

Abstract
β-Amyloid (Aβ) acquires toxicity by self-aggregation. To identify and characterize the toxic form(s) of Aβ aggregates, we examined in vitro aggregation conditions by using large quantities of homogenous, chemically synthesized Aβ1–40 peptide. We found that slow rotation of Aβ1–40 solution reproducibly gave self-aggregated Aβ1–40 containing a stable and highly toxic moiety. Examination of the aggregates purified by glycerol-gradient centrifugation by atomic force microscopy and transmission electron microscopy revealed that the toxic moiety is a perfect sphere, which we call amylospheroid (ASPD). Other Aβ1–40 aggregates, including fibrils, were nontoxic. Correlation studies between toxicity and sphere size indicate that 10- to 15-nm ASPD was highly toxic, whereas ASPD <10 nm was nontoxic. A positive correlation between the toxicity and ASPD >10 nm also appeared to exist when Aβ1–42 formed ASPD by slow rotation. However, Aβ1–42-ASPD formed more rapidly, killed neurons at lower concentrations, and showed ≈100-fold-higher toxicity than Aβ1–40-ASPD. The toxic ASPD was associated with SDS-resistant oligomeric bands in immunoblotting, which were absent in nontoxic ASPD. Because the formation of ASPD was not disturbed by pentapeptides that break β-sheet interactions, Aβ may form ASPD through a pathway that is at least partly distinct from that of fibril formation. Inhibition experiments with lithium suggest the involvement of tau protein kinase I/glycogen synthase kinase-3β in the early stages of ASPD-induced neurodegeneration. Here we describe the identification and characterization of ASPD and discuss its possible role in the neurodegeneration in Alzheimer's disease.
A 40- to 42-residue peptide named β-amyloid (Aβ) is a major constituent of senile plaques in Alzheimer's disease (AD) (1). Although multiple pathways have been suggested to lead to AD, recent advances indicate a causal link between Aβ and AD (2), and this idea is supported further by findings that vaccination against Aβ ameliorates behavioral deficits in transgenic mice (3, 4, 5). Among various in vivo Aβ species, Aβ1–42 generally is considered as the primary vehicle of toxicity, whereas Aβ1–40, a major species under physiological conditions, is considered less harmful and more resistant to the formation of oligomers than Aβ1–42 (6). However, it remains controversial which Aβ species contributes predominantly to AD pathogenesis, because both in vitro and in vivo studies have confirmed toxicity of Aβ1–40 aggregates (7, 8).
It has been widely accepted that toxicity of Aβ requires aggregation of native Aβ monomers (9, 10, 11). Besides fibrils, several types of nonfibrillar aggregates have been reported: Aβ1–40 oligomers from dimers–hexamers (6, 12, 13, 14); a mixture of Aβ1–42 oligomers named Aβ-derived diffusible ligands (ADDLs), ranging from trimers–hexamers up to 24-mers (15); and fibril intermediates named protofibrils (PFs) (16, 17). All of these aggregates are mixtures of Aβ oligomers with a variety in oligomer size, and precise morphological analysis of each Aβ aggregate is difficult. In other words, the heterogeneity of Aβ aggregates in terms of Aβ species and oligomer size makes it difficult to reach a consensus about the oligomerization state of Aβ causing the pathogenicity. It is yet to be determined whether all of these in vitro oligomers actually exist in vivo and induce neuronal damage, or whether a particular oligomer is primarily responsible for AD.
To identify and characterize the toxic forms of Aβ aggregates, we first examined in vitro aggregation conditions by using the chemically synthesized Aβ1–40 and found that slow rotation of Aβ1–40 solution reproducibly gave Aβ1–40 aggregates containing a stable and highly neurotoxic moiety. Purification of the toxic moiety revealed that it is a perfect sphere, which we call amylospheroid (ASPD). Other moieties, including fibrils, were nontoxic. Second, we applied the above conditions to Aβ1–42 and compared Aβ1–42-ASPD with Aβ1–40-ASPD. Finally, we used lithium to examine whether there is a link between ASPD toxicity and activation of glycogen synthase kinase-3β (GSK-3β) (18), which also is known as tau protein kinase I (TPKI) (19). Here, we describe the identification and characterization of ASPD and discuss its potential involvement in AD.
Materials and Methods
Synthesis of Aβs. Aβ1–40 and Aβ16–20 were synthesized and purified in Mitsubishi Kagaku Institute of Life Sciences by solid-phase fluorenylmethoxycarbonyl chemistry (>50% yield). Amino acid analysis, analytical HPLC, and matrix-assisted laser desorption ionization–time-of-flight mass spectrometry confirmed the purity of the products. The purified Aβs were lyophilized, dissolved in 35% CH3CN in 0.1% trifluoroacetic acid, distributed into Eppendorf tubes (0.1 μmol), lyophilized, and kept at –20°C until used.
Preparation of Toxic Aβ1–40 Aggregates by Slowly Rotating Aβ1–40 Solution. Aβ1–40 was synthesized or purchased from Bachem or U.S. Peptide (Rancho Cucamonga, CA). Toxic Aβ1–40 aggregates were prepared as follows: Aβ1–40 was suspended in 0.22-μm-filtered Milli Q water (700 μM), incubated at 4°C for 30 min, and diluted with 0.22-μm-filtered Dulbecco's PBS without Ca2+ and Mg2+ (PBS) (Nacalai Tesque, Kyoto) to 350 μM. Aβ1–40 solution (350 μM) was rotated slowly at 37°C for 5–7 days by using a rotating cultivator. We describe toxic Aβ1–40 aggregates prepared by slowly rotating Aβ1–40 solution (350 μM in 50% PBS) as SR350–Aβ1–40. In some experiments, the concentration of Aβ1–40 was reduced to 1 or 50 μM, which we describe as SR1–Aβ1–40 and SR50–Aβ1–40, respectively. In experiments using Aβ16–20 (KLVFF) (20) or iAβ5 (LPFFD) (21) (Peptide Institute, Osaka), Aβ1–40 solutions were mixed with a 10-fold molar excess of the pentapeptides at the onset of slow rotation.
