

Abstract
Spectroscopy (UV–Vis, 1H NMR, ESR) and electrochemistry revealed details of the structure of the Cu(II)–TRH (pyroglutamyl–histidyl–prolyl amide) complex. The 1H NMR spectrum of TRH has been assigned. NMR spectra of TRH in the presence of Cu(II) showed that Cu(II) initially binds TRH through the imidazole. TRH analogs, pGlu-His-Pro-OH, pGlu-(1-Me)His-Pro-amide, pGlu-His-(3,4-dehydro)Pro-amide, pGlu-His-OH, pGlu-Glu-Pro-amide, and pGlu-Phe-Pro-amide provided comparison data. The stoichiometry of the major Cu(II)–TRH complex at pH 7.45 and greater is 1:1. The conditional formation constant (in pH 9.84 borate with 12.0 mM tartrate) for the formation of the complex is above 105 M−1. The coordination starts from the 1-N of the histidyl imidazole, and then proceeds along the backbone involving the deprotonated pGlu-His amide and the lactam nitrogen of the pGlu residue. The fourth equatorial donor is an oxygen donor from water. Hydroxide begins to replace the water before the pH reaches 11. Minority species with stoichiometry of Cu–(TRH)x (x = 2–4) probably exist at pH lower than 8.0. In non-buffered aqueous solutions, TRH acts as a monodentate ligand and forms a Cu(II)–(TRH)4 complex through imidazole nitrogens. All the His-containing analogs behave like TRH in terms of the above properties.
Keywords: Thyrotropin-releasing hormone, NMR, Divalent copper, Peptide, ESR, Imidazole
1. Introduction
Thyrotropin-releasing hormone (also called thyrotropin-releasing factor), pyroglutamyl-histidyl-prolinamide, is a tripeptide in the central nervous system that improves functional recovery after neurologic dysfunctions, such as brain trauma and epilepsy [1]. Produced in the hypothalamus, TRH has been found to exist in significant quantities in other brain regions in rats [2,3], which implies other bioactivities. The effects of regulating body temperature [4] and stimulating hepatic blood flow [5] have been reported recently.
TRH exerts its effects through interactions with a membrane-bound receptor which has been found to be a member of the G-protein coupled receptor family [6]. Perlman et al. [7] modeled TRH binding to its receptor. They claim that the carbonyl and NH of the pyroglutamyl moiety, the imidazole and the C-terminal amide are all involved in the interaction with receptor. At the same time, these sites are potential electron donors for metal ions in that C═O, –NH–, and imidazole nitrogen atoms are known to be electron-rich. Ogawa et al. [8] found that synaptic membranes carrying TRH receptors lose their function after pretreatment with metal ions in the absence of TRH, indicating irreversible changes. Moreover, of the divalent ions they studied, Ni(II) was found to decrease TRH–receptor binding, while Cu(II) and Zn(II) were found to increase the binding at physiological serum concentrations (20 μM). The authors suggested that metal ions are important modulators of TRH activity.
A few studies have been carried out on the coordination of TRH and analogs with biologically active metal ions, e.g. Cu(II) and Ni(II), using techniques including potentiometric titration and spectroscopic methods such as UV–Vis, CD and ESR [9,10]. In the case of coordination with Cu(II) at pH 9.5–10, there was a major disagreement on the number of coordinated nitrogens (two nitrogens in [9], three nitrogens in [10]). While both works reported 1:1 stoichiometry of the complex, in [14] the authors also claimed a major 1:2 metal–ligand binding stoichiometry when ligand was in fivefold excess.
TRH–metal binding is important in another context. The sensitive and selective HPLC-based method for peptides is well established, based on the reversible electrochemistry of the Cu(III)/Cu(II) couple in polydentate peptide complexes, or biuret complexes [11]. The concentration of the neuropeptide TRH can be determined by the above mentioned means [12]. In both cases, the interaction of TRH with its receptor and the determination of TRH concentration, it is important to understand the metal complex structure. In this aspect, the role of pH should draw significant attention.
We also briefly studied the Cu(II) complexes of five TRH analogs in order to relate the nature of the coordination to the functional groups in the peptide side chain. These analogs are (abbreviations listed in parentheses):
| pGlu-His-OH | (pEH) |
| pGlu-His-Pro-OH | (TRH-OH) |
| pGlu-(1-Me-His)-Pro-amide | (MeHis2-TRH) |
| pGlu-His-(3,4-dehydro-Pro)-amide | (deHPro3-TRH) |
| pGlu-Glu-Pro-amide | (Glu2-TRH) |
| pGlu-Phe-Pro-amide | (Phe2-TRH) |
The data support the view that the most prevalent complex in neutral and basic solutions has 1:1 stoichiometry. The four equatorial donor atoms are the imidazole 1-N, and the amide nitrogens resulting from the deprotonation of the amide bonds between pGlu and His and the lactam of pGlu as well as a water oxygen.
2. Experimental
2.1. Chemicals
TRH and all its analogs studied (all from Bachem, USA) were used without further purification, but purities were taken into account in preparation of solutions. All solutions were made in purified water using a Milli-Q A10 Synthesis water purification system (Millipore, USA) except for those used in NMR experiments in which D2O (Cambridge Isotope Lab, USA) was used. The carbonate buffer is a mixture of equi-molar sodium carbonate and bicarbonate (both from EM Science, USA), each at half of the concentration specified, i.e., given concentrations are for total carbonate. Borate buffers were all prepared with Na2B4O7·10H2O (Sigma, USA) in 0.25 M KNO3 (J.T. Baker, USA). The borate concentration was 0.050 M except for the pH effect experiments, in which case the concentration was 0.10 M. The pH values were adjusted with 0.60 M boric acid or 4 M NaOH (both from EM Science, USA), and measured with an Accumet® pH meter (Fisher Science, USA), standardized before each use. In ESR experiments, pH values were measured with pH test strips (Fisher Science, USA) of the pH 5.5 and 9.0 solutions. These two pH values thus have one fewer significant figure. Na2HPO4 (0.126 M)–citric acid (0.037 M) was used as a weakly acidic buffer. Copper-peptide solutions were prepared by adding the desired amount of stock solutions to buffer in the order of peptide then copper sulfate. Molar ratios were 1:1 (Cu(II):peptide) unless otherwise specified.
