

Abstract
In this study we date the introduction of HIV-2 into the human population and estimate the epidemic history of HIV-2 subtype A in Guinea-Bissau, the putative geographic origin of HIV-2. The evolutionary history of the simian immunodeficiency virussooty mangabey/HIV-2 lineage was reconstructed by using available database sequences with known sampling dates, and a timescale for this history was calculated by using maximum likelihood methods. The date of the most recent common ancestor of HIV-2 subtype A strains was estimated to be 1940 ± 16 and that of B strains was estimated to be 1945 ± 14. In addition we used coalescent theory to estimate the past population dynamics of HIV-2 subtype A in a rural population of Guinea-Bissau. Parametric and nonparametric estimates of the effective number of infections through time were obtained for an equal sample of gag, pol, and env sequences. Our estimates of the epidemic history of HIV-2 subtype A in Guinea-Bissau show a transition from constant size to rapid exponential growth around 1955–1970. Our analysis provides evidence for a zoonotic transfer of HIV-2 during the first half of the 20th century and an epidemic initiation in Guinea-Bissau that coincides with the independence war (1963–1974), suggesting that war-related changes in sociocultural patterns had a major impact on the HIV-2 epidemic.
The AIDS epidemic is clearly recognized as a viral zoonosis (1–3). Phylogenetic analysis indicates that multiple interspecies transmissions from simian species have introduced two genetically distinct types of HIV into the human population: HIV-1, closely related to simian immunodeficiency virus (SIV) from chimpanzees (SIVCPZ), and HIV-2, closely related to SIV from sooty mangabeys (SIVSM). Whereas HIV-1 group M subtypes (A–D, F, H, J, and K) are spread globally, HIV-2 subtypes are mainly restricted to West Africa and can be categorized as epidemic subtypes (A and B) and nonepidemic subtypes (C–G) (4–7). Biological reasons have been invoked to explain the difference in global epidemiology between HIV-1 and HIV-2, such as lower HIV-2 viral loads that correlate with a lower transmissibility (8–10). However, attempts to compare the history of the epidemics at their respective geographic origins are still lacking.
A useful strategy for investigating the epidemic history of HIV combines molecular clock analysis, to estimate the timescale of the epidemic, and coalescent theory, to infer the demographic history of the virus (11). Various molecular clock calculations have dated the most recent common ancestor (MRCA) of HIV-1 group M around 1930 ± 15 (12–14). HIV-1 group M has subsequently spread globally, generating the pandemic observed today. Here, we investigate the epidemic history of HIV-2 to test previously suggested hypotheses of its origin. We provide the first estimated dates of cross-species transmissions of HIV-2. Our results indicate a transfer of HIV-2 subtypes A and B from sooty mangabeys to humans during the first half of the 20th century. In addition, we estimate the epidemic history of HIV-2 before its identification and provide genetic evidence for a change in epidemiology of HIV-2 during 1955–1970.
Methods
Molecular Clock Analysis. A collection of 33 SIV/HIV-2 sequences with known isolation dates was retrieved from the HIV database (http://hiv-web.lanl.gov/content/index). Partial gag and env sequences were aligned according to their amino acid alignments, gaps were removed, and both alignments were concatenated, resulting in a total alignment of 1,107 nucleotides. A partition homogeneity test, also known as the incongruence length difference test (15), indicated no significant incongruence between the sequence data of both gene regions (P = 0.086). It should be noted that a threshold of 0.05 may be too conservative for this test (16, 17), and P values < 0.001 have been proposed as an indication against combining data (16). Therefore, the gene regions were combined, which generally improves phylogenetic accuracy when P > 0.01 (14). A suitable model of evolution was determined by hierarchical model testing (18), and phylogenetic trees were constructed by using different algorithms (neighbor-joining, quartet puzzling, and maximum likelihood heuristic search with tree bisection–reconnection branch swapping). A majority-rule consensus tree for the inferred phylogenies was obtained by using PAUP* V. 4.0b8 (kindly provided by David Swofford, Florida State University, Tallahassee), and branch lengths were reestimated under the Tamura–Nei model of evolution and a discrete γ-distribution to account for rate heterogeneity among sites. Different outgroups external to the SIV/HIV-2 lineage were explored, but plotting the number of transitions and transversions against genetic distances allowed consistent observation of saturation (no saturation was observed without these outgroups). Therefore, the phylogeny was rooted by using a maximum likelihood approach. All possible root positions were evaluated under the single rate dated tip (SRDT) model (19), and the root that yielded the highest likelihood was retained. One thousand bootstrap replicates were generated to calculate the support for the nodes in the tree. All alignments and phylogenies are available from the authors on request.