Purification of ASPD from SR350–Aβ1–40. ASPD was isolated by eliminating fibrils from SR350–Aβ1–40 by filters with 0.65-μm and 30-kDa molecular sieves. For further purification, SR350–Aβ1–40 (0.11 ml) was centrifuged at 86,000 × g for 16 h (TLS55; Beckman Coulter) at 4°C in a 15–30% linear glycerol gradient. Ten-drop fractions were collected from the bottom. The pellet was suspended in 50% PBS (0.11 ml). Fractionated samples were examined immediately by immunoblotting, toxicity assays, and transmission electron microscopy. As a control, 50% PBS was centrifuged and analyzed similarly.
Preparation and Purification of Aβ1–42-ASPD. Toxic Aβ1–42 aggregates were prepared from Aβ1–42 (Bachem) in essentially the same way as from Aβ1–40, by slowly rotating Aβ1–42 solutions (1–0.01 μM in 50% PBS) at 4°C for 8–10 h. Aβ1–42-ASPD was purified from SR0.1–Aβ1–42 by using glycerol-gradient centrifugation as described above.
Transmission Electron Microscopy of ASPD. The isolated ASPD was negatively stained with 4% uranyl acetate solution on carbon-coated grids, analyzed immediately by JEOL JEM-1200EX/Top at 100 kV with an anticontamination system under liquid nitrogen, and photographed by using a minimum-dose system to prevent radiation damage. The size of ASPD was determined by NIH IMAGE or by measuring the diameter at the equator with a micrometer. Both methods gave similar results. In the case of unstained ASPD, ASPD was mixed with type VII agarose (1.0%; Sigma), fixed with 4% glutaraldehyde/PBS for 2 h, and embedded in Epon 812 resin (22). A horizontal thin section was made from the resin, and a cross section was made from a horizontal section, adjacent to the first one, reembedded in the resin. Both sections were examined by JEOL JEM-1230 at 100 kV.
Fluid-Phase Imaging of ASPD by Atomic Force Microscopy. The isolated ASPD was deposited on freshly cleaved mica (Nihon Veeco KK, Tokyo) that was modified with 3-aminopropylethoxysilane (23) and left for 30 min. Imaging was performed under 50% PBS by a multimode microscope with a Nanoscope III controller in a tapping-mode fluid cell (Digital Instruments, Santa Barbara, CA) by using A-scanner with 700-nm maximum lateral scan area. Silicon nitride probes coated with 0.5% polyethyleneimine (0.15 M borate buffer), attached to a 100-μm triangular cantilever (force constant of 0.08 N/m; Olympus, New Hyde Park, NY), were operated at 9-kHz resonant frequency with 0.5-Hz scan rates. Images were captured at 256 × 256 pixels.
Toxicity Assays. Primary cultures from rat septum regions, which include septal and basal forebrain cholinergic neurons, were prepared, and the medium (400 μl per well) was replaced with serum-free medium after 3 days in vitro (24). After 5 days in vitro, cultures were treated with either slowly rotated Aβ1–40 solution (SR–Aβ) or ASPD. After a 16-h treatment, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) activity, a toxicity indicator (25, 26), was determined by measuring the absorbance at 570 nm. The background value was obtained by lysing cells with 0.1% Triton X-100 treatment for 10 min. Toxicity was estimated as inhibition of MTT activity compared with the control value obtained upon vehicle addition. After a 40-h treatment, cultures were rinsed and exposed for 30 min to propidium iodide (5 μM) and calcein-AM (0.2 μM) in 20 mM Hepes (pH 7.3) containing 130 mM NaCl, 5.4 mM KCl, 5.5 mM glucose, and 2 mM CaCl2 at 37°C. Dye uptake was measured by using Zeiss Axiovert 200M with CoolSNAP HQ (Roper Scientific, Trenton, NJ) and analyzed with MetaMorph (Molecular Devices). Cells with shrunken or fragmented nuclear staining were counted as dead cells. The total cell number was assessed by Hoechst 33258 (1 μg/ml) staining after 10% formalin fixation. At least 1,200 cells were selected randomly from four different fields. Apoptotic activity was expressed as the ratio of apoptotic cells to the total cell number.
TPKI/GSK-3β Kinase Activity. Cultures were disrupted after the treatment, and TPKI/GSK-3β activity was determined (24).
Immunoblotting. Samples were separated on NOVEX 10–20% Tris/N-[tris(hydroxymethyl]glycine gels (Invitrogen). Immunoreactive bands with 4G8 (1 μg/ml; Signet Laboratories, Dedham, MA) were detected (27) by using SuperSignal West Femto substrates (Pierce).
Statistics. We examined the statistical significance of differences between groups by applying Games–Howell post hoc tests, using STATVIEW 5.0 (SAS Institute, Cary, NC).
Results
Preparation and Identification of Spherical Aβ1–40 Aggregates (ASPD) as a Highly Toxic Moiety Among Aβ1–40 Aggregates. We chemically synthesized Aβ1–40 to obtain large quantities of homogenous peptide as starting material. After purification and characterization, Aβ1–40 dissolved in 50% PBS to 350 μM was allowed to self-aggregate under various conditions, and the toxicity of each Aβ1–40 solution was estimated by MTT activity by using primary cultures from rat septum regions. Initially, Aβ1–40 solutions were nontoxic. When Aβ1–40 solutions (Fig. 1A) were rotated slowly for 5–7 days, they showed potent toxicity at 0.1 μM. Without slow rotation, Aβ1–40 solution remained nontoxic even at 5 μM (Fig. 1 A). Therefore, we prepared toxic Aβ1–40 aggregates by slowly rotating Aβ1–40 solutions (350 μM in 50% PBS); the aggregates thus prepared are designated as SR350–Aβ1–40.
