

Abstract
A method of analyzing DNA microarray data based on the physical modeling of hybridization is presented. We demonstrate, in experimental data, a correlation between observed hybridization intensity and calculated free energy of hybridization. Then, combining hybridization rate equations, calculated free energies of hybridization, and microarray data for known target concentrations, we construct an algorithm to compute transcript concentration levels from microarray data. We also develop a method for eliminating outlying data points identified by our algorithm. We test the efficacy of these methods by comparing our results with an existing statistical algorithm, as well as by performing a cross-validation test on our model.
Use of DNA microarrays, wherein the expression levels of thousands of genes can be monitored simultaneously (1, 2), has become widespread in biochemical research. Accurate interpretation of microarray data remains a significant challenge, however. The raw data exhibit large fluctuations, the origin of which is unclear, and most algorithms for extracting quantitative expression levels from the data are either empirical or statistical in nature.
The central component of all DNA microarray technologies is an array of different oligonucleotides (between 20 and several hundred bases long) deposited onto a single substrate. When a fluorescently labeled nucleic acid sample is washed over the substrate, it hybridizes to those probes that are complementary to it. Subsequent detection of the fluorescent signal allows one to quantify the presence of various sequences (and thus the expression levels of various genes) within the sample. One class of DNA microarrays is exemplified by Affymetrix (Santa Clara, CA) GeneChips (2, 3), wherein each transcript is probed by multiple, short oligomers (typically 20- to 25-mers) or probes. Typically, ≈20 perfect match (PM) probes and an equal number of mismatch (MM) probes correspond to each transcript. Each PM probe exactly complements a short region of the transcript, referred to as the target. The corresponding MM probe is identical to the PM probe except at its centermost base, which is noncomplementary to the transcript. Typical algorithms that use microarray data to infer quantitative transcript expression levels begin by subtracting the MM intensity from the corresponding PM intensity (with adjustments to the MM value if MM > PM) (4). The expression level is then taken to be a weighted average of those differences obtained from all the pairs that probe the given transcript. One advantage of these genechips over other technologies, such as arrays spotted with a single, longer probe for each transcript, is that these chips can provide an absolute measure of gene expression (i.e., transcript copies per cell), whereas spotted arrays typically measure up- or down-regulation relative to some control condition (1).
Although many techniques have been developed to identify trends in the expression levels inferred from DNA microarray data (5), less attention has been devoted to methods of obtaining accurate expression levels from the raw data. One promising method, developed by Li and Wong (6), assumes probe-dependent target-binding strengths determined through statistical analysis of data. In addition, several studies have been carried out to identify sources of noise present in microarray data (7, 8). However, to date there has been little attempt (9) to analyze raw microarray data by constructing an algorithm based on the underlying physical principles of the hybridization process.
In this article we present such an analysis. That is, we develop a model based on calculated hybridization free energies and the rate equation for duplex formation and melting. Using publicly available data (see www.affymetrix.com/analysis/download_center2.affx), we demonstrate a clear correlation between calculated hybridization free energies and observed hybridization intensities, and show that the data can be well modeled by using the static equilibrium solution of the rate equation and incorporating the correlation. We further present a method of systematically identifying and removing outlying data points from the analysis, and compare our results both with the known answers for a controlled, publicly available data set and the results of an existing statistical algorithm. Finally, we discuss possible causes for variations observed among data sets from nominally equivalent experiments.
Experiments and Data
All the data shown and analyzed in this article are taken from the publicly available results of experiments carried out at Affymetrix (see www.affymetrix.com/analysis/download_center2.affx and ref. 10). In particular, the data are the results of a series of controlled ``spike-in'' experiments, in which a transcript group comprised of known concentrations (each between 0 and 1,024 pM), of 14 human genes is spiked into a background consisting of a labeled mixture of mRNA from a human pancreatic tissue source. (None of the spike-in genes were expressed in the tissue source.) This mixture is then hybridized onto Affymetrix U95A GeneChips. Fourteen different transcript groups, each containing different concentrations, of the various spike-in genes (following an experimental design known as a Latin square), are then each hybridized onto a genechip. The net result is that each of the genes is spiked into one of the transcript groups at each of the following concentrations: 0, 0.25, 0.5, 1, 2, 4,... 1,024 pM. That is, data are collected for all 14 genes, each at 14 different concentrations. Sixteen distinct PM–MM probe pairs interrogate each transcript. Finally, each of the measurements for a given gene at a given concentration was replicated between 2 and 12 times. The raw data provided by Affymetrix contain intensities for each PM and MM probe (the intensities provided corresponding to the 75th percentile of the intensity of the scanned pixels associated with the probe site). We performed no normalization of these data before the analysis discussed below. Because two of the genes in the experiment (407_at and 36889_at) had some defective probes, only 12 of the 14 genes are used in our analysis. Note that throughout this article we use Affymetrix notation to identify genes.