Rates of evolution and divergence times with 95% confidence limits were estimated under the SRDT constraint in PAML (20) by using the Tamura–Nei model of nucleotide substitution with different combinations of parameter heterogeneity among gag and env alignments (21). The varied parameters were the substitution rate, nucleotide frequencies, the transition/transversion bias (κ1), the A-G/C-T transition bias (κ2), and the discrete γ-distribution. Separate analyses of the gag and env alignments (complete parameter heterogeneity) were not informative enough to reliably estimate separate evolutionary rates for the two genes. Evolutionary rates also were estimated under codon substitution models with a single nonsynonymous/synonymous substitution rate (dN/dS) ratio and a discrete class of dN/dS ratios among sites (22). Incorporating a discrete distribution of three dN/dS site classes in codon substitution models is analogous to a discrete γ-distribution of rates among sites in nucleotide substitution models. The molecular clock was tested by a likelihood ratio test between the SRDT model and the general unconstrained branch length model (different rates model; ref. 19).
Demographic History Inferences. Genealogies for a set of 73 pol, gag, and env sequences, sampled from the same 73 patients, were reconstructed in PAUP* V. 4.0b8 by using a maximum likelihood approach. Tree-likelihood values were computed under the Hasegawa–Kishino–Yano (HKY) model of evolution with γ-distributed rate heterogeneity among sites, as determined by hierarchical model testing (18). Because of the large data sets used, coestimation of the tree topology and model parameters was not possible, so the model parameters were estimated on an initial neighbor-joining tree. Tree topologies then were evaluated by using a heuristic search approach that implemented both tree bisection–reconnection and nearest-neighbor interchange perturbations. Finally, the branch lengths were reestimated for all possible rooted trees under the molecular clock constraint, keeping the model parameters fixed, and the maximum likelihood root was retained. The molecular clock hypothesis was rejected for all three genealogies (P < 0.01).
Estimates of demographic history were obtained by using GENIE 3.0 (23). Nonparametric estimates of effective population size against time were calculated by using generalized skyline plots (24). Demographic parameters were estimated by maximum likelihood under a piecewise expansion growth model (1):
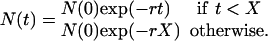 |
[1] |
This model describes the population size at time t in the past [N(t)], the population size at present [N(0)], the exponential growth rate (r), and the transition time (X). The population size before transition is equal to N(0)exp(-rX). Model fitting was evaluated by likelihood ratio testing of the parametric maximum likelihood estimates. The exponential model was rejected in favor of the piecewise expansion model for the env and pol phylogenies (P < 0.05). For gag, the piecewise expansion model gave the best fit, but this was not significantly better than the exponential model. Evolutionary rates were obtained by setting the MRCAs of the subtype A trees at 1940 as estimated from the SIVSM/HIV-2 phylogeny. Approximate 95% confidence intervals for the parameters were estimated by using the likelihood ratio test statistic.
Results
To investigate the origin of HIV-2, we reconstructed a phylogenetic tree of the SIVSM/HIV-2 lineage based on partial gag and env SIV sequences from sooty mangabeys (Cercocebus torquatus atys), HIV-2 subtypes A–D and F, and SIV strains from macaques (Macaca arctoides and Macaca nemestrina) that were acquired by cross-species transmission from sooty mangabeys (Fig. 1). A timescale for the HIV-2 epidemic was calculated by using a model that includes the isolation dates of noncontemporaneous samples. Although the molecular clock was rejected by likelihood ratio testing (P < 0.01), simulations have shown that even when the clock is rejected, the confidence limits of the substitution rate sometimes still may include the true rate (25). In addition, a site stripping for clock detection (SSCD) procedure based on likelihood ratio reduction indicates that the rate variability did not bias the estimated dates (P.L., unpublished work). Estimates obtained under various models accounting for heterogeneous parameters among gag and env alignments and under various codon substitution models gave very similar results (Table 1). According to this timescale (Fig. 1), the MRCAs for subtypes A and B (each represented by multiple strains and supported by maximal bootstrap support) are dated to 1940 ± 16 and 1945 ± 14, respectively.
Fig. 1.