Fig. 1.

ASPD as a neurotoxin in Aβ1–40 aggregates. (A)Aβ1–40 solutions (350 μM) were aggregated, and their toxicity was estimated by MTT assay (n = 6). Aβ1–40 solution was rotated in Eppendorf tubes (•), glass tubes (▵), carbon-coated tubes (○), gold-coated tubes (⊞), and silicon-coated tubes (▴). Aβ1–40 solution without rotation also is shown ( ). Hereafter, we designated toxic Aβ1–40 aggregates formed by slowly rotating Aβ1–40 solution (350 μMin50% PBS) as SR350–Aβ1–40. (B) The structure and toxicity of Aβ1–40 aggregates formed by slow rotation. At the indicated time, aliquots were removed from SR350–Aβ1–40 for transmission electron microscopy and MTT assay (n = 9) (5 μM). (C) Aβ1–40 solutions [350 μM (SR350–Aβ1–40), 50 μM (SR50–Aβ1–40), and 1 μM (SR1–Aβ1–40)] each were rotated slowly for 7 days, and the toxicity was assessed (n = 7). Thioflavine T assay (20 μM; Sigma; excitation at 445 nm and emission at 485 nm; ref. 48) determined fibril content in freshly dissolved Aβ1–40 and in each SR–Aβ1–40 (n = 6). (D) SR350–Aβ1–40 was prepared with or without KLVFF or LPFFD. The toxicity was determined by MTT assay (n = 9) or propidium iodide staining (Right). Image of SR350–Aβ1–40 with LPFFD is shown (bar, 100 nm). Also shown are SR350–Aβ1–40 (○) with KLVFF (•) and with LPFFD (). Data represent the mean ± SE. *, Significant difference from the control. The results suggest that toxicity of SR350–Aβ1–40 is associated with the spherical structures (B, 4 h), which we call ASPD.
). Hereafter, we designated toxic Aβ1–40 aggregates formed by slowly rotating Aβ1–40 solution (350 μMin50% PBS) as SR350–Aβ1–40. (B) The structure and toxicity of Aβ1–40 aggregates formed by slow rotation. At the indicated time, aliquots were removed from SR350–Aβ1–40 for transmission electron microscopy and MTT assay (n = 9) (5 μM). (C) Aβ1–40 solutions [350 μM (SR350–Aβ1–40), 50 μM (SR50–Aβ1–40), and 1 μM (SR1–Aβ1–40)] each were rotated slowly for 7 days, and the toxicity was assessed (n = 7). Thioflavine T assay (20 μM; Sigma; excitation at 445 nm and emission at 485 nm; ref. 48) determined fibril content in freshly dissolved Aβ1–40 and in each SR–Aβ1–40 (n = 6). (D) SR350–Aβ1–40 was prepared with or without KLVFF or LPFFD. The toxicity was determined by MTT assay (n = 9) or propidium iodide staining (Right). Image of SR350–Aβ1–40 with LPFFD is shown (bar, 100 nm). Also shown are SR350–Aβ1–40 (○) with KLVFF (•) and with LPFFD (). Data represent the mean ± SE. *, Significant difference from the control. The results suggest that toxicity of SR350–Aβ1–40 is associated with the spherical structures (B, 4 h), which we call ASPD.
To identify the toxic moiety in SR350–Aβ1–40, we examined the structure–toxicity relationship of Aβ1–40 aggregates during the course of slow rotation. Aβ1–40 aggregates first detected by transmission electron microscopy consisted of spherical structures 3–20 nm in diameter without any detectable fibrils (Fig. 1B, 4 h). At 27 h, short, straight fibers and PF-like structures were detected, which disappeared when 6- to 10-nm-wide fibrils were formed at 75 h, whereas spherical structures continued to exist (Fig. 1B). MTT assay indicates that the toxicity of SR350–Aβ1–40 reached a plateau after 4 h of slow rotation, when spherical structures, but not fibrils, were present.
Next, Aβ1–40 solutions at 350 μM (SR350–Aβ1–40), 50 μM (SR50–Aβ1–40), and 1 μM (SR1–Aβ1–40) each were rotated slowly for 7 days. Below a critical concentration (≈100 μM) (28), the fibril formation in each SR–Aβ1–40 was reduced drastically (Fig. 1C Right). However, with a decrease in the proportion of fibrils to other Aβ1–40 aggregates in SR–Aβ1–40, toxicity became much stronger (Fig. 1C Left), as shown by a decrease in IC50 (0.2 μM in SR350–Aβ1–40, 0.01 μM in SR50–Aβ1–40, and 0.001 μM in SR1–Aβ1–40). The kinetic studies further support a lack of toxicity in fibrils.
We inhibited the fibril formation by pentapeptides containing the Aβ16–20 motif, which break β-sheet interactions required for the fibril formation. As reported previously, addition of Aβ16–20 (KLVFF) (20) or iAβ5 (LPFFD) (21) at the onset of slow rotation led to ≈70% reduction in fibrils in SR350–Aβ1–40 in thioflavine T assay. However, toxicity of SR350–Aβ1–40 was unchanged (Fig. 1D). Slow rotation of the pentapeptides alone did not yield any toxicity. These results (Fig. 1 B–D) strongly suggest that Aβ1–40 aggregates other than fibrils are responsible for the toxicity in SR–Aβ1–40. We obtained similar results by using Aβ1–40 from Bachem or U.S. Peptide.
In contrast to the fibril, the spherical structure (Fig. 1B, 4 h) was not affected by the pentapeptides (Fig. 1D) and was present in SR–Aβ1–40 even under conditions in which fibrils were rarely detected owing to low Aβ1–40 concentrations. These results suggest that the spherical structure is the toxic moiety in SR–Aβ1–40. In the following text, this spherical structure, a candidate neurotoxin in SR–Aβ1–40, is designated as ASPD.
