

Abstract
We have designed an autocatalytic cycle based on the highly efficient 10–23 RNA-cleaving deoxyribozyme that is capable of exponential amplification of catalysis. In this system, complementary 10–23 variants were inactivated by circularization, creating deoxyribozymogens. Upon linearization, the enzymes can act on their complements, creating a cascade in which linearized species accumulate exponentially. Seeding the system with a pool of linear catalysts resulted not only in amplification of function but in sequence selection and represents an in vitro selection experiment conducted in the absence of any protein enzymes.
Demonstrating molecular self-amplification is essential for understanding origins and can potentially foment biotechnology applications, especially in diagnostics. In modern biology, molecular replication is dominated by cycles in which nucleic acids encode and are replicated by protein enzymes. However, the discovery and subsequent engineering of nucleic acid catalysts raises the possibility that nucleic acid-based, autocatalytic cycles might be designed.
In this regard, von Kiedrowski (1) and Zielinski and Orgel (2) showed that oligonucleotide palindromes can serve as templates for the ligation of shorter oligonucleotide substrates, and thus for their own reproduction. Variations on this theme have led to proof that short oligonucleotide templates are capable of semi-conservative (cross-catalytic) replication (3), and that substrates as short as single nucleotides can be used (4–6). However, because of the strong interactions that exist between complementary strands, the products formed in these ligation-based systems bind strongly to one another and the reaction kinetics of these systems are limited to parabolic, rather than exponential, growth (7, 8).
Various strategies have been implemented to avoid the problem of product inhibition. For example, Li and Nicolaou (9) used cyclic pH changes and the addition of excess complementary oligonucleotide to ensure the release of ligated products from a template. The von Kiedrowski lab (10) facilitated product release by template immobilization and denaturation. Cycles of immobilization and denaturation resulted in the “SPREAD” (surface-promoted replication and exponential amplification) of oligonucleotides beyond an initial, immobilized population. Those experiments have yielded one of the first convincing demonstrations of the possibility of primordial Darwinian sequence selection. Most recently, Paul and Joyce (11) designed an autocatalytic cycle based on the R3C RNA ligase ribozyme. The self-reproducing ligase used “half-ribozymes” as substrates and was capable of limited exponential growth. This replicator appears to have neatly avoided the problem of product inhibition by refolding and forming more stable intramolecular contacts within the newly ligated product. However, the replicator did not show extensive exponential growth, as substrate self-association resulted in the accumulation of stable complexes that could not bind to the ribozyme catalyst.
We have designed an autocatalytic deoxyribozyme cycle based on cleavage rather than ligation (Fig. 1). The fast (kcat = ≈0.1 min–1), highly efficient (kcat/Km > 109), and programmable 10–23 deoxyribozyme (12) was circularized, resulting in its inactivation. The inactive circles can nonetheless still be used as substrates by a linear, complementary 10–23 deoxyribozyme. Circularization of both complementary deoxyribozymes effectively eliminates catalytic activity, but poises the system so that even a single ring-opening event can potentially result in a cascade of cleavage reactions. As anticipated, the conformational change from circular substrate to linear catalyst does indeed drive exponential accumulation of products. This system most closely resembles protein-based proteolytic cascades such as those involved in blood clotting (13, 14) and apoptosis (15), in which inactive zymogens, proenzymes, and procofactors are activated via cleavage. For this reason, the inactive deoxyribozyme circles have been dubbed “deoxyribozymogens.” Because the autocatalytic cleavase cycle avoids the accumulation of longer products with greater base-pairing potential, we hypothesized that it should be capable of robust exponential growth, and thus might serve as a model system for exploring how nucleic acid autocatalysis might lead to sequence evolution. As a demonstration, the cleavase cascade was used to select optimal deoxyribozymes from a random sequence population in the complete absence of proteins.
Fig. 1.