Timescale for the SIVSM/HIV-2 lineage. HIV-2 subtypes and sooty mangabey and macaque strains are indicated at the tips of the reconstructed phylogeny. Branch lengths were estimated under the SRDT model, which allowed us to impose the estimated timescale under the phylogenetic tree (19). Numbers at the nodes indicate the percentage of bootstrap samples (of 1,000) in which the right cluster is supported (only values >80% are shown).
Table 1. Evolutionary rate analysis under various substitution models.
| Substitution model | Model description | Evolutionary rate, substitutions per year per site | MRCA of subtype A |
|---|---|---|---|
| Nucleotide | Complete homogeneity | 1.53 × 10-3 ± 4.70 × 10-4 | 1940 ± 15.85 |
| Different rates | 1.40 × 10-3 ± 4.43 × 10-4 | 1940 ± 15.90 | |
| Different rates and frequencies | 1.36 × 10-3 ± 4.61 × 10-4 | 1939 ± 17.50 | |
| Different rates, frequencies, and κ1 and κ2 | 1.39 × 10-3 ± 5.12 × 10-4 | 1939 ± 18.92 | |
| Different rates, frequencies, κ1 and κ2, and γ | 1.37 × 10-3 ± 4.40 × 10-4 | 1939 ± 16.36 | |
| Codon | Single dN/dS among sites | 4.89 × 10-3 ± 1.52 × 10-3* | 1940 ± 15.89 |
| Discrete distribution of dN/dS among sites | 5.07 × 10-3 ± 1.30 × 10-3* | 1941 ± 12.75 |
The model description for the nucleotide substitution models refers to the parameter heterogeneity allowed between the gag and env parts of the concatenated alignment. The only model that fitted the data significantly better was the one allowing for different rates between the genes (P < 0.05), which was the model used in all further estimates. For the codon substitution models, the model descriptions specify the dN/dS among sites. Substitution rates and dates for the MRCA of HIV-2 subtype A were estimated under the SRDT constraint.
Under codon substitution models the rates are expressed in number of substitutions per year per codon site and therefore are not directly comparable to the nucleotide substitution rates under nucleotide substitution models.
To investigate the epidemic history of HIV-2, we estimated the demographic history of subtype A, the principal contributor to the HIV-2 epidemic, in a rural population of Guinea-Bissau. The study data set resulted from serological screening of 2,774 subjects, representative of a population of ≈7,000 living in an area 30 km from the regional center of Canchungo (Fig. 2a; ref. 26). Because of the surprisingly high HIV-2 seroprevalence in this region (26), it has been implicated as the possible nucleus of the HIV-2 epidemic (27). We reconstructed genealogies based on an equal sample of gag, pol, and env sequences (28). To choose a suitable parametric model that describes effective population size through time, nonparametric estimates were obtained by using generalized skyline plots (Fig. 2; refs. 23 and 24). A recent period of exponential growth can be clearly discerned in all plots, whereas the early history of the epidemic seems to be characterized by constant population size. This behavior can be modeled by using a piecewise expansion model (see Methods). This model provided a good fit to the demographic signal in the data, as evaluated by likelihood ratio testing (see Methods). In Fig. 2, the maximum likelihood parametric estimates under the piecewise expansion model were superimposed onto the skyline plots.
Fig. 2.

Demographic history of HIV-2 subtype A in a rural population situated near the regional center of Canchungo. (a) Map of Guinea-Bissau pinpointing the location of the study population (26, 28). (b–d) Coalescent results for the env (b), pol (c), and gag (d) genes. The skyline plots, representing nonparametric estimates, are indicated by a gray line, whereas the parametric estimates under the piecewise expansion model are indicated by a superimposed black line. Both nonparametric and parametric results show the change in the effective number of infections on a natural timescale (23).
To rescale the demographic estimates into a real timescale and transform the population parameters into their natural units, the substitution rates of the genes can be used. Because the population sample contains only contemporary sequences, we used the date for the MRCA of the HIV-2 subtype A clade in the SIVSM/HIV-2 phylogeny to calibrate the substitution rates for the gag, pol, and env genealogies. Substitution rates and population parameters are listed in Table 2. Based on the results for pol and env, for which the piecewise expansion model gave a significantly better fit, the transition time should be situated in 1955–1970. The exponential growth rate for the effective number of HIV-2 subtype A infections in this population is estimated to be ≈0.20 yr-1 (range: 0.16–0.28 yr-1). Although direct comparison of population parameters for HIV data should be treated with caution (24), the HIV-2 growth rate in our studied population seems to be faster than the growth rate of HIV-1 group M in the Democratic Republic of Congo (11).
