

Abstract
Nitric oxide (NO) is an antiviral effector of the innate immune system. Viruses that can interfere with NO synthesis may be able to replicate more rapidly than viruses that cannot limit NO synthesis. We show that the adenovirus E1A protein inhibits NO production by decreasing expression of the inducible NO synthase (NOS2). The amino-terminal portion of E1A decreases transactivation of the NOS2 5′-flanking region, limiting the DNA binding activity of NF-κB and inhibiting NOS2 expression. E1A is thus able to deactivate a critical component of the host defense against viral infection. Viral inhibition of NO production is a mechanism that may enable certain viruses to evade the host innate immune system.
Keywords: NF-κB, IFN, transcription, peroxynitrite, histone acetyltransferase
Adenoviruses are nonenveloped DNA viruses that cause a variety of respiratory, ocular, gastrointestinal, and urogenital disorders (1). Adenovirus infection in immunocompromised hosts can cause severe illness with high morbidity. In immunocompetent hosts, the immune response to adenovirus infection consists of an early innate immune response, characterized by the rapid appearance of macrophages and natural killer cells, followed by a delayed but highly specific acquired immune response, characterized by T lymphocyte and B lymphocyte responses. The innate and acquired immune responses to adenovirus infection limit viral replication and clear infected cells.
Adenoviruses have evolved multiple mechanisms for inhibiting host immune responses (2–4). Viral encoded, double-stranded RNA molecules block the activation of protein kinase R, an enzyme that normally inhibits viral and host protein synthesis by phosphorylating eukaryotic initiation factor eIF-2 (5–7). The adenovirus early region 3 (E3) encodes a set of proteins, some of which regulate the host immune system (8, 9). The adenovirus E3 19-kDa glyco-protein is an integral membrane protein that interferes with presentation of viral antigens to host immune cells by retaining class I MHC heavy chain in the endoplasmic reticulum, and also by interfering with tapasin processing of viral peptides before MHC I binding (10–12). Apoptosis of infected cells is limited by the adenovirus E3 14.7-kDa protein, which inhibits tumor necrosis factor α (TNF-α)-induced apoptosis, and by the adenovirus E3 10.4/14.5-kDa (RIDα/RIDβ) complex, which inhibits TNF- and Fas-induced apoptosis. Adenovirus E3 14.7-kDa limits activation of NF-κB by interacting with IκB kinase-γ (8, 13–16).
Adenovirus polypeptides encoded by the E1A gene also mediate adenoviral immune suppression (17). The adenovirus serotypes 2/5 E1A transcription unit encodes by alternatively splicing two overlapping polypeptides, a 243-aa (12S) polypeptide and a 289-aa (13S) polypeptide. Both polypeptides share an amino-terminal sequence (N), a conserved region 1 (CR1), a conserved region 2 (CR2), and a carboxyl terminal region (C). The 13S E1A also contains a 46-aa conserved region 3 (CR3) fused to the carboxyl end of the CR2 region (18). These domains of E1A interact with a variety of host proteins, regulating cellular and viral transcription, remodeling chromatin, modulating acetylation, and controlling the cell cycle (17, 19).
The domains of E1A modulate host gene transcription and cell replication to create an environment favorable for viral replication. The amino-terminal and CR1 domains of E1A can interact directly with transcriptional coactivators such as CBP/p300 or P/CAF (20). These coactivators normally regulate transcription of inflammatory response genes by recruiting chromatin-remodeling enzymes that acetylate histones, leading to recruitment of a SWI/SNF complex, and ultimately leading to formation of a preinitiation complex (PIC) which includes RNA polymerase II (21–26). E1A inhibition of coactivator activity might thus interfere with PIC formation and transcription of inflammatory genes. The CR1 and CR2 domains of E1A interact with the retinoblastoma (Rb) protein and other pocket proteins, inducing the host cell to enter the S phase of the cell cycle, thus optimizing the environment for viral DNA replication.
E1A also regulates the activity of specific transcription factors that are critical components of the host antiviral response. For example, E1A blocks IFN-γ gene activation by directly interacting with signal transducer and activator of transcription-1 and IFN regulatory factor-1 (27, 28). Furthermore, E1A interferes with I-κB and NF-κB signal transduction (29–31). However, other studies show that E1A is capable of activating NF-κB under certain circumstances, thereby driving expression of intercellular adhesion molecule-1 and IL-8 (32, 33). Many of these transcription factors affected by E1A regulate expression of the inducible nitric oxide synthase (NOS2).