Design of linear and circular 10–23 deoxyribozymes. (a) Active linear deoxyribozymes LA and LB and inactive circular deoxyribozymes CA and CB. The 10–23 catalytic core is shown in gray with substrate-binding arms shown in blac.k Complementary arms are denoted by upper and lowercase letters. The cleavage site, consisting of two ribose residues, is shown in outline. (b) Schematic for the autocatalytic 10–23 deoxyribozyme cleavase cycle. Cleavage of inactive CB by active LA produces active LB, which can in turn cleave inactive CA to reproduce active LA.
Materials and Methods
Oligonucleotide Synthesis and Circularization. All oligonucleotides were synthesized on a expedite 8909 synthesizer (Perkin–Elmer) by using standard DNA and RNA phosphoramidite chemistry. All synthesis reagents were purchased from Glen Research (Sterling, VA).
The linear deoxyribozymes LA (UCGGACAGGCTAGCTACAACGAGAGTGACA-p) and LB (UGTCCGAGGCTAGCTACAACGAGTCACTCA-p) were chemically synthesized with terminal RNA residues (underlined) and a 3′ phosphate (denoted p) to more closely mimic the product of the cleavage reaction, which produces a 2′,3′ cyclic phosphate.
Circular deoxyribozymes were synthesized from linear permutations of LA and LB bearing a 5′ phosphate (denoted p) (pre.CA, p-AGCTACAACGAGAGTGACAUCGGACAGGCT; pre.CB p-AGCTACAACGAGTCACTCAUGTCCGAGGCT, where underlined residues are RNA). Circularization reactions were carried out with T4 DNA ligase (NEB, Beverly, MA) and a splint oligonucleotide (splint.a TGTAGCTAGCCTGT; splint.b GCTCCGATCGATGT). The concentration of the linear substrate was 100 nM, and the splint oligonucleotide was present in 1.2-fold molar excess. The reactions were allowed to proceed for 16 h at room temperature and the circular products (50–70%) were isolated on denaturing (7 M urea) 20% polyacrylamide gels. After gel purification, CA and CB were treated with exonuclease VII to remove any remaining linear nucleic acids. Where appropriate, the constructs were radiolabeled before circularization via an exchange reaction with T4 polynucleotide kinase and γ-32ATP (>7,000 Ci/mmol; ICN).
Circular pools C.E1 and C.E2 were synthesized from linear precursor pools bearing three randomized nucleotides, a 5′ phosphorylated ribouridine and a 3′ riboadenosine (pre.CE1, p-UCGGACAGGCTAGCTACAACGAGANNNACA; pre.ce2, p-UGNNNGAGGCTAGCTACAACGAGTCACTCA, where N denotes randomized positions). Circularization reactions were in this instance performed with T4 RNA ligase (Promega) (16). The circular constructs were purified as described above.
Reaction Conditions and Deoxyribozyme Kinetics. All reactions were equilibrated at 70°C in 50 mM Tris, pH 8.0, 0.1% SDS, and slow-cooled to 23°C before initiation by the addition of 30 mM MgCl2. Reactions were stopped by the addition of 95% formamide loading dye containing a 2-fold excess of EDTA over the concentration of magnesium. Circular and linear species were separated on denaturing (7 M urea) 20% acrylamide gels and quantitated with a PhosphorImager (Molecular Dynamics).
Multiple turnover reactions were conducted with substrate concentrations at least 10-fold greater than that of the enzyme. The value kobs was determined by fitting three time points that occurred within the first ≈10% of the reaction. The values kcat and Km were determined by fitting a plot of kobs versus substrate concentration.
In Vitro Selection. The selection was carried out by using the circular pools C.E1 and C.E2. All rounds were conducted in reaction buffer (Tris, pH 8.0/0.1% SDS, 10 μl final volume), and initiated by the addition of 30 mM MgCl2.
The initial round of selection contained 40 pmol of each circular pool and 4 pmol of each linear pool. The linear pool was catalytically active and could initiate the autocatalytic cycle. After initiation with MgCl2, the reaction was allowed to proceed for 48 h. A fraction of this reaction (1 μl) was then transferred to another tube containing an additional 40 pmol of each circular pool. This reaction (round 2) was again initiated by the addition of MgCl2. The process was repeated for an additional two rounds, which should have resulted in a vast dilution of the original, linear pools. These subsequent rounds (rounds 3 and 4) contained only 20 pml of each pool. The incubation times for each round were 48, 72, 27, and 15 h.