Table 2. Parametric estimates (with 95% confidence intervals) under the piecewise expansion model for the three genealogies.
| Parameter | env | pol | gag |
|---|---|---|---|
| Substitution rate | 4.20 × 10-3 | 2.46 × 10-3 | 1.22 × 10-3 |
| Transition time | 1960 (1955-1963) | 1967 (1962-1970) | 1951 (NA-1957) |
| Growth rate, yr-1 | 0.201 (0.167-0.236) | 0.243 (0.197-0.286) | 0.198 (0.164-0.240) |
| Current population | 10,109 (5,138-21,218) | 6,183 (3,166-11,896) | 31,636 (13,829-888,070) |
The estimates for the population parameters under the piecewise expansion model are based on the evolutionary rate calibrated by the 1940 date for the MRCA of HIV-2 subtype A. NA, not applicable.
Discussion
The main goal of this study was to provide a timescale for the HIV-2 interspecies transmissions and to investigate the epidemic history in its putative geographical origin. According to our findings, the MRCAs for both subtypes A and B are situated in 1940 ± 16 and 1945 ± 14, respectively, which can be considered as upper limits for the interspecies transmissions. Although subtypes A and B have no simian counterpart, there is evidence that these subtypes are a result of independent transmission events from sooty mangabeys to humans (4). Therefore, the common ancestor of subtypes A and B can be considered as the lower bound for individual cross-species transmissions (1889 ± 33).
The temporal setting of interspecies transmissions reveals that the origin of HIV-2 is not as enigmatic as previously suggested (29). Seroprevalence data suggest that HIV-2 originated in Guinea-Bissau and that the virus's rapid spread in this state did not start until 1960–1970 (27). Because the sooty mangabey subspecies C. torquatus atys seems to have become extinct in this area during the last half century, a natural SIV transfer from that species has been questioned in this time frame (29). However, our dating results place the cross-species transmissions of the dominant subtypes in the first half of the 20th century, indicating that HIV-2 was present before the earliest retrospective serological evidence. At that time, the sooty mangabeys probably had a wider geographical spread (29), and therefore our dating is consistent with a natural transfer hypothesis.
Despite different evolutionary constraints on the pol, gag, and env genes, the violation of the molecular clock assumption, and potential recombination events in the evolutionary history, the demographic estimates are surprisingly consistent. As in previous studies on the origin of HIV-1 group M, recombination might be the major cause of the loss of a molecular clock, and estimated dates may be associated with larger variances when recombination is frequent (30, 31). The quantitative effects of recombination on coalescent-based estimates have not yet been determined. However, the consistency of our results among the three genome regions studied is reassuring. The demographic history of HIV-2 in its putative region of origin, like that of HIV-1 (11), is characterized by a period of low endemicity, followed by an exponentially increasing number of infections. A low baseline of infected individuals before the transition around 1955–1970 explains why retrospective evidence of HIV-2 infections is limited to the early 1960s. Our estimates specifically relate to the epidemiology of HIV-2 subtype A in the rural population of Guinea-Bissau that was sampled and should not be extrapolated to the HIV-2 epidemic as a whole.
Our demographic estimates suggest that an event enabled HIV-2 subtype A to switch to epidemic growth sometime around 1955–1970. An initiation of the epidemic at this time coincides with the time frame of the independence war (1963–1974) in Guinea-Bissau, a former Portuguese colony, and it has recently been hypothesized that this war in Guinea-Bissau played a critical role in the early dissemination of HIV-2 (27). There is evidence that both sexual and blood-borne HIV-2 transmission markedly increased during this period (27). Epidemiological linkage of HIV-2 with Portugal, established during the presence of the colonial army, was recognized when the first reported cases of HIV-2 in Europe were Portuguese veterans who had served in the army during the independence war (27, 32). In Guinea-Bissau, HIV-2 seroprevalence was found to be consistently higher in older age groups (26, 27). This pattern can be explained by a cohort effect for a generation that was sexually active at the time of the war of independence (27). Equally importantly, HIV-2 transfusion cases related to this period have been reported, suggesting that the virus had entered the blood supply in Guinea-Bissau by 1966 (33). Increased unsterile injections during 1950–1970 have been hypothesized as being the principal agent in the emergence of epidemic HIV in Africa (6). For Guinea-Bissau in particular, it has been recorded that army-trained doctors launched massive inoculation campaigns at the clinic of Canchungo (34). Because this is the exact location from which the population samples discussed here were drawn, it would not be surprising if the fast epidemic growth rates we estimated (Table 2) reflect parenteral transmission patterns (35). The effective population size before transition is on the order of 10, indicating that the MRCA of subtype A (dated to 1940) existed some time after the cross-species transmission event, at which, by definition, only a single person was infected. Because we estimate that the cross-species transmission occurred in the first half of the 20th century, there was sufficient time between the zoonosis and the MRCA of subtype A for the number of infections to increase.
