

Abstract
Neurexins are a large family of proteins that act as neuronal cell-surface receptors. The function and localization of the various neurexins, however, have not yet been clarified. Beta-neurexins are candidate receptors for neuroligin-1, a postsynaptic membrane protein that can trigger synapse formation at axon contacts. Here we report that neurexins are concentrated at synapses and that purified neuroligin is sufficient to cluster neurexin and to induce presynaptic differentiation. Oligomerization of neuroligin is required for its function, and we find that beta-neurexin clustering is sufficient to trigger the recruitment of synaptic vesicles through interactions that require the cytoplasmic domain of neurexin. We propose a two-step model in which postsynaptic neuroligin multimers initially cluster axonal neurexins. In response to this clustering, neurexins nucleate the assembly of a cytoplasmic scaffold to which the exocytotic apparatus is recruited.
Synapses are highly specialized cellular junctions that join individual neurons into a functional network. The appropriate differentiation of pre- and postsynaptic terminals is controlled by bi-directional signaling between the synaptic partners1-3. One family of postsynaptic proteins that might contribute to the induction of synapse formation between CNS neurons are the neuroligins4,5. Non-neuronal HEK293 cells that ectopically express neuroligin-1 or neuroligin-2 trigger the formation of functional presynaptic elements in contacting axons in vitro6. The extracellular domain of neuroligin presented at the cell surface of the non-neuronal cells is sufficient for this activity, suggesting that it may signal through an axonal receptor that triggers the induction of presynaptic differentiation.
The identity of this axonal receptor is unknown. Leading candidates are the beta-neurexins, which are neuronal plasma membrane proteins that interact with neuroligins in vitro4,7. Consistent with a role for beta-neurexins in neuroligin-induced synapse formation, recombinant beta-neurexin can block the synapse-inducing activity of neuroligin-expressing HEK293 cells6, though a similar blocking effect would be expected for any protein interacting with the extracellular domain of neuroligin.
A major concern regarding the role of neurexins as presynaptic neuroligin receptors is that there is currently no direct evidence for a synaptic function or localization of neurexins in the vertebrate CNS. Moreover, it is unclear how neuroligin-binding to beta-neurexin could trigger the recruitment of the presynaptic exocytotic machinery to the cell–cell contact sites. An attractive hypothesis is that the cytoplasmic tail of neurexins may recruit scaffolding molecules via a PDZ-binding motif8,9. A specific signaling function resulting from such interactions with neurexins, however, has not yet been observed in a cellular context.
Here we investigated the molecular mechanism of neuroligin-induced synapse formation. We found that endogenous neurexins localize to synapses and that overexpression of neuroligin-1 stimulates synapse formation and promotes recruitment of neurexins to newly forming synapses. We also show that neuroligin activity does not require any additional factors in the target cell, as purified recombinant neuroligin reconstituted into lipid bilayers induced presynaptic differentiation. In addition, the formation of neuroligin multimers is necessary for its activity, indicating a role for lateral clustering of neurexin by neuroligin multimers in the induction of presynaptic terminals. Strikingly, direct antibody-mediated clustering of beta-neurexin is sufficient to trigger recruitment of synaptic markers. This synaptogenic activity of beta-neurexin requires the cytoplasmic tail of the protein, which contains the PDZ interaction sites for scaffolding proteins. We propose that the clustering of beta-neurexin molecules by neuroligin multimers in a target cell and interaction of beta-neurexins with axonal scaffolding proteins are two essential (possibly sequential) steps by which cognate cell–cell interactions can trigger the assembly of presynaptic active zones.
RESULTS
To investigate the function of neurexins at the synapse, we first focused on the neurexin ligand neuroligin-1, which has a synapse-promoting activity. We dissected the structural requirements for this activity and then investigated how they relate to the interactions between neuroligin and neurexin.
Neuroligin mutants with reduced synaptogenic activity
The synapse-promoting activity of neuroligin can be measured in a mixed culture assay in which axons emerging from pontine explants form functional presynaptic terminals on HEK293 cells transfected with neuroligin-1 expression constructs6. We used this assay to identify structural elements in neuroligin that are essential for its activity. In an initial screen (see Supplementary Fig. 1 online), we generated chimeric proteins between the neuroligin ectodomain and its structural homologue acetylcholinesterase (AChE). Using this approach, we identified three sequence elements that are essential for neuroligin function: one located in the center of the domain (mutated in NLG/AChE-2) and two within the carboxy (C)-terminal portion of the domain (NLG/AChE-4 and NLG/AChE-6) (Fig. 1a and Supplementary Fig. 1).
Figure 1.
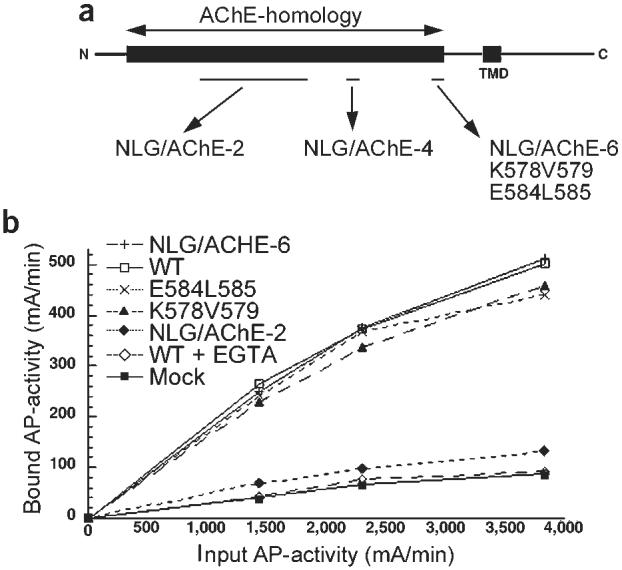
Synaptogenic activity of neuroligin. (a) Schematic representation of inactive neuroligin mutants. Within the sequences mutated in the chimeric mutant NLG/AChE-6, two inactive alanine substitution mutants were isolated (K578A/V579A and E584A/L585A). See Supplementary Fig. 1 online for details. (b) Binding of a neurexin–alkaline phosphatase fusion protein to HEK293 cells expressing neuroligin mutants. NLG/AChE-2 shows background levels of neurexin-binding, as do mock-transfected cells. All other constructs show calcium-dependent neurexin binding at levels similar to wild-type neuroligin-1.
Within these regions, we performed alanine-scanning mutagenesis and assayed the synaptogenic activity of the mutants. The mutagenesis yielded two mutants within the C-terminal region, K578A/V579A and E584A/L585A, that showed expression levels comparable to the wild-type protein, but that had strongly reduced synaptogenic activity, suggesting that these residues are essential for the function of neuroligin at the plasma membrane (Fig. 1a and Supplementary Fig. 1).