After each serial transfer, the remaining 9 μl from each round was quenched by the addition of an equal volume of 95% formamide loading dye containing a 2-fold excess of EDTA. Circular and linear species were separated on denaturing (7 M urea) 20% acrylamide gels, and the amounts of nucleic acid present in individual bands were quantitated via fluorescent staining with SyberGold (Molecular Probes) and a Fluorimager (Molecular Dynamics).
The linear species from round 4 were extracted from the gel for cloning and sequencing. After gel extraction, linear DNA molecules were treated with T4 polynucleotide kinase to remove the 2′,3′ cyclic phosphate (17) and then radiolabeled with an excess of γ-32ATP (>7,000 Ci/mmol; ICN) in standard buffer. Primer binding sites were ligated to the 3′ or 5′ end of a portion of the purified, radiolabeled pool by using T4 RNA ligase (L3, p-UUUCTGAGACGTAGACAGCACGAT-c3; L5, TCGTACTACTAGCATCGTTATGGAAA; the underlined residues are RNA; p, a 5′ phosphate; and c3, a three-carbon alkyl blocking group). The ligated products were purified on denaturing (7 M urea) 8% acrylamide gels and amplified by using primers specific for either C.E1 or C.E2 (for C.E1, P5.E1 CGTTATGGA A ATCGGACAGGC and P3.E1 ATCGTGCTGTCTACGTCTCAG; for C.E2, P5.E2 CGTACTACTAGCATCGTTATGG and P3.E2 TGAGTGACTCGTTGTAGC). After amplification, PCR products were cloned by using the Topo TA cloning kit (Invitrogen) and sequences were acquired via standard dideoxy, cycle sequencing methods on a CEQ 2000XL automated sequencer (Beckman Coulter). To obtain sequences from round 0, the circularized pools were treated with RNase U2 before the ligation of primer binding sites, PCR amplification, and sequencing.
Reaction Simulations. The following reactions were used to model the autocatalytic cycle:
| [1] |
| [2] |
| [3] |
| [4] |
| [5] |
| [6] |
where reactions 1 and 2 represent the catalyzed reactions; reactions 3 and 4 represent the background rates of hydrolysis; and reactions 5 and 6 represent cleavage reactions catalyzed by the circular (as opposed to linear) deoxyribozymes. For reactions 5 and 6, we used second-order rate constants based on the initial rates of substrate cleavage by the noncleavable enzymes circle CA and CB. Simulations were conducted by using the program SIGMAPLOT 7.0 (SPSS, Chicago) and the following set of differential equations:
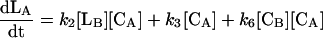 |
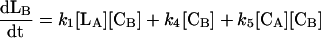 |
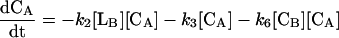 |
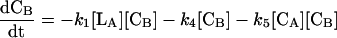 |
where
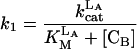 |
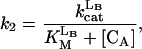 |
and k3, k4, k5, and k6 are defined by Eqs. 3, 4, 5, and 6, respectively. The initial concentrations of CA and CB have been set at 160 nM. The concentrations of trigger LB are listed in the Fig. 3c legend.
Fig. 3.