Nitric oxide (NO) is an antiviral effector of the innate immune system (34). Many viruses are capable of inducing NOS2 expression and NO synthesis in cells and animals (35–38). Adenoviral infection induces NOS2 expression and NO synthesis in mice (39). NO inhibits replication of a wide variety of DNA and RNA viruses in cells and animals, although the effects of NO on adenoviruses are unknown (35–38).
A variety of cytokines produced during viral infection can stimulate NOS2 expression by triggering transcription factors that transactivate the NOS2 5′-flanking region (40–44). Transcription factors that regulate NOS2 expression include NF-κB, IFN regulatory factor-1, and signal transducer and activator of transcription-1α (45–51). Coactivator and accessory factors that regulate NOS2 transcription include the high-mobility group protein HMG I(Y) and CCAAT/enhancer binding protein (52–54).
NO derived from NOS2 can inhibit viral replication (34, 38, 55). However, whether virus infection can inhibit NOS2 expression is unknown. Because the adenovirus E1A can disrupt signal transduction pathways regulating inflammatory responses, we hypothesized that adenoviruses inhibit NOS2 expression. In particular, we explored the role of the adenovirus E1A protein in the regulation of NOS2 transcription.
Materials and Methods
Cell Culture and Reagents. The mouse macrophage cell line RAW 264.7 (American Type Culture Collection TIB-71) was grown in DMEM supplemented with 10% FBS. Actinomycin D, cyclo-hexaimide, and 4-hydroxytamoxifen (4-HT) were purchased from Sigma–Aldrich. Puromycin was purchased from Calbiochem. Lipopolysaccharide (LPS, Escherichia coli) was from Sigma. Antibodies to NF-κB p65, p50, IκBα, and NOS2 (M-19) were purchased from Santa Cruz Biotechnology. The murine NOS2 cDNA and murine NOS2 promoter–luciferase construct and its deletion mutants have been described (41).
The E1A expression plasmids were gifts of Richard Goodman (Oregon Health Sciences University, Portland, OR) and of Debabrata Chakravarti (University of Pennsylvania, Philadelphia) (20). The plasmid E1A289 expresses E1A amino acids 1–289, also called 13S E1A. The plasmid E1A243 expresses E1A residues 1–139 fused to residues 185–289, also called E1A 12S. Plasmid E1AC79 expresses E1A amino acids 1–79; plasmid E1AC109 expresses E1A amino acids 1–109; plasmid E1AC139 expresses E1A amino acids 1–139; plasmid E1AC223 expresses E1A amino acids 1–223; and plasmid E1AC249 expresses E1A amino acids 1–249. Plasmid E1AN25 expresses E1A amino acids 25–289; plasmid E1AN76 expresses E1A amino acids 76–289; plasmid E1AN120 expresses E1A amino acids 120–289; plasmid E1AN132 expresses E1A amino acids 132–289; and plasmid E1AN186 expresses E1A amino acids 186–289.
Plasmid pBabe puro E1A-ER was constructed by inserting a PCR-amplified E1A cDNA (encoding 243 amino acid residues) into pBabe puro estrogen receptor (ER) vector, a generous gift from Alan Friedman (The Johns Hopkins University School of Medicine).
Adenovirus Infection. The adenovirus type 5 and an E1A deletion mutant Ad/dl312 were generous gifts from Jeffrey Rade (The Johns Hopkins University School of Medicine). RAW cells were infected by adenoviruses at various multiplicities of infection (mois) for 3–6 h without FBS, then washed and resuspended in complete medium.
Cell Lines and Cell Culture. To transfect RAW cells transiently, RAW cells were cultured until 80% confluency was attained. Plasmid DNAs were transfected into RAW cells by using LipofectAMINE–PLUS according to the manufacturer's instructions (Life Technologies, Carlsbad, CA). Cell lysates were collected 48 h after transfection and luciferase activity was measured in a luminometer by using a kit (Promega). To establish stably transfected RAW cells, the pBabe puro E1A-ER plasmid was transfected into RAW cells by using LipofectAMINE–2000 (Life Technologies). Transfected cells were treated with puromycin (2 μg/ml) for 48 h after the transfection, and puromycin-resistant single colonies were selected from the pools and expanded. RAW stable cell lines were maintained under 2 μg/ml puromycin. E1A-ER fusion protein expression was induced with 4-HT (200 nM) for 6 h before adding LPS. Cell lysates were harvested, fractionated by SDS/PAGE, and immunoblotted with antibody to murine NOS2 (M-19 from Santa Cruz Biotechnology) or antibody to E1A (Pharmingen). The blots were visualized with a chemiluminescence detection system (Amersham Biosciences).
