Engl. J. Med. 350:1872-1880. [DOI] [PubMed] [Google Scholar]
- 41.Steffens, C. M., E. Z. Managlia, A. Landay, and L. Al Harthi. 2002. Interleukin-7-treated naive T cells can be productively infected by T-cell-adapted and primary isolates of human immunodeficiency virus 1. Blood 99:3310-3318. [DOI] [PubMed] [Google Scholar]
- 42.Stevenson, M., T. L. Stanwick, M. P. Dempsey, and C. A. Lamonica. 1990. HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J. 9:1551-1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suerbaum, S., and P. Michetti. 2002. Helicobacter pylori infection. N. Engl. J. Med. 347:1175-1186. [DOI] [PubMed] [Google Scholar]
- 44.Sundrud, M. S., V. J. Torres, D. Unutmaz, and T. L. Cover. 2004. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc. Natl. Acad. Sci. USA 101:7727-7732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Timmerman, L. A., N. A. Clipstone, S. N. Ho, J. P. Northrop, and G. R. Crabtree. 1996. Rapid shuttling of NF-AT in discrimination of Ca2+ signals and immunosuppression. Nature 383:837-840. [DOI] [PubMed] [Google Scholar]
- 46.Unutmaz, D., F. Baldoni, and S. Abrignani. 1995. Human naive T cells activated by cytokines differentiate into a split phenotype with functional features intermediate between naive and memory T cells. Int. Immunol. 7:1417-1424. [DOI] [PubMed] [Google Scholar]
- 47.Unutmaz, D., V. N. KewalRamani, S. Marmon, and D. R. Littman. 1999. Cytokine signals are sufficient for HIV-1 infection of resting human T lymphocytes. J. Exp. Med. 189:1735-1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Unutmaz, D., P. Pileri, and S. Abrignani. 1994. Antigen-independent activation of naive and memory resting T cells by a cytokine combination. J. Exp. Med. 180:1159-1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vella, A. T., S. Dow, T. A. Potter, J. Kappler, and P. Marrack. 1998. Cytokine-induced survival of activated T cells in vitro and in vivo. Proc. Natl. Acad. Sci. USA 95:3810-3815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vinion-Dubiel, A. D., M. S. McClain, D. M. Czajkowsky, H. Iwamoto, D. Ye, P. Cao, W. Schraw, G. Szabo, S. R. Blanke, Z. Shao, and T. L. Cover. 1999. A dominant negative mutant of Helicobacter pylori vacuolating toxin (VacA) inhibits VacA-induced cell vacuolation. J. Biol. Chem. 274:37736-37742. [DOI] [PubMed] [Google Scholar]
- 51.Willhite, D. C., and S. R. Blanke. 2004. Helicobacter pylori vacuolating cytotoxin enters cells, localizes to the mitochondria, and induces mitochondrial membrane permeability changes correlated to toxin channel activity. Cell. Microbiol. 6:143-154. [DOI] [PubMed] [Google Scholar]
- 52.Zack, J. A., S. J. Arrigo, S. R. Weitsman, A. S. Go, A. Haislip, and I. S. Chen. 1990. HIV-1 entry into quiescent primary lymphocytes: molecular analysis reveals a labile, latent viral structure. Cell 61:213-222. [DOI] [PubMed] [Google Scholar]
- 53.Zack, J. A., A. M. Haislip, P. Krogstad, and I. S. Chen. 1992. Incompletely reverse-transcribed human immunodeficiency virus type 1 genomes in quiescent cells can function as intermediates in the retroviral life cycle. J. Virol. 66:1717-1725. [DOI] [PMC free article] [PubMed] [Google Scholar]