Time courses of autocatalytic cleavage. (a) Formation of LB from CB. Reactions contained 200 nM CA and 200 nM 32P-labeled CB except for  , which contained only 200 nM 32P-labeled CB. The symbols for added trigger or antisense oligonucleotides are: ▪, no added trigger; •, 5 nM LA;
, which contained only 200 nM 32P-labeled CB. The symbols for added trigger or antisense oligonucleotides are: ▪, no added trigger; •, 5 nM LA; , 5 nM LB;, 5 nM LA; and
, 5 nM LB;, 5 nM LA; and  , 50 nM antisense. (b) Formation of LA from CA. Reactions contained 200 nM CB and 200 nM 32P-labeled CA. The symbols for added trigger oligonucleotides are: □, no added trigger; ○, 5 nM LA; and ▵, 5 nM LB. (c) Formation of LA from CA with increasing concentrations of trigger LB. The symbols for trigger oligonucleotides are:
, 50 nM antisense. (b) Formation of LA from CA. Reactions contained 200 nM CB and 200 nM 32P-labeled CA. The symbols for added trigger oligonucleotides are: □, no added trigger; ○, 5 nM LA; and ▵, 5 nM LB. (c) Formation of LA from CA with increasing concentrations of trigger LB. The symbols for trigger oligonucleotides are: , no trigger; ♦, 1 nM; ▾, 5 nM; ▪,20 nM; ▴, 50 nM; and •, 100 nM. The overlaid curves were theoretically derived based on the kinetic schema described in Materials and Methods and the rates are given in the text. (d) Denaturing (7 M urea) polyacrylamide gel analysis of an autocatalytic cleavage reaction time course. These particular data contributed to c, no trigger or 50 nM trigger. Circular (C) and linear deoxyribozymes (L) are indicated.
, no trigger; ♦, 1 nM; ▾, 5 nM; ▪,20 nM; ▴, 50 nM; and •, 100 nM. The overlaid curves were theoretically derived based on the kinetic schema described in Materials and Methods and the rates are given in the text. (d) Denaturing (7 M urea) polyacrylamide gel analysis of an autocatalytic cleavage reaction time course. These particular data contributed to c, no trigger or 50 nM trigger. Circular (C) and linear deoxyribozymes (L) are indicated.
Results and Discussion
Deoxribozyme Cascade. To determine whether it would be possible to generate a deoxyribozyme cascade, we initially designed noncleavable versions of both circle CA and circle CB that contained no RNA residues and assayed these noncleavable constructs for their ability to cleave their circular counterparts that did contain RNA linkages. When 200 nM noncleavable, circular enzyme (CA) was incubated with 200 nM cleavable, circular substrate, the rate of ring opening was found to be 1 nM·h–1, >250-fold slower than if the catalyst were linear (LA). Similarly, noncleavable CB was found to cleave CA at a rate of 0.2 nM·h–1, >1,000-fold slower than LB and similar to the rate of background hydrolysis (≈3 × 10–4 h–1 and 200 nM circular deoxyribozyme).
The sequence of the substrate-binding arms of the 10–23 deoxyribozyme has been shown to influence catalysis (12, 18), therefore, the activities of the linear deoxyribozymes were assayed. As shown in Fig. 2, the kinetic analysis of the reaction of the linear deoxyribozyme LA with CB gave a KM of 260 ± 9 nM and a kcat of 2.4 ± 0.03 h–1 and the analysis of LB with CA gave a KM of 320 ± 7 nM and a kcat of 3.6 ± 0.03 h–1. When LA and LB were assayed with linear substrates that had the same sequences as the circular deoxyribozymes, higher kcats and lower KMs were observed (LA with linear substrate gave a KM of 85 ± 9 nM and a kcat of 16 ± 0.6 h–1, whereas that of LB gave a KM of 43 ± 7nManda kcat of 21 ± 1.0 h–1, data not shown). Overall, whereas the catalytic efficiencies of the linear deoxyribozymes were similar to those previously reported (12, 19, 20), circularization decreased substrate utilization.
Fig. 2.

Kinetic analysis of pairs of circular and linear deoxyribozymes. • indicates reaction of LB with CA; ▪ indicates reaction of LA with CB. Initial velocities were determined by the best linear fit through three time points within the first ≈10% of the reaction. The concentrations of the circular deoxyriboxzymes (CA and CB) were 20, 40, 80, 160, 320, 640, and 1,280 nM. The concentrations of the linear deoxyribozyme catalysts (LA and LB) varied from 2 to 10 nM, but were always at least 10-fold less than that of their circular substrates. Each point represents the average of three rate measurements. Lines represent the best fit to the Michaelis–Menten equation.