2.2. Instruments and procedures
Room temperature was 20 ± 2 °C during all experiments.
UV–Vis spectroscopic instruments consisted of an HP 8453 spectrophotometer with HP 8453E operating software. Spectra of copper–peptide complexes in 1 cm quartz cuvettes were measured over 400–1000 nm with a 0.5 s integration time, using deionized water as a solvent blank in all cases. The wavelengths of maximum absorption (λmax) were identified by taking the first derivative with PeakFit (Version 4 for Win32 by Jandel Software, USA). The complex called “Cu(H2O)62+” was CuSO4 in aqueous solution; the Cu(OH)42− complex was prepared by adding dropwise 4 M NaOH to CuSO4 aqueous solution until the light blue Cu(OH)2 precipitate cleared up and a deep blue solution was obtained; the Cu(II)–tartrate complex was a mixture of 1.0 mM CuSO4 and 6.0 mM sodium tartrate in buffer; and the Cu(II)–peptide complexes were mixtures of 1.0 mM corresponding peptide and 1.0 mM CuSO4 in buffer.
TRH spectra of 1H–1H 2-D and 1H–13C 2-D NMR were recorded on an AVANCE 500 NMR spectrometer from Bruker (Billerica, MA, USA). TRH spectra of 1H, 13C, DEPT (Distortionless Enhancement by Polarization Transfer) NMR and 1H spectra of Cu(II) titration experiments were carried out on an AVANCE 300 NMR spectrometer from Bruker. The temperature was controlled at 0 °C. In copper titration experiments, the concentration of TRH was 4.0 mM in 0.20 M carbonate prepared in D2O, with different concentrations of CuSO4.
Electron spin resonance spectroscopy experiments were done with a Bruker Elexsys-E580 FT/CW spectrometer equipped with a Bruker ER 4118X-MS3 split ring resonator. Continuous wave ESR spectra were recorded with a quality factor Q of about 1600, a field-sweep of 600 G, microwave power of 6.3 mW and modulation amplitude of 5–7 G. All Cu(II)–TRH sample solutions were in 20%/80% glycerol/water (the glycerol was added as a cryoprotectant). The pH of each solution was adjusted with 2 M NaOH aqueous solution. All ESR experiments were carried out in the X-band (9.75 GHz) at a controlled temperature of 80 K.
Electrochemical instruments consisted of a DT-29 glassy carbon/glassy carbon ring-disk electrode, an ASR-2 analytical rotator, an RDE-3 potentiostat (all from Pine Instrument Co., Grove City, PA, USA), and a WaveTek 852 filter (WaveTek, San Diego, CA, USA) set for low pass with 5 Hz cutoff frequency. A DT2802 chromatography board (Data Translation, Inc., USA) digitized the filtered analog signal to a personal computer. RRDE data were collected using Ez-Chrom chromatography software (Scientific Software, Inc., San Raman, CA, USA). A platinum grid counter electrode and a BAS Ag/AgCl reference electrode (3 M NaCl) completed the electrochemical cell.
Complex concentrations as a function of total metal and ligand concentrations were calculated using Math-cad 2001 professional (Mathsoft, USA). Molecular simulation was carried out with the software CAChe (Fujitsu, Ltd., Japan) version 6.1.1. Structures were first checked for correct valences and hybridizations, then optimized using Allinger’s standard MM3 force field model [13]. The following factors were taken into account when optimizing: bond stretch, bond angle, dihedral angle, improper torsion, van der Waals interaction, electrostatics interaction, hydrogen bond, torsion stretch, and bend-bend interactions.
3. Results and discussion
In basic Cu(II) solutions, for example pH 9.83 carbonate with tartrate in excess of Cu(II) ion, addition of TRH generates a bluish purple color with maximum absorption wavelength (λmax) about 590 nm (Table 1). This wavelength is substantially lower than that of CuSO4 aqueous solution (Cu(H2O)62+)or Cu(II)–tartrate complex, which are at 810 and 725 nm, respectively. Among the peptides studied, the characteristic bands are at similar wavelengths, except for Phe2-TRH and Glu2-TRH. The half wave potential is also a robust indicator for the formation of the biuret complex. By coordination with peptides, the redox couple Cu(III)/Cu(II) has its oxidation potential significantly lowered to typically 0.5–0.8 V. Based on the distinguishable behavior of the two non-histidyl TRH analogs, we can conclude that for these N- and C-termini blocked peptides, His is necessary for the formation of electrochemically active biuret complexes with Cu(II) ion. DeHPro3-TRH, TRH-OH and pGlu-His all behave like TRH, suggesting that the N-teriminal Pro-NH2 does not play an important role in the binding.
Table 1.