Neurexin binding is not sufficient for neuroligin-1 activity
Neuroligin-1 was originally purified as a neurexin-binding protein by affinity chromatography4,7, and neurexins are therefore candidate neuroligin receptors in synapse formation. We reasoned that the inactive neuroligin mutants might lose activity because of reduced affinity for a neurexin in pontine axons. To quantitatively assay neurexin–neuroligin interactions, we performed binding assays with a soluble neurexin-alkaline phosphatase fusion protein (neurexin-AP) on neuroligin-1 transfected cells (Fig. 1b). Mock-transfected HEK293 cells do not show significant neurexin-AP binding, whereas wild-type neuroligin-1 expressing cells bound neurexin-AP in a calcium-dependent manner. The chimera NLG/AChE-2, which lacks synaptogenic activity, showed a complete loss of neurexin binding, suggesting that the interaction with neurexin is necessary for the synapse-inducing function of neuroligin. However, soluble neurexin-AP bound equally well to HEK293 cells expressing wild-type neuroligin-1 and the inactive mutants NLG/AChE-6, K578A/V579A and E584A/L585A (Fig. 1b). This suggests that neuroligin activity might require neurexin binding but that its synaptogenic activity also depends on additional interactions or properties of the protein.
Lack of neuroligin-neurexin adhesion in neuroligin mutants
In trans-synaptic adhesion complexes, neurexin and neuroligin must interact as membrane proteins in opposing cell membranes. To directly assay adhesion between neuroligin-1 and neurexin presented on cell membranes, we performed cell aggregation assays with PC12 cells that had been separately transfected with epitope-tagged neurexin-1β and wild-type neuroligin-1. Neurexin-expressing cells showed robust calcium-dependent aggregation with cells expressing wild-type neuroligin that was comparable to aggregation of N-cadherin expressing cells (Supplementary Fig. 2 online). Within the cell aggregates, neurexin-1β and neuroligin-1 were strongly concentrated at sites of cell–cell contacts and strings of alternating neuroligin and neurexin expressing cells were observed frequently, further supporting the notion that the adhesive interactions are exclusively heterophilic (Fig. 2a).
Figure 2.

Inactive neuroligin mutants are impaired in neuroligin/neurexin mediated cell adhesion. (a) Aggregates of HA-tagged neuroligin-1 expressing cells (green) and VSV-tagged neurexin-1β expressing PC12 cells (red) show localization of neuroligin-1 and neurexin at cell contacts. The asterisks indicate three HA–neuroligin expressing cells forming contacts with adjacent VSV–neurexin expressing cells. Scale bar is 7 μm. (b) An aggregation index N0 (number of particles at time 0) divided by Nt (number of particles at time t) was determined after 0, 20, 40, 60 and 80 min of incubation. Mutants E584A/L585A and K578A/V579A both showed a significant loss of aggregation with neurexin-1β expressing cells. NRXsp4 indicates neurexin-1β with an insertion at site 4 that abolishes the binding to neuroligin. (c) Surface expression of wild-type neuroligin-1 and neuroligin mutants. Western blots of an aliquot of the total cell lysates and the surface biotinylated neuroligin proteins are shown. The biotin-modified NLG/swap reproducibly showed an aberrant migration on SDS-PAGE; the lower band is most likely due to proteolytic cleavage that occurred during the isolation of the biotinylated protein. In the graph, means and standard errors of the mutant neuroligin protein levels at the cell surface are compared to wild-type neuroligin surface levels (n =4 independent experiments). See Supplementary Fig. 2 online for additional data.
We next assayed adhesion between cells expressing neurexin-1β and cells expressing mutant neuroligins. Only background levels of aggregation were observed for the chimeric neuroligin mutants that were deficient in neurexin binding, such as the NLG/AChE swap mutant in which the extracellular AChE-homologous domain of neuroligin is replaced by the homologous sequences of mouse acetylcholinesterase. Strikingly, mutants K578A/V579A and E584A/L585A showed a dramatic loss of aggregation with neurexin-1β expressing cells (Fig. 2b) despite their wild-type level of interaction with soluble neurexin-1β (Fig. 1b). The observed differences in aggregation were not due to lower expression levels of the mutant proteins since western-blotting analysis of surface-biotinylated proteins indicated comparable protein levels at the plasma membrane of the transfected cells (Fig. 2c). Therefore, the amino acids K578/V579 and E584/L585 are essential for strong adhesion between neuroligin- and neurexin-expressing cells, but not for binding to individual neurexin molecules in soluble form.
Oligomerization of neuroligin is required for activity
To understand the functional importance of the residues K578/V579 and E584/L585 in neuroligin-1, we generated a structural model of neuroligin-1 based on the mouse AChE crystal structure (Fig. 3a)10. In this model, amino acids K578/V579 and E584/L585 are located in an alpha helix at the base of the AChE-homologous domain of neuroligin-1 (Fig. 3b). AChE forms tetramers by parallel association of two primary dimers with each other. Importantly, the helix in AChE corresponding to the mutated helix in neuroligin represents exactly the primary dimerization interface of the molecule, and mutations in this site have been shown to perturb AChE oligomerization10,11. According to the model for neuroligin-1, these amino acids are perfectly positioned to mediate lateral interactions with other neuroligin molecules through a second helix in the protein (Fig. 3b). Strikingly, in our original screen for essential regions in neuroligin-1, we had also isolated mutations in this second helix that led to a loss of synaptogenic activity (mutant NLG/AChE-4; Fig. 1a). These results suggest that interactions between these two helices are critical for neuroligin-1 function and that the inactivating mutations K578A/V579A and E584A/L585A perturb lateral (cis) interactions between individual neuroligin-1 molecules.
Figure 3.

Oligomerization of the neuroligin-1 AChE-homologous domain. (a) Mouse AChE crystal structure10 (blue) and modeled neuroligin-1 structure (green). Regions mutated in NLG/AChE-4 and NLG/AChE-6 are part of a four-helix bundle at the base of the AChE-homologous domain of neuroligin-1 (located at the left of the drawing). (b) In the crystal structure, AChE forms tetramers assembled from two dimers10. Two views of analogous, predicted neuroligin-1 dimers. Regions mutated in NLG/AChE-4 (highlighted in light blue) and NLG/AChE-6 are part of a four-helix bundle; residues E584/L585 and K578/V579 are marked in purple. (c) Analysis of neuroligin oligomers. Left panel, purified recombinant extracellular domain of neuroligin-1 could be cross-linked into dimers and tetramers. Higher-order complexes of wild-type neuroligin with different electrophoretic mobility were detected by native gel electrophoresis and western blotting (middle panel, arrowheads). Right panel, oligomerization of neuroligin was also detected by co-immunoprecipitation of differentially tagged neuroligin forms. Wild-type neuroligin was precipitated with myc antibodies and any co-precipitating HA-tagged wild-type or mutant neuroligin protein was detected by immunoblotting with anti-HA antibodies. Expression levels of all transfected constructs were monitored by western blotting of total cell lysates and were similar (data not shown and Supplementary Fig. 1).