Fluctuations and Hybridization Energies
Because target fragments complementary to each of the 16 PM probes representing any given transcript are present with virtually identical concentration in the sample solution, one might expect that in any experiment the intensities of the 16 spots containing these probes should be close in magnitude. In practice, however, significant variations in intensity are reproducibly observed among probes within the probe set interrogating a given target (3). One possible cause for these variations is the tendency of certain probes to hybridize more strongly with their complementary targets than others. In this article we study and exploit this possibility.
Because the hybridization rate increases as the free-energy differential associated with the reaction becomes more negative, some of the probe-to-probe variations observed in the data might be ascribable to sequence-dependent variations in these free energies. We show below that this indeed is the case and then incorporate these systematic differences in hybridization rates into algorithms designed to infer transcript concentrations from microarray data.
Studying the dependence of measured microarray intensities on target–probe hybridization free energies requires first determining those free energies. In the absence of detailed experimental results on hybridization in the confined geometry of microarrays, we use an existing nearest-neighbor model (11) developed for calculating free energies in solution. Obviously this model represents an approximation for the microarray geometry, where (at least) the entropic contributions to free-energy changes clearly differ from those in solution. Because the entropies, in microarrays, of both the initial, confined, single-strand probe and the final, hybridized, probe–target double strand are reduced relative to their values in solution, however, one hopes that the two reductions are close enough to make the approximation reasonable. The fact that such a model provides a reasonable fit to the experimental data provides some a posteriori justification for its use.
In nearest-neighbor models, the hybridization free energy of any base pair depends not only on whether that pair is a C-G or an A-T but also on which base pairs occupy the neighboring positions along the strand. Table 2 of ref. 11 gives the changes in enthalpy and entropy, and hence free energy (at 310 K), associated with each of the 10 independent stackings of two base pairs along an oligonucleotide chain in a 1 M solution. To calculate the hybridization free energy for any sequence, one sums the contributions for each of these stacked pairs along the chain and adds a correction (see table 2 of ref. 11) for the base pairs terminating the sequence at each end.
One now can check whether measured microarray intensities in fact increase monotonically with increasingly negative free-energy changes. Although such a trend is not obvious in the data obtained in a single measurement of the 16 probes representing any single gene, it becomes apparent once one aggregates the Latin-square data from all the genes at a given concentration. Fig. 1 shows such aggregated data for all the experiments performed on all 12 genes, the different curves corresponding to different sample concentrations. The clear upward trend in the figure demonstrates the expected increase in hybridization intensity with increasingly negative free energy.† Note, however, that none of the curves in Fig. 1 for concentrations <256 pM are strictly monotonic. It is unclear whether this nonmonotonic behavior is due to statistical fluctuations in the relatively sparse Latin-square data set, the approximations inherent in our calculations of free energies, or other causes.
Fig. 1.

Observed hybridization intensity as a function of calculated hybridization free energy, –ΔG, for spike-in target concentrations between 0 and 1,024 pM. Each data point shows the average measured intensity for all probes with a calculated free energy that lies within a given energy bin at a particular target concentration. The centers of the 10 energy bins range between 26 and 38 kcal/mol, and their widths are all 1.33 kcal/mol. The average intensities are plotted at the values of the bin centers. An increase in observed hybridization intensity with increasing ΔG is clearly observable. The lines simply connect the data points.