Summary of spectroscopic and electrochemical properties of Cu(II) complexes of TRH and analogs
| λmax (nm)
|
ɛappa (cm−1 M−1)
|
E1/2 (mV)
|
||||
|---|---|---|---|---|---|---|
| Ligands | Carbonate | Borate | Carbonate | Borate | Carbonate | Borate |
| H2Ob | 810 | 17 | n/t | |||
| OH−b | 637 | n/t | n/t | |||
| Tartrate | 729 | 701 | 51 | 36 | N/A | N/A |
| pGlu-His | 595 | 585 | 64 | 65.7 | n/t | 780 |
| TRH | 595 | 588 | 58.7 | 59.6 | 780 | 785 |
| TRH-OH | 596 | 588 | 55.1 | 65.9 | 778 | n/t |
| MeHis2-TRH | 590 | 587 | 58.9 | 66.8 | 802 | n/t |
| deHPro3-TRH | 590 | 588 | 55.5 | 61.4 | 787 | n/t |
| Glu2-TRH | 730 | ppt | 54.4 | N/A | N/A | n/t |
| Phe2-TRH | 730 | ppt | 51.6 | N/A | N/A | n/t |
N/A, no detectable signal available; n/t, not tested; ppt, precipitation.
Obtained by dividing the absorbance at λmax by Cu(II) concentration.
Not in buffer.
3.1. The binding stoichiometry
Tripeptides typically bind Cu(II) ion with 1:1 stoichiometry [14,15]. The tertiary histidyl-prolinamide is, however, a weak donor [16], thus TRH may act as a dipeptide. The molecular binding stoichiometry was determined using “the method of continuous variation”, or Job’s plot (Fig. 1). The total concentrations of Cu(II) and TRH were held constant, while the relative ratios varied for each sample, from 0:1 (all TRH) to 1:0 (all Cu(II)). For pH higher than 8.0, a sixfold excess of tartrate over Cu(II) was included at the same concentration in each sample to prevent excess Cu(II) ion from precipitating. Cu(II) not bound to peptide absorbs light, so measured absorbances need to be corrected to give a measure of the concentrations of Cu(II)–peptide complex. Assuming that the molar absorptivity of unbound Cu(II) is constant, we have CCu(total) = CCu-TRH + CCu-other. At a certain wavelength
Fig. 1.

Job’s plots of Cu(II)–TRH (1:1) complex at different pH, all 2 mM total concentration. (a) Recorded in borate buffer containing 12.0 mM KNaTartrate. (b) In borate buffer, except for * which is non-buffered aqueous solution.
The first term on the right side is just the absorbance of a sample with no peptide (“blank”). Thus, by subtracting a blank absorbance from the observed absorbance, a quantity Anet(= Atotal − CCu(total)·ɛCu-other) is produced that is directly proportional to CCu-TRH·Anet values are then plotted against Cu(II) fraction in the total. The left half of each plot represents excess TRH, and the right half of the plot represents excess Cu(II). The extrapolations of both sides intersect at a Cu(II):peptide ratio indicating the complex stoichiometry.
The Job’s plots revealed 1:1 stoichiometry from pH 8.15 to 11.90, as shown in Fig. 1(a). The λmax and absorbance at this wavelength are also very similar from pH 8.15 to pH 9.91. Fig. 1(b) shows Job’s plots at lower pH values. A 1:4 stoichiometry predominates in non-buffered aqueous system (pH ranged between 5.2 and 5.7). The λmax was 592 nm. No other reports about this stoichiometry have been found in the literature. At both pH 7.45 and 7.10 the apex is not well defined. Also, in both cases the apparent binding stoichiometry involves non-integral TRH per Cu(II) ion: 1.5 (3:2) at pH 7.10 and 1.1 (11:9) at pH 7.45. The deduction from these results at pH lower than 8 is that: (1) another component co-exists with the 1:1 complex; (2) binding is probably incomplete. The second component is a multi-peptide binding mode. A Cu–(TRH)2 complex has been reported to account for ~70% of all Cu(II) between pH 7.5 and 10.0 when ligand was at fivefold excess [10]. Our spectroscopic data cannot determine the actual order of TRH in the multi-peptide binding mode. It could be 1:2, 1:3 or 1:4. Also, according to Fig. 1(b), at pH 7.45 the fraction of the second component is not as much as in the reference cited. It decreases with increasing pH, from a substantial fraction of the entire complex at pH 7.10, to a negligible amount at pH 8.15. At pH 7.45 it only represents a small fraction. Considering the pH dependence of the total apparent stoichiometry, deprotonation is involved in the formation of the 1:1 complex.
It draws interest that the binding of Cu(II) to TRH through amide groups can occur at physiological pH. Metal ions affect TRH–receptor interaction and TRH bioactivity. These observations were related to the tertiary conformation of TRH upon chelation [8,17]. The biuret complex of Cu(II) and TRH should play a significant role if the observed 1:1 binding exists at the much lower concentrations of both TRH and Cu(II) in biological environment. The molar absorptivity of the d–d transition investigated is too low at physiological concentrations to make measurements. Knowledge of the formation constant is required to predict the extent of coordination. In principle, our Job’s plots contain information on the formation constants. However, in practice this method only works at a concentration range where binding is not complete. At the sub-milimolar concentrations adopted in typical visible spectroscopic measurements, the binding of Cu(II) and TRH is completed to a great extent, shown by the well-defined sharp apexes in Job’s plots of pH 8.15–11.90. The observed formation constant Kobs,
is estimated as no lower than 105 M−1, but more accurate determination is not possible from these data.