Having established that the circularized deoxyribozymogens were catalytically impaired yet could still act as substrates, a mixture of the circles CA and CB were incubated and the reaction was monitored for the production of linear species (Fig. 3). There was a lag phase of ≈3 h followed by an exponential growth phase. This phenomenon was observed irrespective of whether the reaction was monitored for the cleavage of CA or CB (Fig. 3a, ▪; Fig. 3b, □), indicating that both linear species were being formed in tandem. Reactions routinely proceeded to ≈80% completion, possibly because of the misfolding of some substrates. In contrast, when each circle was incubated separately very little linear product was formed, consistent with the measured rates of background hydrolysis in buffer 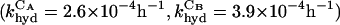 .
.
To demonstrate that the predicted cross-catalytic reactions were occurring, cleavage reactions were initiated by the addition of either linear LA or linear LB at 1 part in 40. As expected, the reaction kinetics were still exponential, but the lag phase was reduced, to <1 h (Fig. 3 a and b, triangles and circles). Increasing amounts of trigger (LB) resulted in the shortening of the lag phase of the reaction until little or no sigmoidicity was observed (Fig. 3c). A simulation of the cascade reaction using the previously determined Michaelis–Menten parameters and background rates of hydrolysis was in good agreement with the observed data, as shown by the overlaid curves (Fig. 3c). In contrast, when only one circle was present, the addition of complementary trigger resulted in a linear rather than an exponential reaction (Fig. 3a, gray diamonds). Additionally, when antisense oligonucleotides that bound to and inhibited the function of both LA and LB were added to the mixture at 1 part in 4, the reaction was inhibited for the duration of the experiment (Fig. 3a, gray squares), and the circles remained unreacted even after 24 h (data not shown). In each instance, the observed reaction kinetics were fully consistent with the engineered cross-catalytic cycle originally shown in Fig. 1b. Just as protease zymogens participate in blood-clotting cascades, cleavage of inactive circularized deoxyribozymogens results in the initiation of an autocatalytic cascade that drives the exponential accumulation of active species.
The kinetic behavior of self-replicating systems can be described by the replicator equation: d[L]/dt]initial = ka[L0]p + kb (7, 8, 11). Although this equation is typically applied to ligationbased systems, it is also applicable to our cleavage-based system where the initial rate of cleavage is proportional to the initial trigger concentration, L0. To show that the reaction was truly exponential and that the deoxyribozymes were turning over, the initial rate of reaction was determined as a function of trigger (LA) concentration (Fig. 4). The linear relationship demonstrated by a plot of [L0]p versus [d[L]/dt]initial, where p = 1 is a hallmark of the exponential character of the cycle. The slope of the line yields a autocatalytic rate constant of 1.5 ± 0.04 h–1, which is in relatively good agreement with the rate of reaction of LA with CB under the subsaturating conditions of the assay (kobs at [160 nM] = 1.1 ± 0.03 h–1). The y intercept, kb, gives the rate for the background reaction of 1.3 ± 0.4 nM·h–1, which is consistent with the value obtained by direct measurement using the noncleavable circle CA (1 nM·h–1 at 160 nM CA and CB).
Fig. 4.

Initial rate of reaction as a function of trigger concentration. Rates were determined by the best linear fit of the first three or four time points from each curve shown in Fig. 3c. The line on the graph represents the best fit of all of the rate data. However, only the first four points were used for the determination of the slope and y intercept as reported in the text.
Recently, Paul and Joyce (11) engineered a ligase ribozyme that was capable of limited self-replication. Unfortunately, strong intermolecular interactions between the half-ribozyme substrates permitted exponential growth to occur only when external template (full-length ribozyme) was seeded into the reaction. Even then, only about one in three template molecules reproduced themselves during the exponential phase of the reaction. However, it is interesting to note that although these two systems function in completely different ways, the autocatalytic efficiency of the exponential portion of the ligase system (ε = ka/kb = 3.3 × 108 M–1) is similar to that of the 10–23 cleavase cycle (ε = 1.2 × 109 M–1).