One can also use the nearest-neighbor hybridization model to compute the melting temperatures of oligonucleotides in solution (11) and then compare the results with experimental observations. Forman et al. (12) measure the degree of hybridization of specific microarray probes as a function of temperature, observing, for example, that a specific 18-mer melts at 338 K (the temperature at which the fraction of hybridized pairs goes to zero, presumably corresponding to the melting of the full-length 18-mers on the given probe spot). The calculated melting temperature for the same probe is 367 K, suggesting that the application of thermodynamic quantities determined for nucleic acids in solution to confined geometries is reasonable.
Calculating the melting temperatures for each of the PM and MM probes for the 12 genes that we analyzed from the Latin-square data set (11, 13), one finds that the variation in melting temperature across the range of free energies represented is ≈20 K, whereas that between a PM–MM probe pair is typically ≈4 K (see Fig. 11, which is published as supporting information on the PNAS web site, www.pnas.org). Thus, incorporating the effects of free energy seems at least as significant as incorporating those of single MMs.
Data Analysis
To understand the dependence of measured intensity on transcript concentration, we plot in Fig. 2 the intensities for a single probe site (probe 1 of gene 37777_at) as a function of spike-in concentration, c, of gene 37777_at. Although the signal increases, as expected, with increasing c, the response between 0.25 and 1,024 pM is decidedly nonlinear. Rather, the signal appears to approach an asymptotic value, presumably as a result of the finite number of probe sites available. [Note that a linear dependence of signal strength on c is implicitly assumed in earlier statistical models (4, 6).] Assuming that the observed fluorescence intensity scales linearly with the number of hybridized probe–target duplexes, one can model the observed intensity by considering the binding and unbinding reactions
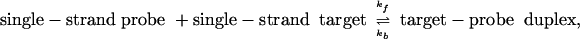 |
where kf and kb are the respective forward and backward rate constants for the reaction, and the concentrations [single-strand probe], [single-strand target], and [target–probe duplex] are given by (np – nB)/Vprobe, (n0 – nB)/Vtotal, and nB/Vprobe, where np, n0, Vprobe, and Vtotal are equal to the number of probe molecules at the given probe site, the number of transcript molecules in the target solution, the volume of the probe site, and the volume of the target solution, respectively. The number of bound target–probe pairs, nB, is determined by the rate equation
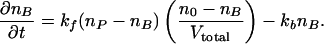 |
[1] |
If one assumes that the system reaches equilibrium (see figures 4 and 5 of ref. 12) and that np << n0 (np ≈ 107 and n0 ≈ 2 × 108 for a 0.25-pM target solution), it follows that nB = npn0/(n0 + ñe), where ñe = kbVtotal/kf. Recasting this equation for nB to yield observed intensity, I, as a function of concentration, c, we obtain
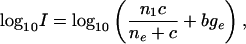 |
[2] |
where we have added a background term bge to account for the hybridization of probes to nucleic acids other than their intended targets. Here c = (no/Vtotal)/(NA/liter) is in mol/liter, NA is Avogadro's number, ne = (ñe/Vtotal)/(NA/liter), and the constant nI in Eq. 2 differs from nP by the proportionality factor that relates nB to the corresponding fluorescence intensity. We have written Eq. 2 in terms of the logarithm of I, as opposed to I itself, so as to maintain comparable sensitivity to multiplicative fold changes over the entire concentration range studied (12 powers of 2 in concentration) when computing least-square best fits of the data to this equation. In Fig. 2, the solid line is a best fit of the data shown to Eq. 2, where ne, nI, and bge are allowed to vary. Fits for all 16 probes of gene 37777_at are shown in Fig. 12, which is published as supporting information on the PNAS web site.
Fig. 2.

Observed hybridization intensity as a function of spike-in target concentration of PM probe one of gene 37777_at. Replicate measurements are shown as distinct data points. The solid line is a best fit of the data to Eq. 2, with nI, ne, and bge adjustable parameters.
In principle, data from each individual probe should follow Eq. 2, with nI varying little from probe to probe. In practice, however, fitting the data for each probe individually results in nI values of nI that range over an order of magnitude. To reduce the effects of such fluctuations, we fit all the data with a single nI. This is accomplished by binning the data over free energy for each concentration and then fitting all the binned data simultaneously. In Fig. 3, for example, observed intensity is plotted as a function of spike-in concentration for each of the free-energy bins shown in Fig. 1, each bin being depicted by a different color. That is, we have averaged the observed intensities of all probes within a given free-energy bin for a given spike-in concentration and plotted this average as a single point.