Yamada et al. [10] reported the formation constants of 1:1 and 1:2 Cu(II)–TRH complexes which agreed with a few other values [18,19]. The average values are logβ1 = −7.70 for MH−2L and logβ2 = −5.42 for MH−2L2 (M – metal, L – ligand, H−2 – two deprotonations). These numbers were obtained from pH titration experiment at tens of mM concentrations and fivefold excess ligand. Under these conditions, the authors saw more than 70% of Cu(II) as a 1:2 complex in the pH range of 7.5–10. Based on their formation constant values, the corresponding conditional formation constants at pH 7.45 were calculated (by multiplying each β by [H+]−2) and used in a calculation of Job’s experiment. In the calculation it was assumed that the 1:1 and 1:2 stoichiometry are the only forms of coordination. The result showed the fraction of 1:2 stoichiometry decreased dramatically with decreasing concentration and peptide:Cu(II) ratio. At the concentrations used in our Job’s experiments, MH−2L (1:1) predominates. When TRH is in excess, no more than 10% of Cu(II) exists as MH−2L2. When Cu(II) is in excess, there is virtually no MH−2L2. Physiologically, Cu(II) is in large excess of TRH. TRH exists in rat brain at nM concentrations [2], while Cu(II) has been determined with values ranging from approximately 40–150 μM [20]. At this excess, TRH forms predominantly a 1:1 complex with Cu(II). As calculated for 5 nM TRH and 50 μM Cu(II), 99.9% of TRH is bound with Cu(II) at 1:1 ratio. In the brain, the Cu(II) activity is certainly less than 50 μM, however, the complex is still predominantly 1:1. For example, with 5 nM TRH and 50 nM Cu(II), about 40% of TRH is bound with Cu(II) at 1:1 ratio. The concentration of the 1:2 species is 10−6 times lower.
For the computations above we used literature values for the binding constants. Our estimate of the formation constant of the 1:1 species is somewhat lower than the one used. The discrepancy is likely due to the difference in Cu(II) activity coefficients. Our measurements involved coordinating ions, nitrate and tartrate, and as such, the conditional constants derived from the data have little meaning in general. Nonetheless, the 1:1 coordination of Cu(II) and TRH cannot be ruled out as an important process occurring biologically.
3.2. The equatorial donors
Billo [21] developed a correlation between the absorption maxima for Cu(II) complexes and the number and type of equatorial donors. With slight modification of the factors, the correlation worked well on multidentate ligands such as peptides [22]. According to this equation, the experimental λmax data (pH 9.84) from the four histidyl-containing peptide complexes in Table 1 are consistent with the NNNO binding mode, in which each Cu(II) ion has three nitrogen atoms and one oxygen atom as equatorial donors. In fact, from pH 7.45 to 9.91, Cu(II)–TRH coordination fell in the NNNO binding region.
Electron spin resonance (ESR) can also give evidence of the number of nitrogen donors in Cu(II) complexes, often of peptides and proteins [23–25]. For Cu(II), the four features due to the parallel component of the hyperfine interaction (A||, with the nucleus of spin 3/2) can often be resolved near the g|| (the parallel component of the axially symmetric g-tensor) region of the spectrum. The magnitude of A|| and g|| are dependent on, among many factors, the ligand environment of Cu(II). Peisach and Blumberg [25] showed the dependence of A|| and g|| values and total charge of Cu(II) complexes on the ligand environment. Generally, as the number of nitrogen donors increases, all the four peaks shift to higher field. The A|| value also increases, from 120 to 220 G when all four oxygen donors are replaced with nitrogen. At the same time, g|| value decreases from 2.4 to 2.1. Millhauser and co-workers [24] reported the binding modes of a Cu(II) complex and the prion protein octarepeats change from OOOO to NNNN in the pH range of 4.10–11.6. Their correlation between A|| and g|| values and the binding mode agreed with Peisach’s.
ESR spectra were obtained for Cu(II) ion in presence of TRH at pH 5.5, 7.45, 9.0 and 10.76. A|| and g|| values were extracted. As shown in Fig. 2 and Table 2, the main binding mode at pH 5.5 is OOOO. There is a minor component other than OOOO at slightly higher field. Due to the ambiguity of the second peak, it is difficult to assign any binding mode to this component. Based on the position of the first peak, one nitrogen binding is estimated. From 7.45 to 10.76, the Cu(II) ion hyper-fine structure did not change significantly, suggesting the same equatorial binding. Shown in Table 2, the A|| and g|| values for all these pH values very well fit the NNNO binding mode. These results also are in good agreement with the visible spectroscopic data.
Fig. 2.
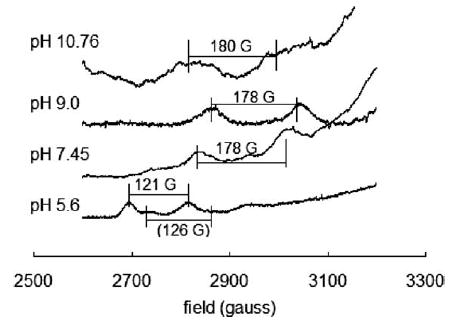
The g|| region of the Cu(II) ESR spectra in the presence of TRH (1.0 mM each). The A|| value at pH 10.76 was obtained by simulating the spectrum with Bruker WINEPR simulation package because the peaks are very broad making direct reading difficult. The A|| value of the “shoulder” component at pH 5.5 was roughly estimated because the second peak was not well defined.
Table 2.
ESR and UV–Vis parameters for Cu(II) signals in presence of TRH at various values of pH
| pH | A||(G) | g|| | λmaxa (nm) | Equatorial binding |
|---|---|---|---|---|
| 5.5 | 121 ± 2 | 2.41 ± 0.01 | 743 | OOOO |
| 7.45 | 178 ± 2 | 2.26 ± 0.01 | 589 | NNNO |
| 9.0 | 178 ± 2 | 2.26 ± 0.01 | 590 | NNNO |
| 10.76 | 175 ± 5 | 2.27 ± 0.01 | 586 | NNNO |
pH 6.24, 7.01, 8.97 and 11.01, respectively.