Chemical cross-linking of purified recombinant neuroligin ectodomain confirmed the formation of dimers and tetramers by the neuroligin extracellular domain (Fig. 3c). To compare oligomerization of full-length, wild-type neuroligin complexes to the mutants, we used native gel electrophoresis which has previously been used to separate different oligomeric complexes of AChE11,12. For wild-type neuroligin-1, we detected two higher molecular weight species which were greatly reduced in the inactive mutants K578A/V579A and E584A/L585A (Fig. 3c). Neuroligin oligomers were also detected by co-immunoprecipitation. Myc-tagged, wild-type neuroligin co-precipitated with hemagglutinin (HA)-tagged wild-type neuroligin, but co-precipitation of HA-tagged mutant neuroligins was significantly reduced (Fig. 3c). Compared to the wild-type protein, only 27 ± 13% and 15 ± 7% (n = 3) was recovered in the immunoprecipitates for the mutants K578A/V579A and E584A/L585A, respectively, further confirming the decreased stability of neuroligin complexes in the mutants. These findings suggest that the formation of stable neuroligin-1 multimers is essential for the synaptogenic activity of the protein as well as for adhesion with neurexin-expressing cells.
Endogenous neurexins are concentrated at synapses
Our structure–function analysis is consistent with a role for neurexins as presynaptic neuroligin receptors. To date, however, there is no direct evidence for a synaptic localization or function of neurexins. To directly analyze the subcellular localization of neurexins, we generated antibodies against the cytoplasmic tail of neurexin-1. Due to the sequence similarities between neurexin family members, this antiserum is likely to recognize the alpha and beta isoforms of neurexin-1, neurexin-2 and neurexin-3, but not proteins in the neurexinIV/CASPR/paranoidin family. Consistent with this notion, affinity-purified antisera recognized two proteins with apparent molecular weights corresponding to alpha- and beta-neurexins in western blots (Fig. 4a). In immunostained dissociated cultures of cerebellar granule cells and hippocampal neurons, the antiserum revealed a punctate distribution of the protein that showed extensive overlap with the synaptic vesicle marker synaptobrevin, demonstrating that endogenous neurexins are indeed concentrated at synapses (Fig. 4e–g and data not shown). In isolated axons emerging from pontine explants neurexins were strongly enriched in growth cones, appropriately localized to mediate initial interactions with neuroligins in the postsynaptic target cell (Fig. 4b–d). In vivo, pontine mossy fiber axons form synapses with cerebellar granule cells in the inner granular layer (IGL) of the cerebellar cortex. Neurexin was detected in the same regions of the IGL as the synaptic vesicle marker synaptobrevin (Fig. 4h–j), consistent with an enrichment at mossy fiber–granule cell synapses. In addition, we also observed an intracellular pool of neurexins in the cell bodies of Purkinje and granule cells (data not shown). The identity of this intracellular compartment and a direct confirmation of neurexin localization in mossy fiber terminals will require ultrastructural studies, which we are currently performing.
Figure 4.

Neurexins are concentrated in growth cones and at synaptic junctions. (a) Western blot of total cerebellar lysate probed with anti-neurexin antibodies. (b–d) Pontine explants were grown in vitro for 3 d and immunostained with affinity-purified anti-neurexin antibodies (b, green in the overlay) and Alexa-conjugated phalloidin (c, red in the overlay) to reveal filamentous actin. (e–g) Dissociated cultures of hippocampal neurons (24 d in vitro) were immunostained for neurexin (e, green in overlay) and synaptobrevin (f, red in overlay). (h–j) Cryostat sections (18 μm) of mouse cerebellum (P21) were immunostained for neurexin (h, green in the overlay) and synaptobrevin (i, red in the overlay). Scale bars are 10 μm (a,e) and 20 μm (h).
Recruitment of neurexin to neuroligin-induced synapses
If the synaptogenic activity of neuroligin-1 were mediated through neurexin in the presynaptic membrane, we would predict a recruitment of endogenous neurexin to the synaptic terminals induced by neuroligin-1. To observe neurexin recruitment to newly forming synapses, we transfected dissociated cerebellar granule cells with a neuroligin-1 expression vector before synapses were formed in culture (at the day of plating). Neuroligin overexpression increased the number of synaptic vesicle clusters formed fivefold, and there was a strong accumulation of endogenous neurexin in neuronal processes at sites of contact with the neuroligin-expressing cells (Fig. 5). Confocal imaging revealed that neuroligin-1 was concentrated at these immature synaptic contacts and that synaptic vesicles and neurexin colocalized at points of contact with neuroligin-expressing cells (Fig. 5e–h). This suggests that neurexins are recruited to newly forming neuroligin-induced synaptic sites.
Figure 5.

Overexpression of neuroligin-1 induces recruitment of neurexin to newly forming synapses. (a–d) Cerebellar granule cells were transfected with HA-tagged neuroligin-1 at the time of plating and were fixed after 3 d in culture. The localization of HA–neuroligin-1 (a, green in overlay), synaptobrevin (b, blue in overlay) and neurexin (c, red in overlay) was analyzed by immunostaining. The strong peri-nuclear neurexin immunoreactivity was consistently observed for young cultures and decreased after maintaining cells for 5–10 d in vitro (data not shown). (e–h) High-magnification view of neuroligin-induced cell–cell contacts showing HA–neuroligin-1 (e, green in overlay), synaptobrevin (f, blue in overlay) and neurexin (g, red in overlay). (i–k) Hippocampal neurons were transfected with HA–neuroligin-1 after 12 d in vitro, maintained for 2 d and immunostained with antibodies against the HA tag to detect neuroligin (i, green in overlay) and antibodies against synapsin (j, red in overlay). (l) Quantitation of synapsin-, PSD95- and GluR2/3-positive puncta in neuroligin-1 overexpressing cells. Hippocampal neurons were transfected at 12 d.i.v. and were analyzed 2 d later. Synaptic puncta were quantitated as described in the Methods. Means and standard errors are given (n ≥ 10 cells each). Scale bars are 15 μm (a,i) and 5 μm (e). See Supplementary Fig. 3 online for additional data.