Morphology and Neurotoxicity of ASPD Isolated from SR–Aβ1–40. ASPD was isolated in the flowthrough fraction of filters with 0.65-μM and 30-kDa molecular sieves, which contained only ASPD ranging from 4 to 18 nm (10.4 ± 2.9 nm on average, n = 101) with no fibrils (Fig. 2A). Based on measurement of the “width” and “length” of each ASPD in Fig. 2 A, simple regression analysis was performed: y/x = 0.982, r2 = 0.869, Δmean (width – length) = –0.1628 and Δσ (width – length) = 0.7. These values support that ASPD is perfectly spherical. ASPD in the resin also gave circular images in both horizontal and cross sections (Fig. 2B), suggesting that ASPD is indeed a sphere.
Fig. 2.

ASPD morphology and toxicity. (A) Transmission electron microscopic images of isolated ASPD (bars, 100 nm in Lower and 20 nm in Inset). The width (x) and length (y) of each ASPD (n = 101) were measured (Upper). (B) Images of horizontal sections (a) and cross sections (b) of ASPD in the resin as described in Materials and Methods. (C) Fluid-phase atomic force microscopy of ASPD as described in Materials and Methods (n = 100). (D) Toxicity of ASPD. (Top and Middle) After a 40-h ASPD treatment (210 nM), cultures were stained with anti-MAP2 (green) and fillipin (blue). ASPD killed ≈40% neurons, with shrunken or fragmented nuclei stained pink with propidium iodide and Hoechst 33258 (Inset). Cell death in the control culture was <3%. (Bottom) ASPD toxicity was estimated by MTT activity (n = 6). Data in D represent the mean ± SE. *, Significant difference compared with control.
To avoid possible effects of sample drying, which was necessary for transmission electron microscopy, the isolated ASPD was visualized in 50% PBS by atomic force microscopy. The shape of the section curve is consistent with a spherical shape of ASPD (Fig. 2C). Furthermore, a statistical analysis was carried out. In atomic force microscopy, the height is obtained precisely, whereas the length or the width is larger than the real value owing to effects depending on the radius of the probe. The radius of the probe (R) used here was estimated to be ≈2.5 nm (data not shown). If the sample is a sphere, its diameter (2r) can be calculated from the measured half-width (D) by using the following equation: 2r = –2R + 2r=-2R+2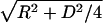 . We measured the height and the half-width of each ASPD and obtained the height/calculated diameter ratio of each ASPD, which was 1.0 on average (n = 100) (Fig. 2C). These results confirmed that ASPD is a sphere, not a disk or a cylinder.
. We measured the height and the half-width of each ASPD and obtained the height/calculated diameter ratio of each ASPD, which was 1.0 on average (n = 100) (Fig. 2C). These results confirmed that ASPD is a sphere, not a disk or a cylinder.
A 40-h treatment of neuronal cultures with the isolated ASPD (210 nM in Aβ1–40 monomer concentration) resulted in ≈40% neurodegeneration, with the remaining neurons displaying disintegrated neurites (Fig. 2D Top and Middle). Shrinkage and fragmentation of nuclei in ASPD-treated neurons stained with propidium iodide and Hoechst 33258 (Fig. 2D Top and Middle Insets) indicated apoptotic degeneration, whereas control cultures retained intact round nuclei. At all concentrations tested (0.35 nM to 0.175 μM; Fig. 2D Bottom), the isolated ASPD caused statistically significant inhibition of MTT activity (Games–Howell post hoc tests, n = 6). The isolated ASPD was stable over 2 months at 4°C and did not change to fibrils during another 7-day slow rotation. These results suggest that ASPD directly induced neurodegeneration.
Purification of the Toxic ASPD by Using Glycerol-Gradient Centrifugation. We purified ASPD further as in Fig. 3A by centrifugation (86,000 × g, 16 h) of SR350–Aβ1–40 in a 15–30% linear glycerol gradient. Immediately after the centrifugation, the apoptotic activity and the Aβ content of each fraction were evaluated, and the specific cell death activity in Fig. 3B was calculated from these values that were obtained in four independent experiments.
Fig. 3.

Purification of toxic ASPD by the glycerol gradient. (A) ASPD and fibrils in SR350–Aβ1–40 were fractionated as shown. Transmission electron microscopic images of representative fractions are shown. Aβ recovery and percentage of the total toxicity in glycerol gradient are indicated. (B) Immunoblottings with 4G8 of each fraction and of freshly dissolved Aβ1–40 are shown (Upper). Fraction 2 induced apoptotic cell death as shown by red arrows in propidium iodide and Hoechst 33258 staining, whereas fraction 11 did not (Lower Inset). Mini-Bradford assay quantitated Aβ content, using freshly dissolved Aβ1–40 as a standard, the concentration of which was determined accurately in advance with a Waters AccQTag system. Comparison of results from mini-Bradford assay with the AccQTag system data by using unfractionated SR350–Aβ1–40 proved a good correlation at concentrations >1 μM. The specific cell death activity was calculated from the Aβ content and the apoptotic activity (n = 16); one unit is defined as the activity inducing 1% apoptotic cell death. The specific cell death activity values of unfractionated SR350–Aβ1–40 and fibrils were 0.37 ± 0.09 and 0.07 ± 0.003, respectively. Data represent the mean ± SE. *, Significant difference compared with control. (C) The particle distribution of each fraction. (D Left) Transmission electron microscopic images of ASPD formed in Aβ1–42 solution (0.1 μMin50% PBS) after an 8-h slow rotation (bar, 20 nm). (Right)Aβ1–42-ASPD was separated by a glycerol gradient as in A. The particle distribution of fraction 2, in which the highest toxicity was recovered in repeated experiments (n = 7), is shown.