Potential nitrogen donors in the TRH molecule include one lactam nitrogen from the pyroglutamyl residue, 1-N (pyridine) and 3-N (pyrrole) of the imidazole ring, two amide nitrogens from the two peptide bonds, and one amide nitrogen at the blocked C-terminus. The oxygen donor could be any one of the carbonyls, a water or a hydroxide. As mentioned above, the Pro residue does not seem to take part in the coordination. The tertiary amide is a weak donor. Furthermore, the nitrogen lone pair and the carbonyl group should be in the same plane [14]. This is extremely difficult for a prolinyl amide. As for the C-terminal amide, formation of a five- or six-membered fused ring structure does not seem possible. At the same time, the spectroscopic properties of TRH, deHPro3-TRH and TRH-OH are very close to each other. All the above argue against the participation of Pro in the binding. Despite all this, TRH does not act as a di-peptide. It forms a 1:1 complex with Cu(II) ion with three nitrogen donors, as do tripeptides. The His residue is known to have an electron-rich side-chain that can act as metal ligand [19,26]. If we do not consider Pro as having a donor, the three nitrogen donors are possibly the 1-N of imidazole, the lactam nitrogen of pGlu, and the amide nitrogen between the pGlu and His residues.
In general, biuret coordination starts at the N-terminal amine nitrogen of the peptide. The peptide amide bonds undergo deprotonation following that to form stable fused ring structures [14,15]. In a TRH molecule, there is no amine group. The C-terminus, known to play a role when the N-terminus is blocked [27], is also blocked. It is of interest to find out how the coordination starts in a peptide with both termini blocked.
Selective broadening of proton and carbon resonances of ligands in NMR spectroscopy by a paramagnetic metal ion, such as Cu(II), can help locate the binding sites. Such ions increase fluctuations in the local magnetic field and reduce the both the longitudinal (T1) and transverse (T2) relaxation times of other nuclei nearby [28], thus altering their NMR signals. As line width is proportional to T2−1, an affected signal could be broadened and shortened, or even eliminated depending on the magnetic ion concentration [29]. By comparing the NMR spectra of ligands in the presence and absence of metal ion, one can deduce the most-affected protons or carbons in the ligand molecules. In a selective broadening experiment, the metal ion is usually held at 1/1000 or less of the ligand concentration. In the case of peptide complexes in basic solutions, however, the binding at this molar ratio is very often not the same as at stoichiometric conditions. The main reason is that the relatively rare Cu(II) ions associate with and dissociate from the peptides rapidly before any peptide amide deprotonation or ring closure occurs [30]. In other words, the peptide acts as a monodentate ligand. The NMR signal changes under this condition will only indicate where the coordination begins, as the N-termini of glycyl di- or tri- peptides [31].
Fig. 3 shows the 1H NMR spectra of intact TRH and Cu(II)-titrated TRH solutions. Assignments of the peaks in both spectra required the TRH spectra of 1H–1H 2-D, 1H–13C 2-D NMR and DEPT (distortionless enhancement by polarization transfer, which records 13C spectra that are edited with respect to the number of protons directly bonded to the carbons). As shown in Fig. 3(b), the imidazole protons respond most sensitively, showing signs of broadening and lowering when Cu(II) is present at only 1/4000 equivalents of TRH. This result suggests that the imidazole acts as the initiating site during the binding, analogous to the primary –NH2 group in most peptides. The two nitrogen atoms are both good donors, while the 1-N does have the advantage to form six-membered ring structure. Therefore, 1-N is thermodynamically favored.
Fig. 3.

1H NMR spectra of TRH in the absence and presence of Cu(II) ion.
To sum up, the NNNO binding is a reasonable structure and consistent with experimental data. Amide deprotonation propagates along the peptide backbone toward pGlu residue once the 1-N of imidazole coordinates with Cu(II). As for the fourth donor, the oxygen, Billo’s equation is not able to distinguish a carbonyl, water or hydroxide. The computational software CAChe helped to verify the feasibility of some possible structures and to exclude some. The structure with a carbonyl oxygen either from the His-Pro peptide bond or from the C-terminal amide yielded structures with no reasonable prolyl C-terminal carbonyl bond length or reasonable tetragonal complex structure. The fourth donor has to be a water or a hydroxide from this point of view. Fig. 4 is an optimized structure by CAChe. The four donors are the deprotonated pGlu lactam nitrogen, the deprotonated pGlu-His peptide amide nitrogen, the His 1-N and H2O. Cu(II)–peptide biuret complexes are square planar [32]. The two axial water ligands are placed 3 Å from Cu(II). Atoms at this distance hardly affect the spectra. The simulation also suggested that there may be a hydrogen bond between the C-terminal amide carbonyl oxygen and the equatorial water proton.
Fig. 4.

CAChe simulation of Cu(II)–TRH complex in the NNNO binding mode. The structure was optimized by the ‘‘beautify’’ function on valence, hybridization and geometry. The 1.763 Å labels the O–H distance of a possible hydrogen bond.
3.3. The effect of pH
As deprotonation is a crucial step in Cu(II) and peptide binding, the pH of the solution is expected to affect significantly the complex formation. Fig. 5 shows the band shift with increasing pH of Cu(II)–TRH in borate buffers. From the Job’s plots and ESR results at various values of pH, we acquired the knowledge that in buffered systems, TRH starts to bind Cu(II) ion in an NNNO mode at pH slightly over 7.0. At least through pH 10.8 the equatorial binding mode is always NNNO. There is no evidence for NNOO binding. The pH range for existence of an NNOO binding mode, if there should be one, is expected to be very narrow, within pH 6–7. This likely results from the cooperativity in the serial binding of the three nitrogen donors.
Fig. 5.

Band shift of Cu(II)–TRH with increasing pH in borate buffer. Regions between dashed lines define the following wavelength ranges: A – NNNO (one imidazole, two deprotonated amides, one water or hydroxide); B – NNNN (two imidazoles, two deprotonated amides); C – NNNN (one imidazole, three deprotonated amides).