Selection of Functional Sequences. We attempted to determine whether the exponential growth of catalytic function could promote the evolution of functional sequences (i.e., change the frequency of functional sequences or “alleles” over time). To do this, two pools, C.E1 and C.E2, based on circles CA and CB, were constructed in which three nucleotides on opposite arms of the deoxyribozymes were randomized (Fig. 5 a and b). The pools were incubated together and allowed to generate linear, active species; the reaction was then transferred to a new tube containing unreacted circles, effectively “seeding” the new reaction. In theory, individual sequences capable of finding their appropriate partner would be expected to amplify exponentially and to predominate over several serial dilutions. The initial round of the selection contained 4 μM of each circular pool and 0.4 μM of each linear, active pool to initiate catalysis. After 48 h, 10% of the round 1 reaction was transferred to another tube containing an additional 4 μM of each circular pool. Serial transfers were conducted for three more rounds of selection with increasingly stringent incubation times of 72, 27, and 15 h. In addition to shortening the incubation time, rounds 3 and 4 were conducted at lower circle concentrations (2 μM each). The progress of the selection was monitored by gel electrophoresis, and the results are shown in Fig. 5c. The fraction of the population that was linear increased after the first round of selection was slightly decreased when the stringency was increased between rounds 2 and 3, and then increased again after round 3.
Fig. 5.

Construction and selection of 10–23 deoxyribozyme N3 pools. (a) Pool design. (b) Hypothesized interaction of linear and circular species during selection. (c) Progress of the selection.
Linear species from rounds 0 and 4 were cloned and sequenced. As can be seen from Fig. 6a, two sequences predominated from the C.E1 pool, the expected complement GTG (13 occurrences) and an alternate sequence GGG (nine occurrences). The GGG might be expected to form a stable duplex with the complement CAC in the C.E2 pool through the formation of a noncanonical A:G pair. On the other randomized deoxyribozyme, the expected complement TCC (20 occurrences) was found, along with the alternate sequences GCC (three occurrences), TCA (three occurrences), and several additional variants. Again, the minor variants that were recovered might still be expected to base-pair in the cross-catalytic cycle by the formation of noncanonical base pairs. If selection of functionality and sequence had occurred, it would be predicted that the “fitness” of the recovered species should be roughly proportional to its representation in the population. We assayed the major and minor variants (GTG and GGG) from C.E1 pool with the parental C.E2 sequence, CB, in an exponential growth assay (Fig. 6b). As predicted, GTG was more functional than GGG.
Fig. 6.

Sequence and functional evolution. (a) Round 0 and round 4 sequences. Sequences for linearized variants are shown with one flanking nucleotide. The randomized region is indicated by the box. (b) Time course of autocatalytic cleavage of selected sequences. Circularized deoxyribozymes containing either GTG (CA) (▪) or GGG (•) were incubated with radiolabeled circle CB. Reactions were set up as described in Materials and Methods and contained 20 nM LB as an initial trigger.
Information transfer and amplification in the absence of protein catalysts has previously been shown. In particular, von Kiedrowski (1) and Sievers and von Kiedrowski (3) have shown that oligonucleotides of defined sequence can replicate themselves by templating the chemical ligation of smaller substrates. However, the accumulation of a given product is ultimately limited by product inhibition (7, 8). Because of this problem, the selection cannot go to completion, although this limitation can be overcome by the stepwise addition of reagents that lead to product release (9, 10) and has been mathematically analyzed to be less limiting in replication schemes involving surfaces (21). In the cleavage-based system that we have developed, all sequences are present in the initial pool and at every round in equal concentrations. However, certain sequences accumulate and predominate in the linearized products over multiple transfer events. As a result, if a sequence is initially a successful catalyst, proportionately more of it will be converted into a linear molecule at later times. These results point at an important distinction to be made when considering early evolution: although individual chemical species can predominate because they drive other species to extinction, it is also possible for chemical species to predominate in the absence of direct competition for resources, purely because they are more fecund.