A combination of phylogenetic, molecular clock, and coalescent analyses forms a powerful framework to construct and test hypotheses about viral epidemics. We have provided, to our knowledge, the first estimates for the date of the MRCA of HIV-2 subtypes and the cross-species transmission events from sooty mangabeys. Our data are consistent with a natural transfer of the epidemic HIV-2 subtypes from sooty mangabeys to humans during the first half of the 20th century. Our genealogical approach strongly supports the major role of the independence war (1963–1974) and associated changes in sociocultural patterns in the transition of HIV-2 from endemic to epidemic behavior. These findings provide general insights on viral zoonosis and outline the important factors to be considered in the interpretation of the epidemiological spread of HIV-2 and other viruses globally.
Acknowledgments
We thank A. Rambaut for clarifying discussions on the SRDT model and rooting phylogenies; Z. Yang for helpful suggestions on models for combined analysis in paml; A. Heredia, A. Hovanessian, and J. M. A. Pereira for providing sampling dates on several HIV-2 strains; K. Robbins for critical comments on an earlier version of the manuscript; and R. Camacho for feedback on the history of HIV-2 in Guinea-Bissau. This work was supported by the Fonds voor Wetenschappelijk Onderzoek (Grant 0288.01), the Flemish Institute for Scientific-Technological Research in Industry (P.L.), and the Wellcome Trust (O.G.P.). M.S. is a postdoctoral fellow with the Fonds voor Wetenschappelijk Onderzoek.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: SIV, simian immunodeficiency virus; MRCA, most recent common ancestor; SRDT, single rate dated tip; dN/dS, nonsynonymous/synonymous substitution rate.
References
- 1.Hahn, B. H., Shaw, G. M., De Cock, K. M. & Sharp, P. M. (2000) Science 287, 607–614. [DOI] [PubMed] [Google Scholar]
- 2.Sharp, P. M., Robertson, D. L. & Hahn, B. H. (1995) Philos. Trans. R. Soc. London B 349, 41–47. [DOI] [PubMed] [Google Scholar]
- 3.Sharp, P. M., Bailes, E., Robertson, D. L., Gao, F. & Hahn, B. H. (1999) Biol. Bull. (Woods Hole, Mass.) 196, 338–342. [DOI] [PubMed] [Google Scholar]
- 4.Chen, Z., Luckay, A., Sodora, D. L., Telfer, P., Reed, P., Gettie, A., Kanu, J. M., Sadek, R. F., Yee, J., Ho, D. D., et al. (1997) J. Virol. 71, 3953–3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao, F., Yue, L., Robertson, D. L., Hill, S. C., Hui, H., Biggar, R. J., Neequaye, A. E., Whelan, T. M., Ho, D. D., Shaw, G. M., et al. (1994) J. Virol. 68, 7433–7447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marx, P. A., Alcabes, P. G. & Drucker, E. (2000) Philos. Trans. R. Soc. London B 356, 911–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yamaguchi, J., Devare, S. G. & Brennan, C. A. (2000) AIDS Res. Hum. Retroviruses 16, 925–930. [DOI] [PubMed] [Google Scholar]
- 8.Popper, S. J., Sarr, A. D., Travers, K. U., Gueye-Ndiaye, A., Mboup, S., Essex, M. E. & Kanki, P. J. (1999) J. Infect. Dis. 180, 1116–1121. [DOI] [PubMed] [Google Scholar]
- 9.Shanmugam, V., Switzer, W. M., Nkengasong, J. N., Garcia-Lerma, G., Green, T. A., Ekpini, E., Sassan-Morokro, M., Antunes, F., Manshino, K., Soriano, V., et al. (2000) J. Acquired Immune Defic. Syndr. 24, 257–263. [DOI] [PubMed] [Google Scholar]