There are signs of multiple TRH binding at lower pH. The histidine is a basic residue. The pKa of protonated imidazole has reported values ranging from 6.0 to 7.1 [33,34]. In about neutral solutions with Cu(II) present, one of the two imidazole nitrogens undergoes deprotonation and binds Cu(II). In this way the TRH acts as a monodentate ligand. This possibly accounts for the donor in non-buffered solutions, in which one Cu(II) ion binds four TRH molecules. From a steric aspect, this seems easier for the 3-N (pyrrole). The non-buffered Job’s experiment of one of the analogs, MeHis2-TRH, should differentiate the two imidazole nitrogens. The MeHis2-TRH molecule bears a methyl group at the 3-N. As a monodentate ligand, the only available nitrogen donor is the imidazole 1-N. If the His 1-N sees substantial steric hindrance, this analog would not bind Cu(II). The Job’s experiment with Me-His2-TRH yielded a similar plot as the one with TRH (see Fig. 6). The λmax was 590 nm. The apparent stoichiometry was about 1:4, too. Obviously, the 1-N of His imidazole is capable of binding Cu(II). The apex of MeHis2-TRH plot was lower than that of TRH. The apparent binding constant for MeHis2-TRH in this case is smaller than that of the native peptide. At this point, we are not able to identify the nitrogen donor when TRH acts as a monodentate ligand. It could be either the 1-N or the 3-N of the imidazole. Fig. 7 shows the CAChe simulation of Cu(II)–(TRH)4 (via imidazole 3-N) and Cu(II)–(MeHis2-TRH)4 (via imidazole 1-N). The relative stabilities of the two divalent complex ions are indicated by their ‘‘current energy’’ calculated by CAChe of the optimized structures. The two numbers are only a few percent different. They show relatively equal stability.
Fig. 6.
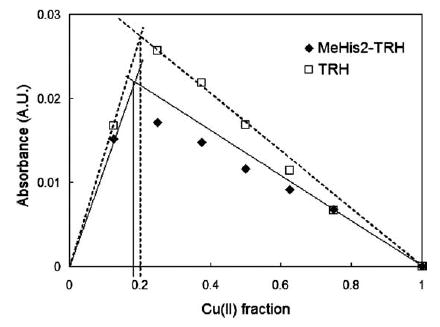
Job’s plots of Cu(II)–TRH and Cu(II)–MeHis2TRH in non-buffered solutions. The perpendicular line indicates Cu(II) fraction 0.20, which corresponds to Cu(II):peptide = 1:4.
Fig. 7.
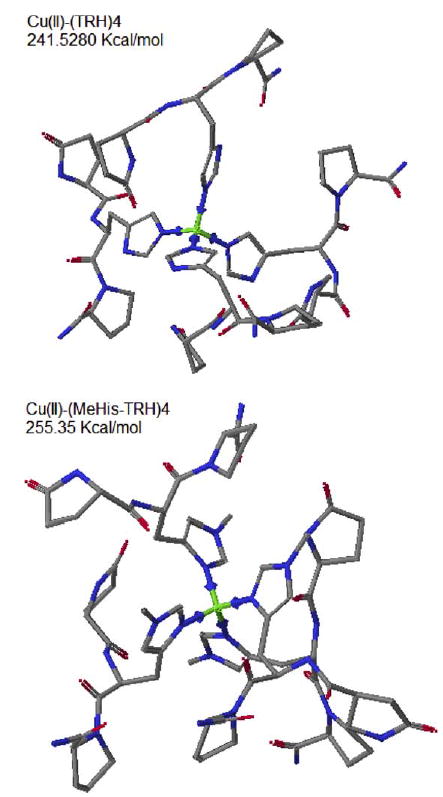
CAChe simulation of Cu(II)–TRH4 (binding at imidazole 3-N) and Cu(II)–(MeHis2TRH)4 (binding at imidazole 1-N). The listed energy values are “current energy” of geometry- and hybridization-beautified structures.
The pH 7.10 Job’s plot reveals a mixture of 1:1 stoichiometry and complexes with higher peptide:Cu(II) ratios. At pH 7.45 the 1:1 complex predominates. This means that in the presence of Cu(II) the pKa of the two amides is not much higher than that of the imidazole. The 1:1 and 1:4 binding modes give very close λmax values: 588 nm for the 1:1 binding and 592 nm for the 1:4 binding. One would deduce that for any transitional binding mode between these two the absorption band is within the range of 588–590 nm. This is why the Cu(II)–TRH d–d band does not shift significantly with increasing pH from 7.01 to 11.01 (<10 nm when Cu(II) and TRH were at 1:1 molar ratio, Fig. 5).
As pH further increases to over 12.0 the band shifts to 564 nm. This sharp shift of absorption band indicates deprotonation. There are two possible deprotonations that could cause band shifting: (1) deprotonation of an amide, which binds Cu(II) as the fourth donor; (2) deprotonation of the 3-N (pyrrole) of the imidazole, which may or may not bind Cu(II). Hypothesis 1 was invalidated by CAChe simulation. The only available amide, the C-terminal amide is not able to reach the equatorial position while keeping reasonable bond angles and lengths. For hypothesis 2, Cu(II)-binding-facilitated proton loss from an imidazole pyrrole nitrogen has been reported widely. It has been reported to occur at various pH values depending on the actual ligand structure, as low as pH 9.6 in some cases with TRH [35,36]. The deprotonated pyrrole nitrogen of imidazole may or may not coordinate with Cu(II) ion [34,37]. However, shown by Fig. 8, all the five His-containing TRH-like peptides including MeHis2-TRH which is not capable of pyrrole deprotonation display very similar pH-dependent band-shifting. This is a strong evidence against hypothesis 2. For TRH the pyrrole nitrogen deprotonation is still possible and consequently may lead to polymerization of Cu(II)–TRH [35] (but the stoichiometry will still be 1:1). However, this deprotonation is not the main cause of the bandshifting at high pH.