Although the results of the current selection were essentially predetermined (the random regions opposed constant regions), the incorporation of larger, overlapping random regions should allow for the direct selection of optimal deoxyribozyme pairs. Ultimately, to the extent that such selections prove feasible, simple molecular ecosystems involving multiple, interdependent deoxyribozymes may evolve, just as McCaskill and coworkers (22, 23) have demonstrated the interdependent evolution of nucleic acids in a protein-mediated replication system.
Although the cleavase cascade is primarily interesting because it demonstrates the potential for sequence evolution, it may also prove possible to engineer a novel detection and amplification system. A number of researchers have exploited the astounding conformational flexibilities exhibited by nucleic acids to design and select “aptazymes,” allosteric nucleic acid enzymes that can be activated or inhibited by molecular effectors ranging from small organic molecules to oligonucleotides to proteins (24, 25, 26, 27). In particular, Sen and coworkers (28, 29) have engineered the 10–23 deoxyribozyme to be responsive to molecule effectors, such as oligonucleotides and ATP. Combining the cleavase cascade with such 10–23 aptazyme “triggers” would potentially allow the exponential amplification of effector-dependent cleavages, and hence the sensitive detection of the effectors themselves. Indeed, by introducing fluorescent reporters and quenchers across from one another on the “arms” of circularized 10–23 variants, the cleavase cascade would be poised to yield an exponential accumulation of fluorescence. Although nucleic acid recognition of effector molecules has previously been transduced to ligation events, the ligated products were detected by subsequent PCR amplification (30, 31). In contrast, a diagnostic cleavase cascade would require no added proteins and potentially no manipulations other than sample addition. An all-nucleic acid cascade also might be coupled with nucleic acid “machines” assisting in the rapid self-assembly and -disassembly of DNA structures (32, 33, 34) or find a use in the world of DNA computation (35), potentially aiding in rapid, sequence-specific readout.
Acknowledgments
We thank Kenneth A. Johnson for his assistance with kinetic models and simulations and the editor and anonymous referees for their valuable comments, including coining the term deoxyribozymogen. This work was supported by a grant from the National Aeronautics and Space Administration Astrobiology Institute.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.von Kiedrowski, G. (1986) Angew. Chem. Int. Ed. Engl. 25, 932–935. [Google Scholar]
- 2.Zielinski, W. S. & Orgel, L. E. (1987) Nature 327, 346–347. [DOI] [PubMed] [Google Scholar]
- 3.Sievers, D. & von Kiedrowski, G. (1994) Nature 369, 221–224. [DOI] [PubMed] [Google Scholar]
- 4.Inoue, T. & Orgel, L. E. (1983) Science 219, 859–862. [DOI] [PubMed] [Google Scholar]
- 5.Inoue, T., Joyce, G. F., Grzeskowiak, K., Orgel, L. E., Brown, J. M. & Reese, C. B. (1984) J. Mol. Biol. 178, 669–676. [DOI] [PubMed] [Google Scholar]
- 6.Achilles, T. & Von Kiedrowski, G. (1993) Angew. Chem. Int. Ed. Engl. 32, 1198–1201. [Google Scholar]
- 7.von Kiedrowski, G. (1993) Bioorg. Chem. Front. 3, 113–146. [Google Scholar]