- 10.De Cock, K. M., Adjorlolo, G., Ekpini, E., Sibailly, T., Kouadio, J., Maran, M., Brattegaard, K., Vetter, K. M., Doorly, R. & Gayle, H. D. (1993) J. Am. Med. Assoc. 270, 2083–2086. [DOI] [PubMed] [Google Scholar]
- 11.Yusim, K., Peeters, M., Pybus, O. G., Bhattacharya, T., Delaporte, E., Mulanga, C., Muldoon, M., Theiler, J. & Korber, B. (2001) Philos. Trans. R. Soc. London B 356, 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Korber, B., Muldoon, M., Theiler, J., Gao, F., Gupta, R., Lapedes, A., Hahn, B. H., Wolinsky, S. & Battacharya, T. (2000) Science 288, 1789–1796. [DOI] [PubMed] [Google Scholar]
- 13.Salemi, M., Strimmer, K., Hall, W. W., Duffy, M., Delaporte, E., Mboup, S., Peeters, M. & Vandamme, A. M. (2001) FASEB J. 15, 276–278. [DOI] [PubMed] [Google Scholar]
- 14.Sharp, P. M., Bailes, E., Chaudhuri, R. R., Rodenburg, C. M., Santiago, M. O. & Hahn, B. H. (2001) Philos. Trans. R. Soc. London B 356, 867–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farris, J. S., Källersjö, M., Kluge, A. G. & Bult, C. (1995) Cladistics 10, 315–319. [Google Scholar]
- 16.Cunningham, C. W. (1997) Mol. Biol. Evol. 14, 733–740. [DOI] [PubMed] [Google Scholar]
- 17.Sullivan, J. (1996) Syst. Biol. 45, 375–380. [Google Scholar]
- 18.Posada, D. & Crandall, K. A. (1998) Bioinformatics 14, 817–818. [DOI] [PubMed] [Google Scholar]
- 19.Rambaut, A. (2000) Bioinformatics 16, 395–399. [DOI] [PubMed] [Google Scholar]
- 20.Yang, Z. (1997) Comput. Appl. Biosci. 13, 555–556. [DOI] [PubMed] [Google Scholar]
- 21.Yang, Z. (1996) J. Mol. Evol. 42, 587–596. [DOI] [PubMed] [Google Scholar]
- 22.Yang, Z., Nielsen, R., Goldman, N. & Pedersen, A. M. (2000) Genetics 155, 431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pybus, O. G., Rambaut, A. & Harvey, P. H. (2000) Genetics 155, 1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strimmer, K. & Pybus, O. G. (2001) Mol. Biol. Evol. 18, 2298–2305. [DOI] [PubMed] [Google Scholar]
- 25.Jenkins, G. M., Rambaut, A., Pybus, O. G. & Holmes, E. C. (2002) J. Mol. Evol. 54, 156–165. [DOI] [PubMed] [Google Scholar]
- 26.Wilkins, A., Ricard, D., Todd, J., Whittle, H., Dias, F. & Paulo Da Silva, A. (1993) AIDS 7, 1119–1122. [DOI] [PubMed] [Google Scholar]
- 27.Poulsen, A. G., Aaby, P., Jensen, H. & Dias, F. (2000) Scand. J. Infect. Dis. 32, 169–175. [DOI] [PubMed] [Google Scholar]
- 28.Grassly, N. C., Xiang, Z., Ariyoshi, K., Aaby, P., Jensen, H., van der Loeff, M. S., Dias, F., Whittle, H. & Breuer, J. (1998) J. Virol. 72, 7895–7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hooper, E. (1999) The River (Little, Brown, Boston), pp. 623–643.
- 30.Schierup, M. H. & Hein, J. (2000) Mol. Biol. Evol. 17, 1578–1579. [DOI] [PubMed] [Google Scholar]
- 31.Schierup, M. H. & Hein, J. (2000) Genetics 156, 879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Piedade, J., Venenno, T., Prieto, E., Albuquerque, R., Esteves, A., Parreira, R. & Canas-Ferreira, W. F. (2000) Acta Trop. 76, 119–124. [DOI] [PubMed] [Google Scholar]
- 33.Mota-Miranda, A., Gomes, H., Marques, R., Serrao, R., Lourenco, H., Santos-Ferreira, O. & Lecour, H. (1995) J. Infect. 31, 163–164. [DOI] [PubMed] [Google Scholar]
- 34.Venter, A. J. (1973) Portugal's Guerilla War: The Campaign for Africa (John Malherbe, Cape Town, South Africa).
- 35.Pybus, O. G., Charleston, M. A., Gupta, S., Rambaut, A., Holmes, E. C. & Harvey, P. H. (2001) Science 292, 2323–2325. [DOI] [PubMed] [Google Scholar]