In hippocampal cultures that had been maintained in vitro for 12 d, endogenous neuroligin-1 was strongly concentrated at synapses (Supplementary Fig. 3). Overexpression of neuroligin-1 in these more mature cultures similarly stimulated synapse formation as in the immature cerebellar granule cells. The number of detectable synaptic vesicle clusters was increased fivefold, and the number of PSD95- and GluR2/3-positive puncta was increased 3–4 fold (Fig. 5i–m and Supplementary Fig. 3). These findings suggest that neuroligin-1 stimulates pre- and postsynaptic differentiation and can recruit neurexins to newly forming synapses.
To test whether neuroligin–neurexin interactions are sufficient to induce synaptic differentiation in the absence of other postsynaptic factors, we purified the AChE-homologous ectodomain of neuroligin-1 attached to a glycosylphosphatidylinositol (GPI) anchor. The recombinant protein was reconstituted into liposomes that were coated onto 5-μm silica beads. This approach allows presentation of the functional domain of neuroligin-1 on a surface with dimensions approximating a neuronal dendrite, in a freely diffusible form (Supplementary Fig. 4 online; for further analysis and detailed methods, see Baksh, M., C.D., E.I. and Groves, J.T., unpublished data). When these neuroligin-coated beads were applied to hippocampal neurons in culture, we observed clustering of neurexin and synaptic vesicle proteins at the bead contact sites (Fig. 6a–d). Synaptic vesicles that accumulated at these sites showed depolarization-induced turnover, as indicated by uptake of an antibody directed against a luminal epitope of synaptotagmin (Fig. 6e–g). These findings show that the AChE-homologous domain of neuroligin-1, as a purified protein, is sufficient to induce the formation of functional synaptic vesicle release sites in axons.
Figure 6.

Induction of neurexin clustering and presynaptic differentiation by purified neuroligin. (a–d) Beads coated with lipid bilayers containing purified GPI–neuroligin were added to 12-d.i.v. hippocampal cultures. After 24 h, cultures were fixed and stained with antibodies for neurexin (a, green in the overlay) and synaptobrevin (b, red in the overlay). (d) A differential interference contrast (DIC) image of the lipid-coated beads. Note that these beads coated with a lipid bilayer have a more irregular, rough surface, whereas uncoated beads (without lipid bilayer) appear as discreet particles with a smooth outline (see Supplementary Fig. 4 online for more details). (e–g) Uptake of synaptotagmin antibodies at axonal specializations induced by neuroligin-coated beads. Cultures were incubated with beads as above and depolarized in the presence of antibodies directed against the luminal domain of synaptotagmin. (e) Staining for bound synaptotagmin antibodies (red in the overlay). (f) A DIC image of the beads. (g) Overlay of synaptotagmin staining and DIC image. (h) Quantitation of synapsin clustering on silica beads. The average intensity of synapsin staining on the bead and in a neighboring area of equal size was measured and the enrichment of staining on beads over the neighboring area was calculated. The graph shows the mean ± s.d. (n = 10 beads each). Beads coated with GPI-anchored neuroligin-1 reconstituted into lipid bilayers (NLG) show a five-fold enrichment of synapsin staining. No enrichment is observed for beads coated with GPI-anchored placental alkaline phosphatase reconstituted into lipid bilayers (PLAP), beads coated only with lipid bilayers (lipid) or uncoated beads (bead). Scale bars are 5 μm (a) and 10 μm (e). See Supplementary Fig. 4 online for more details.
This activity of neuroligin was highly specific and was critically dependent on membrane anchoring of the protein. Uncoated beads, beads coated only with lipid bilayers and beads coated with bilayers containing GPI-anchored placental alkaline phosphatase did not induce any accumulation of synaptic vesicle markers (Fig. 6h and Supplementary Fig. 4). Soluble recombinant neuroligin that was not anchored in a bilayer did not show activity when applied at a concentration of up to 100 μg/ml (data not shown).
Synaptic vesicle recruitment by clustering of neurexins
Our findings suggest that complexes of neuroligin-1 in the postsynaptic membrane induce presynaptic differentiation by recruitment of neurexin to cell–cell contact sites. To test whether neurexin clustering triggers the recruitment of synaptic vesicles, we transfected neurexin-1β with an extracellular vesicular stomatitis virus (VSV) epitope tag into cultured hippocampal neurons. Overexpressed VSV–neurexin-1β was distributed all along the axonal plasma membrane of the transfected cells (data not shown). Rather than using neuroligin-coated beads, we directly clustered neurexin with anti-VSV antibodies that had been multimerized with a secondary antibody. Oligomerized antibodies induced patches of neurexin and an accumulation of synaptic vesicle proteins (Fig. 7a–c). Recruitment of synaptic vesicle markers occurred downstream of neurexin-1β in the transfected cell, and postsynaptic markers such as GluR2/3 were absent from such clustering-induced sites (Supplementary Fig. 5 online). Addition of anti-VSV antibodies that had not been multimerized by secondary antibodies did not show vesicle accumulation (Supplementary Fig. 5), suggesting that clustering of at least four neurexin molecules is required to induce recruitment of synaptic vesicles.
Figure 7.

Clustering of neurexin is sufficient to induce recruitment of synaptic vesicles. (a–i) Pre-clustered anti-VSV multimers were added to 12-day-old hippocampal cultures transfected with VSV–neurexin (a–c), VSV–neurexin-delta-C (d–f) and VSV–L1 (g–i). Antibody clustered proteins (a, d and g, green in the overlay) and the distribution of synapsin (b, e and h, red in overlay) are shown. The integrity of all cells and the cellular processes was controlled by DIC microscopy to ensure that the cells remained intact during the procedure. Scale bar, 10μm. (j) Quantitation of synapsin accumulation in response to clustering of membrane proteins with antibodies. The percentage of recognizable antibody-induced clusters that showed accumulation of synapsin was determined (n ≥ 140 puncta from ≥ 4 cells for each construct). The mean ± s.d. for cells expressing VSV–neurexin (NRX), VSV–neurexin lacking the cytoplasmic tail (ΔC), VSV–N-cadherin (N-cad) and VSV–L1CAM (L1) are shown. See Supplementary Fig. 5 online for additional data.
This activity was specific to neurexins and was not observed when other cell adhesion molecules were clustered by antibodies. We tested L1 and N-cadherin, both fused to the same VSV epitope-tag. Oligomerized VSV antibodies produced patches of L1 and N-cadherin, but did not induce an accumulation of presynaptic markers, indicating that the recruitment of vesicles depends on specific interactions downstream of neurexin (Fig. 7g–i and data not shown).