- 8.Von Kiedrowski, G., Wlotzka, B., Helbing, J., Matzen, M. & Jordan, S. (1991) Angew. Chem. Int. Ed. Engl. 30, 423–426. [Google Scholar]
- 9.Li, T. & Nicolaou, K. C. (1994) Nature 369, 218–221. [DOI] [PubMed] [Google Scholar]
- 10.Luther, A., Brandsch, R. & von Kiedrowski, G. (1998) Nature 396, 245–248. [DOI] [PubMed] [Google Scholar]
- 11.Paul, N. & Joyce, G. F. (2002) Proc. Natl. Acad. Sci. USA 99, 12733–12740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Santoro, S. W. & Joyce, G. F. (1998) Biochemistry 37, 13330–13342. [DOI] [PubMed] [Google Scholar]
- 13.Butenas, S. & Mann, K. G. (2002) Biochemistry (Moscow) 67, 3–12. [DOI] [PubMed] [Google Scholar]
- 14.Jones, K. C. & Mann, K. G. (1994) J. Biol. Chem. 269, 23367–23373. [PubMed] [Google Scholar]
- 15.Zimmermann, K. C., Bonzon, C. & Green, D. R. (2001) Pharmacol. Ther. 92, 57–70. [DOI] [PubMed] [Google Scholar]
- 16.Pan, T. & Uhlenbeck, O. C. (1992) Biochemistry 31, 3887–3895. [DOI] [PubMed] [Google Scholar]
- 17.Schurer, H., Lang, K., Schuster, J. & Morl, M. (2002) Nucleic Acids Res. 30, e56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cairns, M. J., Hopkins, T. M., Witherington, C., Wang, L. & Sun, L. Q. (1999) Nat. Biotechnol. 17, 480–486. [DOI] [PubMed] [Google Scholar]
- 19.Ota, N., Warashina, M., Hirano, K., Hatanaka, K. & Taira, K. (1998) Nucleic Acids Res. 26, 3385–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santoro, S. W. & Joyce, G. F. (1997) Proc. Natl. Acad. Sci. USA 94, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.von Kiedrowski, G. & Szathmary, E. (2000) Selection 1, 173–179. [Google Scholar]
- 22.Ellinger, T., Ehricht, R. & McCaskill, J. S. (1998) Chem. Biol. 5, 729–741. [DOI] [PubMed] [Google Scholar]
- 23.Wlotzka, B. & McCaskill, J. S. (1997) Chem. Biol. 4, 25–33. [DOI] [PubMed] [Google Scholar]
- 24.Soukup, G. A. & Breaker, R. R. (1999) Proc. Natl. Acad. Sci. USA 96, 3584–3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang, J. & Breaker, R. R. (1997) Chem. Biol. 4, 453–459. [DOI] [PubMed] [Google Scholar]
- 26.Robertson, M. P. & Ellington, A. D. (2000) Nucleic Acids Res. 28, 1751–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robertson, M. P. & Ellington, A. D. (2001) Nat. Biotechnol. 19, 650–655. [DOI] [PubMed] [Google Scholar]
- 28.Wang, D. Y. & Sen, D. (2001) J. Mol. Biol. 310, 723–734. [DOI] [PubMed] [Google Scholar]
- 29.Wang, D. Y., Lai, B. H. & Sen, D. (2002) J. Mol. Biol. 318, 33–43. [DOI] [PubMed] [Google Scholar]
- 30.Fredriksson, S., Gullberg, M., Jarvius, J., Olsson, C., Pietras, K., Gustafsdottir, S. M., Ostman, A. & Landegren, U. (2002) Nat. Biotechnol. 20, 473–477. [DOI] [PubMed] [Google Scholar]
- 31.Robertson, M. P. & Ellington, A. D. (1999) Nat. Biotechnol. 17, 62–66. [DOI] [PubMed] [Google Scholar]
- 32.Seeman, N. C. (2003) Nature 421, 427–431. [DOI] [PubMed] [Google Scholar]
- 33.Yurke, B., Turberfield, A. J., Mills, A. P., Jr., Simmel, F. C. & Neumann, J. L. (2000) Nature 406, 605–608. [DOI] [PubMed] [Google Scholar]
- 34.Eckardt, L. H., Naumann, K., Pankau, W. M., Rein, M., Schweitzer, M., Windhab, N. & von Kiedrowski, G. (2002) Nature 420, 286. [DOI] [PubMed] [Google Scholar]
- 35.Benenson, Y., Paz-Elizur, T., Adar, R., Keinan, E., Livneh, Z. & Shapiro, E. (2001) Nature 414, 430–434. [DOI] [PubMed] [Google Scholar]