Fig. 3.

Observed hybridization intensity as a function of spike-in target concentration for the energy-binned data shown in Fig. 1. Units of energy are kcal/mol. Solid lines show the best fits of all the plotted data fit simultaneously to Eq. 2 (see Data Analysis).
We fit each of the curves in Fig. 3 to Eq. 2, the fits being constrained so that nI is held constant for all of the curves, whereas both ne and bge are allowed to vary from curve to curve. The results of this fitting procedure are shown as the solid lines in Fig. 3. The best-fit value for nI was found to be 9,494 (in the same units of fluorescence intensity as the data), whereas the best fits for ne and the background bge are plotted as functions of ΔG, the negative of the calculated hybridization free energy, in Fig. 4 a and b, respectively.
Fig. 4.

Best-fit values of ne (a) and bge (b) obtained from the fits shown in Fig. 3 vs. calculated ΔG. Solid lines are best fits to the plotted results (see Data Analysis).
From equilibrium thermodynamics, one would expect that ne = e–ΔG/RT. The best-fit straight line through the data in the semilog plot of Fig. 4a is shown as a solid line. However, equating the slope of this line to 1/2.3RT yields a temperature of 2,130 K, approximately seven times the true temperature. Possible reasons for this discrepancy are addressed in Discussion.
In Fig. 4b, we see that the background bge depends strongly on ΔG. One expects the probes that have the most negative hybridization free energies with their complementary targets to hybridize most strongly with background targets that are not quite complementary. Hence, assuming that bge results from background oligonucleotides in the test solution binding to the probe sites, and further assuming that all different sequences are represented roughly equally within these background oligonucleotides, one can readily understand why those probes with the largest ΔG values exhibit the strongest background signals. The background intensity is well described by the sum of a constant and a term that scales exponentially with ΔG. The best fit of this type is shown as a solid line in Fig. 4b. Assuming a nonspecific background of this form, the normalization of the background intensity (of microarray data not taken under the controlled conditions of these Latin-square measurements) could be quantified through the use of probe sites that are known not to bind specifically to any mRNA in the target mixture (14–17).
Combining the best-fit value of nI from the fits of Fig. 3 with the expressions for ne and bge derived from the fits in Fig. 4, we model the observed intensity as a function of ΔG and transcript concentration by Eq. 2 with bge = 127 + 6 × 10–5e0.423ΔG, ne = 10–6.505–0.102ΔG, and nI = 9,494.
To determine the concentration, c, of a given transcript from the data obtained from a single genechip, we plot the observed PM-probe intensities for that transcript as a function of calculated probe hybridization free energy. We then perform a least-squares fit of this data to Eq. 2, using the above values of nI, ne, and bge, with c as the only adjustable parameter and the constraint c ≥ 0. In Fig. 5 this procedure is illustrated on a data set for gene 36311_at at a spike-in concentration of 256 pM. The observed intensities of the 16 PM probes for this transcript are plotted as a function of the calculated ΔG values. The black line is a best fit of this data to Eq. 2. The best-fit value of c is 92 pM.
Fig. 5.

Observed hybridization intensity as a function of calculated ΔG for the probe set of gene 36311_at. The data were collected on a single GeneChip with a spike-in target concentration of 256 pM. The dotted line is a best fitof all the data to Eq. 2, with concentration c the only adjustable parameter; the best fit c is 92 pM. The solid line is a best fit to only the filled data points, the hollow ones having been identified as statistical outliers (see Data Analysis); the best fit c in this case is 151 pM, closer to the known spike-in value of 256 pM.
To improve the analysis, it is desirable to identify those data points that have either anomalously low or high intensities. Such points can be identified by their large distance from the best-fit curve of the model just described. In particular, we calculate the mean distance from the 16 data points for a given transcript to the best-fit curve, and the standard deviation of these distances. We next identify all the data points with distances from the best fit that exceed the mean distance by at least 1 standard deviation. We discard these data points and then refit the remaining data. In Fig. 5, the two points plotted as hollow squares have been identified as outliers based on this criterion. When the data are refit without these points, the best-fit value for c is 151 pM, significantly closer to the known spike-in value of 256 pM. The elimination of outlying data points in this way is the final step of our analysis algorithm.