When a neurexin mutant lacking the cytoplasmic tail (which contains its PDZ-interaction domain) was clustered with oligomerized anti-VSV antibodies, the mutant protein formed patches at the cell surface similar to wild-type neurexin, but these patches did not accumulate synaptic vesicle proteins (Fig. 7d–f). Whereas synapsin accumulation was observed in 92 ± 3% of all clusters of wild-type VSV–neurexin, synapsin was only detected in 9 ± 4% of the clusters of the tail-deleted VSV–neurexin mutant comparable to what was seen with N-cadherin (10 ± 2%) and L1 (10 ± 3%) (n ≥ 140 puncta from ≥ 4 cells for each construct, Fig. 7j). This result supports the idea that the synaptogenic activity of neurexin critically depends on protein interactions with the cytoplasmic domain of neurexin, suggesting that extracellular clustering of neurexin leads to intracellular recruitment of scaffolding or signaling proteins in the axonal cytoplasm.
DISCUSSION
In our analysis of the molecular mechanism of neuroligin-induced synapse formation, we observed that overexpression of neuroligin stimulates pre- and postsynaptic differentiation in cultured hippocampal neurons, suggesting that neuroligin is a limiting component of the postsynaptic machinery involved in synapse formation. Neuroligin activity depends on its interaction with neurexins. We found that (i) endogenous neurexins are concentrated in synaptic terminals, (ii) postsynaptic multimers of neuroligin-1 are sufficient to trigger the recruitment of neurexin to newly forming synaptic sites and (iii) clustering of neurexin induces recruitment of synaptic vesicles.
The neurexin family of proteins was first identified as high-affinity receptors for the venom alpha-latrotoxin13. Although an involvement of neurexins in the latrotoxin response of neuronal cells is now well documented14,15 the subcellular localization and normal function of the neurexins are still unknown16-18. Using a newly generated neurexin antibody that recognizes most neurexin isoforms, we show here that neurexins are concentrated at synapses. Neurexin immunoreactivity was not completely restricted to synapses, so we cannot determine whether the additional non-synaptic neurexin pool consisted of specific non-synaptic isoforms, or whether most neurexins are localized to both synapses and non-synaptic regions of the plasma membrane. In either case, our data show that at least a subset of neurexin family members is concentrated at CNS synapses.
Neuroligin was originally isolated as a splice-specific ligand of beta-neurexins by affinity chromatography4. Using a functional in vitro assay, we previously demonstrated that neuroligins have a synaptogenic activity, but it remained unclear whether this activity of neuroligin was mediated through a neurexin6. We now provide strong evidence that neurexin functions as a neuroligin receptor in synapse formation: overexpression of neuroligin in neurons induced recruitment of neurexins to newly forming terminals and promoted the formation of synaptic specializations. This activity of neuroligin-expressing cells can be mimicked by purified neuroligin presented to axons in a fluid lipid bilayer on a bead. Mutations in neuroligin that inhibit neurexin binding or adhesion with neurexin-expressing cells reduce the synapse-promoting activity of the protein.
Moreover, our results indicate that lateral clustering has a critical role in neuroligin–neurexin signaling. Clustering of beta-neurexin in axons with multimerized antibodies is sufficient to trigger the accumulation of synaptic vesicles, whereas crosslinking of neurexin with monomeric antibodies is ineffective. This suggests a highly cooperative process in which a minimum number of beta-neurexins (at least four) must be clustered to result in productive signaling (Supplementary Fig. 6 online). These findings support a striking mechanistic analogy between the induction of synaptic differentiation in CNS neurons and synapse formation in the immune system where receptor clustering represents a crucial event in T-cell activation19.
How can the clustering of beta-neurexin trigger the assembly of presynaptic terminals? We found that for beta-neurexin to be active it must have an intact cytoplasmic C-terminal tail, which contains a PDZ-binding motif. This domain is known to interact with the cytoplasmic scaffolding molecules CIPP, CASK/Lin-2 and Mint/Lin-10/X11α8,9,20 and may interact with other proteins that have not yet been identified. The clustering of beta-neurexin monomers in the axon by neuroligin tetramers may either recruit specific downstream signaling components to the beta-neurexin at the forming contacts or activate signaling through components that are ‘pre-bound’ to beta-neurexin. In either case, the small neurexin clusters would provide the minimal nucleation site for assembling a presynaptic protein scaffold and, subsequently, the secretory apparatus (see model in Supplementary Fig. 6). Most of the cytoplasmic neurexin binding proteins are multivalent and can generate a scaffold with additional neurexin binding sites8,21. Thus, the initial complexes could be enlarged by recruitment of more beta-neurexins, and, subsequently, additional neuroligins, forming an expanding cell–cell contact. Such a model might also account for the fact that clustering of ephrinB by EphB receptors induces the recruitment of adapters and signaling molecules resulting in a retrograde signal transmitted into the ephrinB-expressing cell22-25.
Our model predicts that a beta-neurexin is localized to the presynaptic terminal. With the immunological reagents currently available to us, we cannot provide direct evidence for a presynaptic localization of beta-neurexin. Here, however, we report several lines of evidence that support such a localization: (i) a pan-neurexin antibody that recognizes alpha- and beta-neurexins shows that neurexin isoforms are concentrated at synapses and in axonal growth cones, (ii) clustering of endogenous neurexins with neuroligin-coated beads induces presynaptic specializations in the absence of postsynaptic elements and (iii) clustering of epitope-tagged beta-neurexin in axons with antibodies triggers the recruitment of synaptic vesicles. It is possible that individual neurexin isoforms show differential distribution over pre- and postsynaptic plasma membrane domains. To address this question, we are currently generating beta-neurexin-specific antibodies, and ultrastructural studies with our pan-neurexin antibodies are underway.
Whereas neuroligin–neurexin signaling is sufficient to promote synaptic differentiation, during development both molecules function in concert with several additional trans-synaptic signaling factors such as WNTs26, cadherins27,28, Ig-domain proteins29-31 and Eph receptors32. One factor whose function at least partially overlaps with that of neuroligin is the Ig-domain protein SynCAM30. Whereas neuroligin–neurexin interactions are heterophilic and therefore provide the synapse-induction process with directionality, SynCAM apparently functions through homophilic interactions. It is noteworthy that neurexin and SynCAM contain binding sites for the same cytoplasmic scaffolding proteins, indicating that they may converge on common downstream effectors. A common cytoplasmic scaffold may thus integrate adhesive and signaling cues from several extracellular pathways and allow for a stepwise assembly of trans-synaptic complexes. Future work should elucidate whether cross-talk between different adhesion and signaling system exists and, if so, how it contributes to the regulation of synapse formation and synaptic specificity in the CNS.