We found that ASPD was distributed into two groups: toxic ASPD recovered in the bottom-most part of the gradient and nontoxic ASPD recovered in the upper part of the gradient (Fig. 3B). In repeated experiments, ASPD with the highest toxicity was found reproducibly in fraction 2 around the migration position of thyroglobulin (669 kDa). The toxic ASPD induced apoptosis in neuronal cultures after a 40-h treatment, whereas nontoxic ASPD in fraction 11 did not (Fig. 3B Inset). ASPD in fraction 2 showed an ≈450-fold-higher toxicity than fibrils and an ≈80-fold-higher toxicity than SR350–Aβ1–40 (Fig. 3B).
Using ASPD purified by the glycerol gradient, we examined whether a correlation exists between the size of ASPD and its toxicity. The particle analysis of each fraction (Fig. 3C) clearly indicates that toxic ASPD consisted of particles >10 nm in transmission electron microscopy and nontoxic ASPD consisted of those <10 nm. ASPD in fraction 3 was <15 nm but exhibited toxicity equal to that of ASPD in fraction 1 (Fig. 3 B and C). Toxicity was absent in the flowthrough fraction of a filter with a 5-kDa molecular sieve size, suggesting a minor contribution to the toxicity from monomer or small oligomers that cannot be detected in transmission electron microscopy. Although there is still a possibility that species present in amounts too small to detect are involved, these results taken together indicate that potent toxicity is correlated best with 10- to 15-nm ASPD.
The toxic ASPD and nontoxic ASPD differed not only in apparent particle size but also in their band pattern in immunoblotting. As in Fig. 3B, at the onset of slow rotation, Aβ1–40 freshly dissolved in 50% PBS gave only a monomeric band, eluting as a single peak at a retention time corresponding to Aβ1–40 dimer in size-exclusion chromatography by using a Superdex 75 column (refs. 12, 17, and 29 and data not shown). After a 7-day slow rotation, SDS-stable oligomeric bands up to tetramers appeared in SR350–Aβ1–40 (Fig. 3B, arrowheads), suggesting a change in the nature of Aβ1–40. After glycerol-gradient centrifugation, the toxic ASPD in fractions 1–3 contained these SDS-stable oligomeric bands, but nontoxic ASPD did not (Fig. 3B Upper).
We applied the conditions established for Aβ1–40 to longer, more hydrophobic Aβ1–42, which generally is considered to be more pathogenic than Aβ1–40. Toxic Aβ1–42 aggregates were prepared by slowly rotating Aβ1–42 solutions (1–0.01 μM in 50% PBS) and were fractionated by using glycerol-gradient centrifugation. ASPD-like spheres with 3- to 20-nm diameters were detected after at least an 8-h slow rotation (Fig. 3D Left); they killed neurons at ≈0.35 nM (40-h treatment). In repeated fractionations, the highest toxicity was recovered reproducibly in fraction 2 (n = 7), ≈70% of which was accounted for by 10- to 14-nm ASPD-like spheres as shown by particle analysis (Fig. 3D Right). The specific cell death activity of Aβ1–42-ASPD in fraction 2 was estimated to be at least ≈100-fold higher than that of Aβ1–40-ASPD in fraction 2. In the case of Aβ1–40, formation of toxic ASPD requires a longer period of slow rotation with decrease in the initial concentration of Aβ1–40: it took 4 h at 350 μM but >30 h at 50 μM. Aβ1–42 at lower concentrations formed ASPD much earlier than Aβ1–40. These results demonstrated that Aβ1–42 forms ASPD, as Aβ1–40, but Aβ1–42-ASPD is formed more rapidly, induces neurodegeneration at lower concentrations, and is more toxic than Aβ1–40-ASPD.
Toxicity of ASPD May Involve TPKI/GSK-3β Activation. We found a rapid increase in TPKI/GSK-3β activity in ASPD-treated neuronal cultures, which reached its maximum (≈2.8-fold) after a 2-h treatment, followed by a decline to a level 50% higher than that of the control (Fig. 4A Upper). TPKI/GSK-3β plays a crucial role in Aβ-triggered neuropathological cascades (19, 24, 30). Lithium has been suggested recently as a possible therapeutic reagent for AD, because of its ability to inhibit TPKI/GSK-3β activity (31, 32). Thus, we examined whether inhibition of TPKI/GSK-3β by lithium protects neurons against ASPD toxicity. Lithium suppressed the ASPD toxicity in a dose-dependent manner with an IC50 of ≈2 mM (Fig. 4B), which is similar to the reported IC50 of lithium on TPKI/GSK-3β activity (33). The addition of 2 mM lithium within 1 h after ASPD treatment protected cells against ASPD toxicity but had no effect 4 h after ASPD treatment (Fig. 4A Lower). Protective effects of lithium were confirmed further by using propidium iodide and Hoechst 33258 staining; in the presence of 2 mM lithium, apoptotic cell death induced by 210 nM ASPD was decreased significantly, nearly to the control level (Fig. 4C). These findings raise the possibility that TPKI/GSK-3β is involved in the early stages of the ASPD-induced neurodegenerative cascades.
Fig. 4.

Lithium suppression of ASPD toxicity. (A Upper) After 210-nM ASPD treatment, TPKI/GSK-3β activity was assayed at the indicated time (n = 6). (Lower) Lithium (2 mM) was added to ASPD-treated culture at the indicated time, and the toxicity was assayed (n = 6). (B) Dose-dependent inhibition of ASPD toxicity by lithium. Effect of lithium on 210-nM ASPD toxicity was examined (n = 6). (C) Lithium protection against ASPD-induced apoptosis. After the treatment, as indicated, propidium iodide and Hoechst 33258 staining was performed (n = 9). Data represent the mean ± SE. Significant difference compared with control (*) or with ASPD-treated culture (**).