Analysis of the Accuracy of the Algorithm
Having used the above algorithm to obtain fitted values of the concentration for each of the spike-in transcripts in each of the Latin-square experiments, we can compare these results with both the known spike-in values of the concentrations and the signal values obtained by using Affymetrix software. For each spike-in concentration (i.e., 0.25–1,024 pM), we plot the median value of the calculated concentrations of all transcript measurements taken at that spike-in concentration (Fig. 6). The red and black squares show the values obtained by using the Affymetrix microarray suite 5 (with default settings for all parameters) and our model, respectively. Because the signal values obtained by using the Affymetrix software provide only relative concentrations rather than absolute ones, we normalized these values as follows before plotting them: First, we divided the median Affymetrix signal value obtained for each spike-in concentration by the known spike-in value itself. (If the analysis were perfect, all these quotients would be equal.) We then calculated the mean of these quotients and rescaled each of the median signal intensities by this mean value. For purposes of comparison, we performed the same normalization procedure on the fits obtained from our model. These normalized median values are shown on log–log plots in Fig. 6. Also shown are best-fit lines through each data set. Ideally, the calculated concentrations should scale linearly with the spike-in concentrations, making the slopes of these lines unity. The best-fit results for our and Affymetrix's analyses are 1.08 and 0.71, respectively.
Fig. 6.

Median value of predicted concentrations of all transcript measurements taken at a given spike-in target concentration vs. that concentration. Results for the analysis discussed in the text and for Affymetrix (microarray suite 5) are plotted as black and red squares, respectively. The solid lines are best fits of the plotted results to power laws (see Analysis of the Accuracy of the Algorithm).
Note that for the spike-in concentration 0.25 pM, the median value of the fits with our model was 0 (the lower limit allowed by our fitting procedure); thus, this point could not be included in the log–log plot above. If we also exclude our results for the spike-in value of 0.5 pM, the best-fit slope to our fitted concentrations becomes 1.01. Given the anomalously low values of our results at 0.25 and 0.5 pM, we believe that our predicted concentrations are not valid for spike-in concentrations <1 pM. (Note that the Affymetrix software predicts that a transcript is present in only 18%, 36%, and 33% of the 0.25-, 0.5-, and 1-pM spike-in experiments, respectively.) These unreliable predictions at low concentration are very likely connected with the fact that the background is comparable with the total signal at the lowest concentrations.
Fig. 7 provides a second measure of how accurately our algorithm predicts the known spike-in concentrations. Specifically, the black error bars show how large a range in concentration around the spike-in concentration is required to encompass half of our fitted values. For comparison, the red error bars show the corresponding result obtained from the Affymetrix software. The ranges are defined to be multiplicatively symmetric about the spike-in concentration; i.e., a range is defined by the smallest numerical factor by which a spike-in concentration must be multiplied at the high end and divided at the low end so as to include 50% of the fitted concentration values. Fig. 7 does not show results for spike-in concentrations of 0.25 and 0.50 pM, because for these concentrations our algorithm yields lower-range values near or equal to 0, and thus infinite error bars on a logarithmic scale. It is clear from Fig. 7 that the Affymetrix analysis yields narrower ranges at low concentrations, our algorithm yields narrower ranges at high concentrations, and the two algorithms yield comparable ranges at the middle concentrations. It would be straightforward, of course, to give more weight to the low-concentration data in determining our values of nI, ne, and bge. This weighting would improve our low-concentration predictions at the expense of those at higher concentrations.
Fig. 7.

Range of predicted concentrations vs. spike-in target concentration. The error bars at each spike-in concentration show the range of predicted concentration values that must be encompassed to include half of the predicted values (see Analysis of the Accuracy of the Algorithm). The black and red error bars show ranges obtained by using nalysis described in the text and with Affymetrix (microarray suite 5), respectively.