METHODS
Neurexin antibodies. For the production of neurexin antibodies, female chickens were immunized with a recombinant neurexin–GST (glutathione S-transferase) fusion protein containing the cytoplasmic tail of neurexin-1. IgY was isolated from egg yolk with the Pierce Eggcellent kit, and antibodies were affinity purified by chromatography on columns carrying the neurexin cytoplasmic tail sequence fused to maltose binding protein. All experiments involving animals were approved by the Institutional Animal Care and Use Committee of the Health Science Division at Columbia University.
Neuronal cultures and transfection. Cerebellar granule cells were isolated from mice at postnatal day 5 or 6 (P5/P6)33. For transfection, cells were resuspended at a density of 500,000 per ml in 150 μl of neurobasal medium containing 2 mM Glutamax, 2% B27 supplement (Invitrogen) and 5 ng/ml BDNF (Preprotech). DNA/lipid complexes were formed by incubation of 0.03 μl Lipofectamine 2000 (Invitrogen) with 0.2 μg of DNA in 100 μl OptiMEM (Invitrogen) and added to the cell suspension. After 45 min incubation at 37 °C and 5% CO2, the cell suspension was plated into one well of an 8-well Lab-Tek Permanox chamber slide coated with polyornithin (Sigma) and Laminin (Invitrogen). After 2–4 h, 200 μl of culture medium containing penicillin and streptomycin (Invitrogen) were added. Cultures were generally maintained in vitro for 4–10 d before analysis.
Hippocampi were removed from embryonic day 18 (E18) rats and treated with trypsin for 20 min at 37 °C, followed by washing and trituration. Dissociated cells were plated at 100,000 cells/cm2 on poly-lysine-coated glass coverslips (Carolina Biologicals) and cultured in neurobasal medium supplemented with 2 mM Glutamax, and 2% B-27. Cells were transfected with Lipofectamine 2000 (Invitrogen) 12–21 d after plating and analyzed 2–4 d after transfection.
Clustering of neurexin by beads or antibodies. Recombinant GPI-anchored neuroligin was expressed in HEK293 cells and isolated from cell lysates by chromatography on NTA-agarose (Qiagen). The protein bound to the column in phosphate-buffered saline containing 350 mM NaCl, 0.5% Triton X-100 and 6 mM imidazole. Unspecifically bound proteins were removed by extensive washing with PBS containing 1 M NaCl and 0.1% Triton X-100. The detergent was then switched to 25 mM octyl-glucopyranoside (Calbiochem), and bound protein was eluted with an imidazole gradient (10–200 mM). Peak fractions containing purified GPI-neuroligin were used for reconstitution into preformed liposomes composed of egg-Phosphatidylcholine (Avanti Polar lipids) by dialysis of octyl-glucopyranoside against PBS. The reconstituted material revealed a single protein band at the predicted size for GPI-neuroligin by silver staining, which showed immunoreactivity with specific antibodies directed against the hexa-histidine tag (data not shown). Purified GPI-anchored placental alkaline phosphatase was purchased from Sigma.
Coating of cleaned 5-μm silica beads with GPI-neuroligin containing liposomes was done using a method developed by J. T. Groves and colleagues (University of California, Berkeley), and beads were added to dissociated cultures of hippocampal neurons. In control experiments, the lipid bilayers were visualized with fluorescently labeled lipids (1,2-Dipalmitoyl-sn-glycerol-3-phosphoetanolamine-N-(Lissamine Rhodamine B Sulfonyl); Avanti Polar Lipids), and the reconstituted protein was visualized in the bilayer by immunostaining. Cells were fixed and analyzed 24–72 h after addition of beads.
Vesicle turnover on beads was assayed 16 h after addition of beads to hippocampal cultures by incubating cultures for 5 min with a pre-warmed, isotonic depolarization solution containing 10 μg/ml of affinity-purified rabbit anti-synaptotagmin lumenal domain antibodies. After incubation, cultures were briefly washed three times with pre-warmed medium, fixed and processed for immunohistochemistry.
Anti-VSV antibody multimers were generated as previously described for EphB ligands34 by pre-incubation of 50 ng/ml anti-mouse Cy-3 antibody (Jackson Laboratories) with 500 ng/ml mouse anti-VSV antibody (Roche) in neuronal culture medium (neurobasal medium containing 2 mM Glutamax and 2% B-27 supplement). Antibodies were added to dissociated hippocampal cultures and analyzed 18–24 h after addition of antibodies.
Image acquisition and quantitation. Images were acquired with a Leica TCS NT or Zeiss LSM510 confocal microscope. Laserpower and photomultipliers were set such that no detectable bleed-through occurred between the different channels. Eight to ten sections were taken from top to bottom of the specimen and brightest point projections were made. Images were analyzed with OpenLab (Improvision) and Metamorph (Universal Imaging) software (see Supplementary Methods online for details) and processed using Adobe Photoshop software.
Neurexin binding assays and cell aggregation assays. The soluble neurexin-1–alkaline phosphatase fusion protein was expressed in HEK293 cells and collected in the culture medium. For binding assays, HEK293 cells in 35 mm dishes were transfected with various neuroligin expression constructs in triplicates. We removed the culture medium 36 h after transfection and added the fusion protein in Hank’s buffered saline containing 2 mM CaCl2,1 mM MgCl2 and 5% fetal bovine serum. After incubation for 90 min at 4 °C, the unbound protein was removed quantitatively, and the bound protein was collected in a solution containing 1% Triton X-100 and 10 mM Tris-HCl, pH 8.0. Alkaline phosphatase activity was measured kinetically with 3 mg/ml para-nitro-phenyl-phosphate (Sigma) as substrate in a solution containing 100 mM Tris-HCl (pH 9.5), 50 mM MgCl2 and 100 mM NaCl.
For aggregation assays, 80% confluent PC12 cells were transfected separately with epitope-tagged neurexin-1 and neuroligin-1 or neuroligin mutants using Lipofectamine 2000 (Invitrogen). The cells were resuspended 36–48 h after transfection, carefully triturated, and mixed at a final density of 5 × 106 cells per ml. Cells were incubated in 1.5-ml microcentrifuge tubes at 37 °C with rotation in a total volume of 0.3 ml HBSS containing 10 mM CaCl2 and 20 mM MgCl2. At 0, 20, 40, 60 and 80 min, an aliquot was removed and aggregation determined by calculating N0 (number of particles at time 0) divided by Nt (number of particles at time t).