Discussion
It is generally accepted that amyloids play a crucial role in neurodegenerative diseases such as AD. Nevertheless, the physiological function of Aβs has not been elucidated. There are several reasons for this. For example, Aβs in the in vivo state are not homogeneous but are mixtures of peptides with different sizes. Although Aβ1–40 is predominant among them (34), accumulated data have indicated that Aβ1–42 is the strongest candidate for the neurotoxic agent. In addition, the aggregation states of these peptides are controversial. Some reports suggest that fibrils are toxic, but others support dimer or oligomer as the real neurotoxin (reviewed in ref. 35). To avoid such ambiguities, we chemically synthesized large amounts of homogeneous Aβ1–40. Although the solution of this preparation was not toxic, we found by chance that slow rotation of Aβ1–40 solution for 5–7 days converted Aβ1–40 to a stable form with potent toxicity (Fig. 1B). Purification and characterization of the toxic moiety provided evidence that a perfectly spherical Aβ1–40 aggregate is the most probable origin of the observed toxicity (Figs. 2 and 3). Other Aβ1–40 aggregates, including fibrils, were nontoxic. Therefore, we designated this toxic Aβ1–40 aggregate as ASPD. Correlation analysis in Fig. 3 indicated that toxicity is most closely associated with 10- to 15-nm ASPD, whereas ASPD <10 nm was nontoxic. We also found that Aβ1–42 forms ASPD, but Aβ1–42-ASPD is formed more rapidly, induces neurodegeneration at a lower concentration (≈0.35 nM), and exhibits ≈100-fold-higher toxicity than Aβ1–40-ASPD. Further analysis is required to elucidate the molecular basis of these differences. Because so far it is largely unknown how ASPD exerts its effect on neurons, we cannot completely rule out the possibility that very minor oligomers that have escaped detection are responsible for the toxicity. However, the data obtained up to the present support a positive correlation between the toxicity and ASPD >10 nm in the case of both Aβ1–40-ASPD and Aβ1–42-ASPD (Fig. 3) as well as when toxic ASPD in fraction 2 of the glycerol gradient was separated further by using size-exclusion chromatography (A.N., M.S., and M.H., unpublished data). Furthermore, the toxicity was not recovered in the flowthrough fraction of a filter with a 5-kDa molecular sieve size, suggesting that the toxicity is not attributable to monomer or small oligomers that cannot be detected in transmission electron microscopy.
PFs and ADDLs are two representative in vitro nonfibrillar neurotoxins detected by transmission electron microscopy or atomic force microscopy. Spherical ASPD is obviously different from curvilinear PFs (4- to 11-nm diameter and <200-nm length) (16, 17). ADDLs are a mixture of Aβ1–42 oligomers from trimers–hexamers up to 24-mers (15, 35). Because toxicity of ADDLs has been assessed in oligomer mixtures, it is difficult to know the precise size of the toxic moiety in ADDLs (15). The major component of ADDLs appeared to be globules of 4.8–5.7 nm in atomic force microscopy with 17- and 22-kDa bands in immunoblotting (15). We could not find any correlation between the toxicity and spheres of ≈5 nm derived from Aβ1–42 or Aβ1–40 in glycerol-gradient fractionation. ASPD showed an apparent size of 9.2 nm on average in atomic force microscopy and contained SDS-stable oligomeric bands up to tetramers. Formation of ADDLs seems to be restricted to Aβ1–42 when fibril formation is suppressed (15), whereas ASPD is formed from either Aβ1–40 or Aβ1–42 irrespective of fibril formation. Thus, ASPD differs from ADDLs in size, the band composition in immunoblotting, and preparation method. Under the conditions examined, Aβ remained nontoxic without slow rotation (Fig. 1 A). Slow rotation has been reported to enhance the aggregation of yeast prion (36) or hemoglobin S (37). Because ASPD is formed below the concentration required for fibril formation and in the presence of β-sheet-breaking peptides (Fig. 1 C and D), Aβ may aggregate into ASPD, at least in part, through a distinct pathway from fibril formation. Thus, it is possible that slow rotation of Aβ solution leads to some as-yet-unknown structural changes in Aβ that induce spherical aggregation, because SDS-stable oligomeric bands of Aβ1–40 are seen only in toxic ASPD. Further studies are required to understand the aggregation process of Aβ into ASPD and the relationship of ASPD with fibrils and other nonfibrillar oligomers.
Recent in vivo as well as in vitro studies support the toxicity of nonfibrillar oligomers and their possible causative role in neuropathology in AD (8, 35, 38), which is consistent with the dissociation between fibril loads and the cognitive decline in AD (39, 40, 41). Because vaccination, in some cases, has protected transgenic mice from memory loss without a significant decrease in Aβ fibril loads (5), Aβ aggregates other than fibrils may be the preferred therapeutic targets for AD (35). However, the nature of Aβ species and the oligomer state responsible for the pathogenesis remain controversial owing to the heterogeneity of Aβ aggregates in terms of Aβ species and oligomer size. Fractionation studies using an in vitro Aβ aggregation system as shown here will help to elucidate the aggregation process and to determine the oligomerization state of Aβ causing the pathogenesis.
In AD brains, intracellular accumulation of nonfibrillar Aβ, especially Aβ1–42 (42, 43), precedes the extracellular fibrillar accumulation of Aβ as senile plaques (1). Recently, LeBlanc et al. (44) have shown that microinjection of Aβ1–42 kills neurons. In their experiments, extracellular treatment of neurons with Aβ1–42 or Aβ1–40 did not affect the viability, despite the reported toxicity. These findings indicate the biological significance of intracellular Aβ accumulation but, at the same time, also raise the question of the source of intracellular Aβ in vivo (44): how does Aβ, which is produced through the secretory pathway, enter the cytosol? In our preliminary experiments, SDS-resistant oligomeric bands could be detected by immunoblotting in cytosolic extracts of neuronal cultures within 1 h after extracellular ASPD treatment (M.H. and M.S., unpublished data). If Aβ really exerts its effect intracellularly, variability in biological activities among in vitro identified Aβ aggregates may be attributed to their differing ability to access the cytosol. Future experiments to examine how ASPD enters the cytosol may provide some clue to the apparent contradiction between intracellular and extracellular Aβ accumulation.