Fig. 8 shows histograms of the concentration values predicted by our algorithm (Upper), and the Affymetrix software (Lower) for all the data at spike-in concentrations of 16 and 256 pM. (Histograms of the 1- and 4-pM data are shown in Fig. 13, which is published as supporting information on the PNAS web site.) At 16 pM, where the distances between the 50% error bars of Fig. 7 are comparable for the two algorithms, the total range of values obtained from the Affymetrix algorithm is narrower than that obtained from ours. Even at 256 pM, where all the concentrations predicted by the Affymetrix algorithm are below the spike-in value, the range of predicted concentrations is significantly narrower than the range predicted by our procedure. One possible reason that the Affymetrix software yields narrower distributions is that it utilizes the difference PM–MM in predicting concentrations, whereas at present our algorithm uses only the PM-probe intensities.
Fig. 8.

Histograms of concentration values predicted by our algorithm (Upper) and with Affymetrix (microarray suite 5)(Lower) for all data at spike-in concentrations of 16 pM (Left) and 256 pM (Right).
As a final test of the efficacy of our methods, we performed a cross-validation study on the Latin-square data. The data set first was divided into three nonoverlapping subsets, each comprising the data from all experiments performed on 4 of the 12 genes. One of the three subsets was arbitrarily chosen as the ``test'' subset, and the data from the other two subsets then were used as the ``training'' subset to determine values of nI, bge, and ne as described above. These values were then used (again, just as described above) to fit the concentrations of the genes of the test subset. This procedure was then repeated twice more, with each of the two remaining subsets used as the test subset. The results for two of three of these tests are combined and displayed in Fig. 9, which shows error bars computed just as for Fig. 7. (The third set was discarded, because the test set included data with both higher and lower hybridization free energies than the training set.) The results shown in Fig. 9 are comparable in accuracy with those derived for the complete data set, demonstrating that as long as there is a fully representative collection of data in the training set, our algorithm is able to produce reasonably accurate predictions for the concentration for genes not contained in that set.
Fig. 9.

Range of predicted concentrations obtained through the cross-validation procedure (see Analysis of the Accuracy of the Algorithm) vs. spike-in target concentration. The error bars are defined by the same criteria as in Fig. 7.
Discussion
In this article we have described algorithms, based on simple physical principles of hybridization, for extracting gene-expression levels from microarray data. The results for the controlled Latin-square spike-in data were comparable in accuracy with those obtained from the statistical Affymetrix algorithm. Specifically, for multiple measurements taken at the same spike-in concentration, our algorithm tended to yield a more accurate median prediction, whereas the Affymetrix software tended to yield a narrower distribution of predicted values. At least in part, the accuracy of our model is limited by the quantity and range of control data available as a training set. More control data, including some at higher spike-in concentrations, would presumably reduce the fluctuations in the data (e.g., Fig. 3) and hence improve the accuracy of the model.
Although the dependence of hybridization strength on free energy clearly accounts for some of the observed variations in probe intensity, there are substantial systematic variations that, equally clearly, have different origins. This may be seen in Fig. 10, in which probe intensity is plotted as a function of ΔG for 11 measurements taken for gene 1091_at at a spike-in concentration of 256 pM. Although there is an overall upward trend with increasing ΔG, the data at 33.17, 34.91, 36.5, and 38.29 kcal/mol are anomalously low. Possible causes for such anomalies include secondary structure of the targets and/or probes and sequence-dependent steric effects associated with the confined geometry. Cross-hybridization would presumably produce anomalously high intensity values, as are observed for some of the other spike-in genes.
Fig. 10.

Observed hybridization intensity as a function of probe hybridization free energy for gene 1091_at at spike-in target concentration of 256 pM. Data points from different replicate measurements are shown in different colors.
Note from Fig. 10 that there are also variations among the 11 equivalent measurements of the same probe, although these are smaller than the probe-to-probe variations. The order of the intensities resulting from the 11 experiments changes little from probe to probe, suggesting that these variations too are largely systematic.