Fig. 8.

Band shift of Cu(II) complexes of TRH analogs with increasing pH in borate buffer. Only His-containing analogs are included.
Another possible deprotonation is the deprotonation of the equatorial water, or replacement of the water by a hydroxide. Billo [21] did not distinguish the contributions of carbonyl, water or hydroxide oxygen in his equation. In a later regression by Sigel and Martin [22], the three types of oxygen donors also had the same contribution to Cu(II) complex absorption. While the above two set of factors agree closely and work relatively well on general Cu(II) complexes of nitrogen-containing ligands, attention should be drawn to the generally different donating abilities of the three oxygen donors, especially the water which is a known weak donor compared to the other two. Prenesti et al. [38] studied over 100 Cu(II) complexes and came up with refined factors. Important to their discussion, they found a bigger contribution from carbonyl and hydroxide (0.39), while their factor for water is in agreement with Billo and Sigel and Martin (0.296). Our absorption data of [Cu(OH−)4]2− (see Table 1) gives a factor of 0.391 for OH− in this complex in agreement with Prenesti et al. The increased contribution factor of hydroxide shifts the calculated Cu(II)–TRH band (NNN(OH−)) to 553 nm. This deprotonation of water ligand does not depend on the actual peptide structure, which is consistent with Fig. 8.
4. Conclusion
TRH and its analogs lack a primary amine group which is generally the initiating site of coordination with Cu(II) ion. In TRH, the imidazole nitrogen initiates complexation. Coordination of Cu(II) ion and TRH starts from 1-N of the imidazole and proceeds along the peptide backbone toward the N-terminus, involving the deprotonated amide between pGlu and His, and the deprotonated lactam amide in the pGlu residue. A water molecule completes the ligand set. Binding in this way occurs at pH as low as 7.10 in buffered systems. There should be considerable Cu(II)–TRH interaction under physiological conditions (pH 7.4), which may affect TRH–receptor interaction. The 1:1 stoichiometry is expected to predominate at physiological concentrations of Cu(II) and TRH.
Acknowledgments
The authors are grateful to the National Institutes of Health for support through Grant GM44842. We are also grateful to Dr. Marco Bonora for his help with ESR data acquisition.
Footnotes
Dedicated to Prof. Rex Shepherd for his many enthusiastic and insightful comments and discussion on inorganic chemistry, teaching of chemistry and life in general
References
- 1.a Kastin AJ, Ehrensing RH, Schalch DS, Anderson MS. Lancet. 1972;2:740. doi: 10.1016/s0140-6736(72)92028-4. [DOI] [PubMed] [Google Scholar]; b Prange AJ, Jr, Breese GR, Cott JM, Martin BR, Cooper BR, Wilson IC, Plotnikoff NP. Life Sci. 1974;14:447. doi: 10.1016/0024-3205(74)90359-2. [DOI] [PubMed] [Google Scholar]; c Prange AJ, Jr, Lara PP, Wilson IC, Alltop LB, Breese GR. Lancet. 1972;2:999. doi: 10.1016/s0140-6736(72)92407-5. [DOI] [PubMed] [Google Scholar]
- 2.Winokur A, Utiger RD. Science. 1974;185:265. doi: 10.1126/science.185.4147.265. [DOI] [PubMed] [Google Scholar]
- 3.A. Winokur, R.D. Utiger, R. Collu, A. Barbeau, J.R. Ducharme (Eds.), Central Nervous System Effects of Hypothalamic Hormones and Other Peptides [Symposium], Raven, New York, 1979, p. 55.
- 4.Bird JA, Clarke L, Symonds ME. Biol Neonate. 1998;73:52. doi: 10.1159/000013960. [DOI] [PubMed] [Google Scholar]
- 5.Hashimoto T, Yoneda M, Kojima K, Murohisa T, Tamano M, Iijima M, Shimada T, Sugaya H, Terano A, Tamori K, Nakade Y, Yokohama S, Nakamura K, Makino I. Jpn Pharmacol Ther. 2001;29:s189. [Google Scholar]
- 6.Yamada M, Monden T, Satoh T, Satoh N, Murakami M, Iriuchijima T, Kakegawa T, Mori M. Biochem Biophys Res Commun. 1993;195:737. doi: 10.1006/bbrc.1993.2107. [DOI] [PubMed] [Google Scholar]
- 7.a Perlman JH, Laakkonen L, Osman R, Gershengorn MC. J Biol Chem. 1994;269:23383. [PubMed] [Google Scholar]; b Perlman JH, Thaw CN, Laakkonen L, Bowers CY, Osman R, Gershengorn MC. J Biol Chem. 1994;269:1610. [PubMed] [Google Scholar]; c Laakkonen LJ, Guarnieri F, Perlman JH, Gershengorn MC, Osman R. Biochemistry. 1996;35 [Google Scholar]