Lithium, one of the most widely used drugs for bipolar disorder, protects neurons against suboptimal conditions or from excitotoxicity-induced lesions (reviewed in ref. 45). Here, we found that lithium may protect neurons in the early phase of ASPD treatment, perhaps through inhibition of TPKI/GSK-3β activation. Possible use of lithium as a therapeutic reagent for AD has been suggested by several authors (31, 32, 33, 46, 47). Considering the important role of TPKI/GSK-3β in AD, more detailed studies on the molecular mechanism of the lithium protection against ASPD may help to design inhibitors of TPKI/GSK-3β as candidate drugs for the treatment of AD.
Acknowledgments
We thank Dr. T. Wakabayashi for comments on ASPD structure; A. Ito, Y. Sakamoto, and C. Aoyama for technical assistance; and Drs. R. L. Veech, Y. Kashiwaya, and D. Masui for discussion. We are grateful to Dr. K. Imahori for critically reading the manuscript. M.H. was supported by a grant from Japan Science and Technology Corp. This work was supported by grants from Mitsubishi Kagaku Institute of Life Sciences, Japan Science and Technology Corp., and the Organization for Pharmaceutical Safety and Research.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Aβ, β-amyloid; AD, Alzheimer's disease; ADDL, Aβ-derived diffusible ligand; PF, protofibril; ASPD, amylospheroid; GSK-3β, glycogen synthase kinase-3β; TPKI, tau protein kinase I; SR–Aβ1–40, slowly rotated Aβ1–40 solution containing Aβ1–40 aggregates; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
References
- 1.Glenner, G. G. & Wong, C. W. (1984) Biochem. Biophys. Res. Commun. 120, 885–890. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe, D. J. (2001) Physiol. Rev. 81, 741–766. [DOI] [PubMed] [Google Scholar]
- 3.Schenk, D., Barbour, R., Dunn, W., Gordon, G., Grajeda, H., Guido, T., Hu, K., Huang, J., Johnson-Wood, K., Khan, K., et al. (1999) Nature 400, 173–177. [DOI] [PubMed] [Google Scholar]
- 4.Janus, C., Pearson, J., McLaurin, J., Mathews, P. M., Jiang, Y., Schmidt, S. D., Chishti, M. A., Horne, P., Heslin, D., French, J., et al. (2000) Nature 408, 979–982. [DOI] [PubMed] [Google Scholar]
- 5.Morgan, D., Diamond, D. M., Gottschall, P. E., Ugen, K. E., Dickey, C., Hardy, J., Duff, K., Jantzen, P., DiCarlo, G., Wilcock, D., et al. (2000) Nature 408, 982–985. [DOI] [PubMed] [Google Scholar]
- 6.Levine, H., III (1995) Neurobiol. Aging 16, 755–764. [DOI] [PubMed] [Google Scholar]
- 7.Hartley, D. M., Walsh, D. M., Ye, C. P., Diehl, T., Vasquez, S., Vassilev, P. M., Teplow, D. B. & Selkoe, D. J. (1999) J. Neurosci. 19, 8876–8884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., Rowan, M. J. & Selkoe, D. J. (2002) Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- 9.Pike, C. J., Burdick, D., Walencewicz, A. J., Glabe, C. G. & Cotman, C. W. (1993) J. Neurosci. 13, 1676–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lorenzo, A. & Yankner, B. A. (1994) Proc. Natl. Acad. Sci. USA 91, 12243–12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardy, J. A. & Higgins, G. A. (1992) Science 256, 184–185. [DOI] [PubMed] [Google Scholar]
- 12.Garzon-Rodriguez, W., Sepulveda-Becerra, M., Milton, S. & Glabe, C. G. (1997) J. Biol. Chem. 272, 21037–21044. [DOI] [PubMed] [Google Scholar]
- 13.Roher, A. E., Chaney, M. O., Kuo, Y. M., Webster, S. D., Stine, W. B., Haverkamp, L. J., Woods, A. S., Cotter, R. J., Tuohy, J. M., Krafft, G. A., et al. (1996) J. Biol. Chem. 271, 20631–20635. [DOI] [PubMed] [Google Scholar]
- 14.Gajewski, T. F., Fields, P. & Fitch, F. W. (1995) Eur. J. Immunol. 25, 1836–1842. [DOI] [PubMed] [Google Scholar]
- 15.Lambert, M. P., Barlow, A. K., Chromy, B. A., Edwards, C., Freed, R., Liosatos, M., Morgan, T. E., Rozovsky, I., Trommer, B., Viola, K. L., et al. (1998) Proc. Natl. Acad. Sci. USA 95, 6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Harper, J. D., Wong, S. S., Lieber, C. M. & Lansbury, P. T. (1997) Chem. Biol. 4, 119–125. [DOI] [PubMed] [Google Scholar]
- 17.Walsh, D. M., Lomakin, A., Benedek, G. B., Condron, M. M. & Teplow, D. B. (1997) J. Biol. Chem. 272, 22364–22372. [DOI] [PubMed] [Google Scholar]
- 18.Ali, A., Hoeflich, K. P. & Woodgett, J. R. (2001) Chem. Rev. 101, 2527–2540. [DOI] [PubMed] [Google Scholar]
- 19.Imahori, K. & Uchida, T. (1997) J. Biochem. (Tokyo) 121, 179–188. [PubMed] [Google Scholar]