Analysis of neuroligin complexes. Recombinant soluble neuroligin was expressed in HEK293 as secreted protein with a C-terminal hexa-histidine tag. The protein was purified on NTA-agarose and eluted with a 10–200 mM imidazole gradient.
For chemical cross-linking, 1 μg of purified recombinant neuroligin-1 was incubated in phosphate-buffered saline for 30 min at ambient temperature with 20 μM BS3 (Pierce). The cross-linker was quenched by addition of Tris-HCl (pH 8.0) to a final concentration of 200 mM and cross-linking products were analyzed by western blot.
For native gel electrophoresis, HEK293 cells on 35-mm culture dishes were transfected with wild-type or mutant neuroligin expression vectors. Thirty-six hours after transfection, cells were washed with ice-cold phosphate-buffered saline and 150 μl of ice-cold lysis buffer was added: 20 mM Tris-HCl (pH 6.8), 50 mM NaCl, 5 mM MgCl2,2 mM CaCl2,2 mM DTT and 1% (w/v) Triton X-100 containing protease inhibitors “complete EDTA-free” (Roche). After 5 min incubation on ice, cell lysates were homogenized by repeated pipetting and centrifuged for 5 min at 14,000 r.p.m. The supernatants, which contained more than 95% of total neuroligin protein, were analyzed by electrophoresis (8 h, 60 V at 4 °C) in 8% poly-acrylamide gels; electrophoresis buffer contained 375 mM Tris-HCl (pH 8.8), 0.5% Triton X-100 with 250 mM Tris-base and 190 mM glycine. Neuroligin complexes were detected by western blot.
For analysis of oligomers by co-immunoprecipitation, myc-tagged wild-type neuroligin-1 was co-transfected into HEK293 cells with HA-tagged wild-type or mutant neuroligin-1. Cells were lysed in 50 mM Tris-HCl (pH 7.4), 100 mM NaCl, 1 mM CaCl2,1 mM MgCl2, 0.5% Triton X-100 and 1 mM DTT containing “complete EDTA-free” protease inhibitor cocktail (Roche) at 4 °C. Insoluble material was pelleted by 5 min centrifugation at 13,200g, and supernatants were incubated with 9E10 anti-myc antibodies for 1 h at 4 °C. Lysates were then re-centrifuged for 5 min and immune complexes in the supernatants were recovered on protein G sepharose (Pharmacia). Immunoprecipitates were washed twice with lysis buffer containing 0.1% Triton X-100 and analyzed by western blotting with anti-HA antibodies (Roche). Expression levels of both transfected proteins in the cell lysates were verified by western blotting with anti-HA and anti-myc antibodies.
Structural modeling. Sequence alignments between neuroligin-1 and mouse AChE were carried out using clustalX35. Homology modeling between aligned sequences was performed with MODELLER36. Figures showing the mouse AChE and modeled neuroligin-1 structures were generated with RIBBONS37.
A more detailed account of the methods and the DNA constructs and antibodies used in this study can be found in Supplementary Methods online.
Supplementary Material
ACKNOWLEDGMENTS
We thank T. Serafini and M. Tessier-Lavigne for their generous support during the early phase of this project, N. Brose and F. Varoqueaux for anti-neuroligin-1 antibodies, and A. Broder for technical assistance. Beads coated with lipid bilayers were generated in collaboration with M. Baksh, S. Pautot, and J. Groves. We also thank J. Dodd, C. Mason, M-M. Poo and members of the Poo lab for support and sharing equipment, and O. Hobert, T. Jessell and M-M. Poo for comments on the manuscript. This work was supported by funds to P.S. from the Searle Scholar Program, the Alfred P. Sloan Foundation, the Esther A. & Joseph Klingenstein Fund and the Christopher Reeve Paralysis Foundation, and a grant from the US National Institutes of Health (NIH) to E.I. (R01MH60771). C.D. was supported by a NIH training grant.
Footnotes
Note: Supplementary information is available on the Nature Neuroscience website.
COMPETING INTERESTS STATEMENT The authors declare that they have no competing financial interests.
References
- 1.Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu. Rev. Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- 2.Garner CC, Zhai RG, Gundelfinger ED, Ziv NE. Molecular mechanisms of CNS synaptogenesis. Trends Neurosci. 2002;25:243–251. doi: 10.1016/s0166-2236(02)02152-5. [DOI] [PubMed] [Google Scholar]
- 3.Scheiffele P. Cell-cell signaling during synapse formation in the CNS. Annu. Rev. Neurosci. 2003;26:485–508. doi: 10.1146/annurev.neuro.26.043002.094940. [DOI] [PubMed] [Google Scholar]
- 4.Ichtchenko K, et al. Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell. 1995;81:435–443. doi: 10.1016/0092-8674(95)90396-8. [DOI] [PubMed] [Google Scholar]
- 5.Song JY, Ichtchenko K, Sudhof TC, Brose N. Neuroligin 1 is a postsynaptic cell-adhesion molecule of excitatory synapses. Proc. Natl. Acad. Sci. USA. 1999;96:1100–1105. doi: 10.1073/pnas.96.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scheiffele P, et al. Neuroligin expressed in nonneuronal cells triggers presynaptic development in contacting axons. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- 7.Ichtchenko K, Nguyen T, Sudhof TC. Structures, alternative splicing, and neurexin binding of multiple neuroligins. J. Biol. Chem. 1996;271:2676–2682. doi: 10.1074/jbc.271.5.2676. [DOI] [PubMed] [Google Scholar]
- 8.Biederer T, Sudhof TC. Mints as adaptors. Direct binding to neurexins and recruitment of munc18. J. Biol. Chem. 2000;275:39803–39806. doi: 10.1074/jbc.C000656200. [DOI] [PubMed] [Google Scholar]
- 9.Kurschner C, Mermelstein PG, Holden WT, Surmeier DJ. CIPP, a novel multivalent PDZ domain protein, selectively interacts with Kir4.0 family members, NMDA receptor subunits, neurexins, and neuroligins. Mol. Cell. Neurosci. 1998;11:161–172. doi: 10.1006/mcne.1998.0679. [DOI] [PubMed] [Google Scholar]