- 8.Ogawa N, Mizuno S, Kishimoto T, Mori A, Kuroda H, Ota Z. Neurosci Res. 1984;1:363. doi: 10.1016/0168-0102(84)90041-5. [DOI] [PubMed] [Google Scholar]
- 9.Formicka-Kozlowska G, Kozlowski H, Jezowska-Trzebiatowska B, Kupryszewski G, Przybylski J. Inorg Nucl Chem Lett. 1979;15:387. [Google Scholar]
- 10.Yamada Y, Nakasuka N, Tanaka M. Inorg Chim Acta. 1991;185:49. [Google Scholar]
- 11.a Chen JG, Woltman SJ, Weber SG. J Chromatogr A. 1995;691:301. doi: 10.1016/0021-9673(94)00843-x. [DOI] [PubMed] [Google Scholar]; b Warner AM, Weber SG. Anal Chem. 1989;61:2664. doi: 10.1021/ac00198a015. [DOI] [PubMed] [Google Scholar]
- 12.Meng R, Xia W, Sandberg M, Stephens R, Weber SG. J Chromatogr. 2004;A doi: 10.1016/j.chroma.2004.12.032. in press. [DOI] [PubMed] [Google Scholar]
- 13.a Lii JH, Allinger NL. J Am Chem Soc. 1989;111:8576. [Google Scholar]; b Lii JH, Allinger NL. J Comput Chem. 1998;1998:9. [Google Scholar]
- 14.Margerum DW. Pure Appl Chem. 1983;55:23. [Google Scholar]
- 15.Margerum DW, Wong LF, Bossu FP, Chellappa KL, Czarnecki JJ, Kirksey ST, Jr, Neubecker TA. Adv Chem Ser. 1977;162:281. [Google Scholar]
- 16.a Bataille M, Formicka-Kozlowska G, Kozlowski H, Pettit LD, Steel I. J Chem Soc Chem Commun. 1984;4:231. [Google Scholar]; b Formicka-Kozlowska G, Kozlowska H, Siemion IZ, Sobczyk K, Nawrocka E. J Inorg Biochem. 1981;15:201. [Google Scholar]
- 17.Tonoue T, Minagawa S, Kato N, Kan M, Terao T, Nonoyama K, Ohki K. Pharmacol Biochem Behav. 1979;10:201. doi: 10.1016/0091-3057(79)90087-x. [DOI] [PubMed] [Google Scholar]
- 18.Formicka-Kozlowska G, Bezer M, Pettit LD. J Inorg Biochem. 1983;18:335. doi: 10.1016/0162-0134(85)85058-3. [DOI] [PubMed] [Google Scholar]
- 19.Farkas E, Sovago I, Kiss T, Gergely A. J Chem Soc Dalton Trans. 1984:611. [Google Scholar]
- 20.a Hanig RC, Aprison MH. Anal Biochem. 1967;21:169. doi: 10.1016/0003-2697(67)90178-9. [DOI] [PubMed] [Google Scholar]; b Andrasi E, Farkas E, Scheibler H, Reffy A, Bezur L. Arch Gerontol Geriat. 1995;21:89. doi: 10.1016/0167-4943(95)00643-y. [DOI] [PubMed] [Google Scholar]
- 21.Billo EJ. Inorg Nucl Chem Lett. 1974;10:613. [Google Scholar]
- 22.Sigel H, Martin RB. Chem Rev. 1982;82 [Google Scholar]
- 23.Burns CS, Aronoff-Spencer E, Dunham CM, Lario P, Avdievich NI, Antholine WE, Olmstead MM, Vrielink A, Gerfen GJ, Peisach J, Scott WG, Millhauser GL. Biochemistry. 2002;41:3991. doi: 10.1021/bi011922x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aronoff-Spencer E, Burns CS, Avdievich NI, Gerfen GJ, Peisach J, Antholine WE, Ball HL, Cohen FE, Prusiner SB, Millhauser GL. Biochemistry. 2000;39:13760. doi: 10.1021/bi001472t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peisach J, Blumberg WE. Arch Biochem Biophys. 1974;165:691. doi: 10.1016/0003-9861(74)90298-7. [DOI] [PubMed] [Google Scholar]
- 26.a Boka B, Myari A, Sovago I, Hadjiliadis N. J Inorg Biochem. 2004;98:113. doi: 10.1016/j.jinorgbio.2003.09.012. [DOI] [PubMed] [Google Scholar]; b Sovago I, Farkas e, Bertalan C, Lebkiri A, Kowalik-Jankowska T, Kozlowski H. J Inorg Biochem. 1993;51:715. doi: 10.1016/0162-0134(93)85004-r. [DOI] [PubMed] [Google Scholar]
- 27.Shi F, Woltman SJ, Weber SG. Anal Chim Acta. 2002;474:1. [Google Scholar]
- 28.Bertini I, Luchinat C. Coord Chem Rev. 1996;150:77. [Google Scholar]
- 29.J.W. Emsley, J. Feeney, L.H. Sutcliffe, High-Resolution NMR Spectroscopy, Pergamon Press, New York, 1965.
- 30.Espersen WG, Martin RB. J Am Chem Soc. 1976;98:40. [Google Scholar]
- 31.Li NC, Scruggs RL, Becker ED. J Am Chem Soc. 1962;84:4650. [Google Scholar]
- 32.a Nolan KB, Soudi AA, Hay RW. Amino Acids Peptides Proteins. 1994;25 [Google Scholar]; b Nolan KB, Soudi AA, Hay RW. Amino Acids Peptides Proteins. 1996;27:282. [Google Scholar]
- 33.Grant G, Ling N, Rivier J, Vale W, Butcher M, Hewitt W. Biochemistry. 1972;11:3070. doi: 10.1021/bi00766a020. [DOI] [PubMed] [Google Scholar]
- 34.Sundberg RJ, Martin RB. Chem Rev. 1974;74:471. [Google Scholar]
- 35.Morris PJ, Bruce Martin R. J Inorg Nucl Chem. 1971;33:2913. [Google Scholar]
- 36.Osz K, Varnagy K, Suli-Vargha H, Sanna D, Micera G, Sovago I. Dalton Trans. 2003;10:2009. [Google Scholar]
- 37.Aiba H, Yokoyama A, Tanaka H. Bull Chem Soc Jpn. 1974;47:1437. [Google Scholar]
- 38.Prenesti E, Daniele PG, Prencipe M, Ostacoli G. Polyhedron. 1999;18:3233. [Google Scholar]