- 20.Tjernberg, L. O., Naslund, J., Lindqvist, F., Johansson, J., Karlstrom, A. R., Thyberg, J., Terenius, L. & Nordstedt, C. (1996) J. Biol. Chem. 271, 8545–8548. [DOI] [PubMed] [Google Scholar]
- 21.Soto, C., Sigurdsson, E. M., Morelli, L., Kumar, R. A., Castano, E. M. & Frangione, B. (1998) Nat. Med. 4, 822–826. [DOI] [PubMed] [Google Scholar]
- 22.Luft, J. H. (1961) J. Biophys. Biochem. Cytol. 9, 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lyubchenko, Y. L., Gall, A. A., Shlyakhtenko, L. S., Harrington, R. E., Jacobs, B. L., Oden, P. I. & Lindsay, S. M. (1992) J. Biomol. Struct. Dyn. 10, 589–606. [DOI] [PubMed] [Google Scholar]
- 24.Hoshi, M., Takashima, A., Noguchi, K., Murayama, M., Sato, M., Kondo, S., Saitoh, Y., Ishiguro, K., Hoshino, T. & Imahhori, K. (1996) Proc. Natl. Acad. Sci. USA 93, 2719–2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shearman, M. S., Ragan, C. I. & Iversen, L. L. (1994) Proc. Natl. Acad. Sci. USA 91, 1470–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu, Y. & Schubert, D. (1997) J. Neurochem. 69, 2285–2293. [DOI] [PubMed] [Google Scholar]
- 27.Ida, N., Hartmann, T., Pantel, J., Schroder, J., Zerfass, R., Forstl, H., Sandbrink, R., Masters, C. L. & Beyreuther, K. (1996) J. Biol. Chem. 271, 22908–22914. [DOI] [PubMed] [Google Scholar]
- 28.Lomakin, A., Chung, D. S., Benedek, G. B., Kirschner, D. A. & Teplow, D. B. (1996) Proc. Natl. Acad. Sci. USA 93, 1125–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang, T. H., Yang, D. S., Plaskos, N. P., Go, S., Yip, C. M., Fraser, P. E. & Chakrabartty, A. (2000) J. Mol. Biol. 297, 73–87. [DOI] [PubMed] [Google Scholar]
- 30.Takashima, A., Noguchi, K., Sato, K., Hoshino, T. & Imahori, K. (1993) Proc. Natl. Acad. Sci. USA 90, 7789–7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong, M., Chen, D. C., Klein, P. S. & Lee, V. M. (1997) J. Biol. Chem. 272, 25326–25332. [DOI] [PubMed] [Google Scholar]
- 32.Sun, X., Sato, S., Murayama, O., Murayama, M., Park, J. M., Yamaguchi, H. & Takashima, A. (2002) Neurosci. Lett. 321, 61–64. [DOI] [PubMed] [Google Scholar]
- 33.Klein, P. S. & Melton, D. A. (1996) Proc. Natl. Acad. Sci. USA 93, 8455–8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shoji, M., Golde, T. E., Ghiso, J., Cheung, T. T., Estus, S., Shaffer, L. M., Cai, X.-D., McKay, D. M., Tintner, R., Frangione, B., et al. (1992) Science 258, 126–129. [DOI] [PubMed] [Google Scholar]
- 35.Klein, W. L., Krafft, G. A. & Finch, C. E. (2001) Trends Neurosci. 24, 219–224. [DOI] [PubMed] [Google Scholar]
- 36.DePace, A. H., Santoso, A., Hillner, P. & Weissman, J. S. (1998) Cell 93, 1241–1252. [DOI] [PubMed] [Google Scholar]
- 37.Briehl, R. W. (1980) Nature 288, 622–624. [DOI] [PubMed] [Google Scholar]
- 38.Bucciantini, M., Giannoni, E., Chiti, F., Baroni, F., Formigli, L., Zurdo, J., Taddei, N., Ramponi, G., Dobson, C. M. & Stefani, M. (2002) Nature 416, 507–511. [DOI] [PubMed] [Google Scholar]
- 39.Dickson, D. W. & Yen, S. H. (1989) Neurobiol. Aging 10, 402–404, 412–414. [DOI] [PubMed] [Google Scholar]
- 40.Terry, R. D., Masliah, E., Salmon, D. P., Butters, N., DeTeresa, R., Hill, R., Hansen, L. A. & Katzman, R. (1991) Ann. Neurol. 30, 572–580. [DOI] [PubMed] [Google Scholar]
- 41.Lue, L. F., Kuo, Y. M., Roher, A. E., Brachova, L., Shen, Y., Sue, L., Beach, T., Kurth, J. H., Rydel, R. E. & Rogers, J. (1999) Am. J. Pathol. 155, 853–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gouras, G. K., Tsai, J., Naslund, J., Vincent, B., Edgar, M., Checler, F., Greenfield, J. P., Haroutunian, V., Buxbaum, J. D., Xu, H., et al. (2000) Am. J. Pathol. 156, 15–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.D'Andrea, M. R., Nagele, R. G., Wang, H. Y., Peterson, P. A. & Lee, D. H. (2001) Histopathology 38, 120–134. [DOI] [PubMed] [Google Scholar]
- 44.Zhang, Y., McLaughlin, R., Goodyer, C. & LeBlanc, A. (2002) J. Cell Biol. 156, 519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jope, R. S. (1999) Mol. Psychiatry 4, 117–128. [DOI] [PubMed] [Google Scholar]
- 46.Stambolic, V., Ruel, L. & Woodgett, J. R. (1996) Curr. Biol. 6, 1664–1668. [DOI] [PubMed] [Google Scholar]
- 47.Alvarez, G., Munoz-Montano, J. R., Satrustegui, J., Avila, J., Bogonez, E. & Diaz-Nido, J. (1999) FEBS Lett. 453, 260–264. [DOI] [PubMed] [Google Scholar]
- 48.LeVine, H., III (1993) Protein Sci. 2, 404–410. [DOI] [PMC free article] [PubMed] [Google Scholar]