- 10.Bourne Y, Taylor P, Bougis PE, Marchot P. Crystal structure of mouse acetylcholinesterase. A peripheral site-occluding loop in a tetrameric assembly. J. Biol. Chem. 1999;274:2963–2970. doi: 10.1074/jbc.274.5.2963. [DOI] [PubMed] [Google Scholar]
- 11.Morel N, et al. Acetylcholinesterase H and T dimers are associated through the same contact. Mutations at this interface interfere with the C-terminal T peptide, inducing degradation rather than secretion. J. Biol. Chem. 2001;276:37379–37389. doi: 10.1074/jbc.M103192200. [DOI] [PubMed] [Google Scholar]
- 12.Bon S, Toutant JP, Meflah K, Massoulie J. Amphiphilic and nonamphiphilic forms of Torpedo cholinesterases: solubility and aggregation properties. J. Neurochem. 1988;51:776–785. doi: 10.1111/j.1471-4159.1988.tb01812.x. [DOI] [PubMed] [Google Scholar]
- 13.Ushkaryov YA, Petrenko AG, Geppert M, Sudhof TC. Neurexins: synaptic cell surface proteins related to the alpha-latrotoxin receptor and laminin. Science. 1992;257:50–56. doi: 10.1126/science.1621094. [DOI] [PubMed] [Google Scholar]
- 14.Tobaben S, Sudhof TC, Stahl B. Genetic analysis of alpha-latrotoxin receptors reveals functional interdependence of CIRL/latrophilin 1 and neurexin 1 alpha. J. Biol. Chem. 2002;277:6359–6365. doi: 10.1074/jbc.M111231200. [DOI] [PubMed] [Google Scholar]
- 15.Sugita S, Khvochtev M, Sudhof TC. Neurexins are functional alpha-latrotoxin receptors. Neuron. 1999;22:489–496. doi: 10.1016/s0896-6273(00)80704-7. [DOI] [PubMed] [Google Scholar]
- 16.Russell AB, Carlson SS. Neurexin is expressed on nerves, but not at nerve terminals, in the electric organ. J. Neurosci. 1997;17:4734–4743. doi: 10.1523/JNEUROSCI.17-12-04734.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Littleton JT, Bhat MA, Bellen HJ. Deciphering the function of neurexins at cellular junctions. J. Cell Biol. 1997;137:793–796. doi: 10.1083/jcb.137.4.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sudhof TC. alpha-Latrotoxin and its receptors: neurexins and CIRL/latrophilins. Annu. Rev. Neurosci. 2001;24:933–962. doi: 10.1146/annurev.neuro.24.1.933. [DOI] [PubMed] [Google Scholar]
- 19.Dustin ML, Colman DR. Neural and immunological synaptic relations. Science. 2002;298:785–789. doi: 10.1126/science.1076386. [DOI] [PubMed] [Google Scholar]
- 20.Hata Y, Butz S, Sudhof TC. CASK: a novel dlg/PSD95 homolog with an N-terminal calmodulin-dependent protein kinase domain identified by interaction with neurexins. J. Neurosci. 1996;16:2488–2494. doi: 10.1523/JNEUROSCI.16-08-02488.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Butz S, Okamoto M, Sudhof TC. A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell. 1998;94:773–782. doi: 10.1016/s0092-8674(00)81736-5. [DOI] [PubMed] [Google Scholar]
- 22.Palmer A, et al. EphrinB phosphorylation and reverse signaling: regulation by Src kinases and PTP-BL phosphatase. Mol. Cell. 2002;9:725–737. doi: 10.1016/s1097-2765(02)00488-4. [DOI] [PubMed] [Google Scholar]
- 23.Cowan CA, Henkemeyer M. The SH2/SH3 adaptor Grb4 transduces B-ephrin reverse signals. Nature. 2001;413:174–179. doi: 10.1038/35093123. [DOI] [PubMed] [Google Scholar]
- 24.Holland SJ, et al. Bidirectional signalling through the EPH-family receptor Nuk and its transmembrane ligands. Nature. 1996;383:722–725. doi: 10.1038/383722a0. [DOI] [PubMed] [Google Scholar]
- 25.Brückner K, Pasquale EB, Klein R. Tyrosine phosphorylation of transmembrane ligands for Eph receptors. Science. 1997;275:1640–1643. doi: 10.1126/science.275.5306.1640. [DOI] [PubMed] [Google Scholar]
- 26.Hall AC, Lucas FR, Salinas PC. Axonal remodeling and synaptic differentiation in the cerebellum is regulated by WNT-7a signaling. Cell. 2000;100:525–535. doi: 10.1016/s0092-8674(00)80689-3. [DOI] [PubMed] [Google Scholar]
- 27.Shapiro L, Colman DR. The diversity of cadherins and implications for a synaptic adhesive code in the CNS. Neuron. 1999;23:427–430. doi: 10.1016/s0896-6273(00)80796-5. [DOI] [PubMed] [Google Scholar]
- 28.Uemura T. The cadherin superfamily at the synapse: more members, more missions. Cell. 1998;93:1095–1098. doi: 10.1016/s0092-8674(00)81452-x. [DOI] [PubMed] [Google Scholar]
- 29.Mayford M, et al. Modulation of an NCAM-related adhesion molecule with long-term synaptic plasticity in Aplysia. Science. 1992;256:638–644. doi: 10.1126/science.1585176. [DOI] [PubMed] [Google Scholar]
- 30.Biederer T, et al. SynCAM, a synaptic adhesion molecule that drives synapse assembly. Science. 2002;297:1525–1531. doi: 10.1126/science.1072356. [DOI] [PubMed] [Google Scholar]
- 31.Yamagata M, Weiner J, Sanes J. Sidekicks. Synaptic adhesion molecules that promote lamina-specific connectivity in the retina. Cell. 2002;110:649–660. doi: 10.1016/s0092-8674(02)00910-8. [DOI] [PubMed] [Google Scholar]
- 32.Dalva MB, et al. EphB receptors interact with NMDA receptors and regulate excitatory synapse formation. Cell. 2000;103:945–956. doi: 10.1016/s0092-8674(00)00197-5. [DOI] [PubMed] [Google Scholar]
- 33.Hatten ME. Neuronal regulation of astroglial morphology and proliferation in vitro. J. Cell Biol. 1985;100:384–396. doi: 10.1083/jcb.100.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stein E, et al. Eph receptors discriminate specific ligand oligomers to determine alternative signaling complexes, attachment, and assembly responses. Genes Dev. 1998;12:667–678. doi: 10.1101/gad.12.5.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson JD, et al. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sanchez R, Sali A. Evaluation of comparative protein structure modeling by MODELLER-3. Proteins. 1997;1(Suppl):50–58. doi: 10.1002/(sici)1097-0134(1997)1+<50::aid-prot8>3.3.co;2-w. [DOI] [PubMed] [Google Scholar]
- 37.Carson M. Ribbons 2.0. J. Appl. Crystallogr. 1991;24:958–961. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.