We noted earlier that, in Fig. 4a, ne shows a much weaker dependence on free energy than is predicted by equilibrium thermodynamics. At least in part, this discrepancy is a consequence of the ``all-or-none'' approximation (18) by which we have modeled the hybridization. This approximation assumes that the probe and target strands are either completely hybridized or completely separate: Partial hybridization is not allowed. The distribution of truncated probe lengths known to be present on lithographically prepared microarrays (12) could also contribute to the observed dependence of ne on free energy, as well as account for the fluctuations observed between replicate measurements. Some of the consequences of the all-or-none model and the distribution of probe lengths are discussed in detail in Supporting Text and Fig. 14, which are published as supporting information on the PNAS web site.
Supplementary Material
Abbreviations: PM, perfect match; MM, mismatch.
Note. After submission of this article, we became aware of concurrent research (19) that addresses similar questions to our work, albeit through different methods.
Footnotes
We have defined ΔG as the negative of the free energy of hybridization; thus, increasingΔG implies a stronger tendency to hybridize. Our ΔG is equal to –ΔG0 in the notation of ref. 11.
References
- 1.Brown, P. O. & Botstein, D. (1999) Nat. Genet. 21, 33–37. [DOI] [PubMed] [Google Scholar]
- 2.Lipshutz, R. J., Fodor, S. P. A., Gingeras, T. R. & Lockhart, D. J. (1999) Nat. Genet. 21, 20–24. [DOI] [PubMed] [Google Scholar]
- 3.Lockhart, D. J., Dong, H. L., Byrne, M. C., Follettie, M. T., Gallo, M. V., Chee, M. S., Mittmann, M., Wang, C. W., Kobayashi, M., Horton, H. & Brown, E. L. (1996) Nat. Biotechnol. 14, 1675–1680. [DOI] [PubMed] [Google Scholar]
- 4.Affymetrix (2001) Statistical Algorithms Reference Guide, Affymetrix Technical Note (Affymetrix, Santa Clara, CA).
- 5.Mount, D. W. (2001) Bioinformatics: Sequence and Genome Analysis (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 519–523.
- 6.Li, C. & Wong, W. H. (2001) Proc. Natl. Acad. Sci. USA 98, 31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Naef, F., Hacker, C. R., Patil, N. & Magnasco, M. (2002) Genome Biol. 3, research0018.1–0018.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tu, Y., Stolovitzky, G. & Klein, U. (2002) Proc. Natl. Acad. Sci. USA 99, 14031–14036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Affymetrix (2001) Array Design for the GeneChip Human Genome U133 Set, Affymetrix Technical Note (Affymetrix, Santa Clara, CA).
- 10.Affymetrix (2001) New Statistical Algorithms for Monitoring Gene Expression on GeneChip Probe Arrays, Affymetrix Technical Note (Affymetrix, Santa Clara, CA).
- 11.SantaLucia, J. (1998) Proc. Natl. Acad. Sci. USA 95, 1460–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forman, J. E., Walton, I. D., Stern, D., Rava, R. P. & Trulson, M. O. (1998) in Molecular Modeling of Nucleic Acids, eds. Leontis, N.B. & SantaLucia, J. (Am. Chem. Soc., Washington, DC), Vol. 682, pp. 206–228. [Google Scholar]
- 13.Peyret, N., Seneviratne, P. A., Allawi, H. T. & SantaLucia, J. (1999) Biochemistry 38, 3468–3477. [DOI] [PubMed] [Google Scholar]
- 14.Hill, A. A., Brown, E. L., Whitley, M. Z., Tucker-Kellog, G., Hunter, C. P. & Slonim, D. K. (2001) Genome Biol. 2, research0055.1–0055.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffmann, R., Seidl, T. & Dugas, M. (2002) Genome Biol. 3, research0033.1–0033.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dai, H. Y., Meyer, M., Stepaniants, S., Ziman, M. & Stoughton, R. (2002) Nucleic Acids Res. 30, e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kepler, T. B., Crosby, L. & Morgan, K. T. (2002) Genome Biol. 3, research0037.1–0037.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cantor, C. R. & Schimmel, P. R. (1980) Biophysical Chemistry Part III: The Behavior of Biological Macromolecules (Freeman, San Francisco).
- 19.Hekstra, D., Taussig, A. R., Magnasco, M. & Naef, F. (2003) Nucleic Acids Res. 31, 1962–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}