Immunocytochemistry. Stably transfected RAW cell lines were plated on glass disks, and E1A-ER expression was activated by 4-HT treatment for 16 h. Cells were washed and fixed in 4% formaldehyde for 30 min on ice. The primary antibody against E1A (Pharmingen) at 500 μg/ml was added to the cells and incubated at room temperature in a wet chamber for 1 h. The cells were washed with PBS Tween 20 again and incubated with secondary Cy-3-conjugated anti-primary antibody at 1:100 dilution. Cells were observed under a fluorescence microscope connected to a PC.
Northern Blotting and Nuclear Run-On Assay. Total cellular RNAs were prepared by TRIzol solution (Life Technologies) according to the manufacturer's recommendation. The total RNAs were fractionated by gel electrophoresis with formaldehyde and transferred to Nytran SPC nylon membranes (Schleicher & Schuell). Equal RNA loading was confirmed by hybridization to ribosomal phosphoprotein probe 36B4 (56). Hybridization probes were labeled with [α-32P]dCTP (Megaprimer, Amersham Biosciences).
New RNA synthesis was measured with a nuclear run-on assay as described (57, 58). RAW cells were harvested, resuspended in lysis buffer that included 0.5% Nonidet P-40, incubated on ice, and then centrifuged to collect nuclei. Nuclei were resuspended and incubated with [32P]UTP, ATP, GTP, and CTP to synthesize RNA probes. The probes were purified and hybridized to dot blots of 5 μg of cDNA for NOS2, GAPDH, and aldolase. The membranes were then washed and autoradiographed.
Electrophoresis Mobility-Shift Assay (EMSA). EMSAs were performed as described (59). Briefly, nuclear extracts were prepared from RAW cells and frozen in liquid nitrogen. EMSAs were performed with 5 μg of nuclear extracts and radiolabeled oligonucleotide containing either a consensus NF-κB binding site (GGCAACTGGGGACTTTCCCTTT), or a fragment from the murine NOS2 promoter (-87 to -52) that contains an NF-κB and an Oct site (TGGGGACTCTCCCTTTGGGAACAGTTATGCAAAATA) (60). The reaction products were fractionated on a 5% acrylamide gel in 0.3× TBE (0.09 M Tris base/0.09 M boric acid/2 mM EDTA, pH 8.3) buffer. The gel was dried and visualized by autoradiography.
Results
E1A Expressed by Adenovirus Inhibits NOS2 Expression. We hypothesized that the adenovirus E1A protein inhibits NOS2 expression, thus explaining one of the mechanisms by which adenoviruses evade the host immune response. To test this hypothesis, we compared the effect of WT adenovirus and an E1A deficient adenovirus mutant on the expression of NOS2.
We first examined the effect of NO on adenovirus replication. HeLa cells were infected with WT adenovirus type 5 and treated with the NO donor DETA-NONOate (diethylenetriamine NONOate) at 0, 0.1, and 1.0 mM, and the number of infected cells was measured with an antibody staining for the adenovirus hexon protein. We found that NO inhibits the replication of the adenovirus (Fig. 1A).
Fig. 1.

Viral E1A decreases NOS2 expression. (A) NO inhibits adenovirus replication. HeLa cells were infected with a WT adenovirus at a moi of 10, and treated with DETA-NONOate (diethylenetriamine NONOate) at 0, 24, and 48 h, and the amount of virus at 60 h was measured by staining cells with antibody to hexon and counting stained cells per high-power field (hpf) (results are presented as mean ± SD; n = 3). (B) Viral E1A decreases NOS2 steady-state protein levels in infected cells. RAW cells were infected with a WT adenovirus or mutant adenovirus lacking E1A (ΔE1A) at a moi of 20 or 200 for 40 h. Cell lysates were immunoblotted for NOS2 (Upper) and E1A (Lower). (This experiment was repeated four times with similar results.) (C) Viral E1A decreases NOS2 steady-state protein levels in infected cells. RAW cells were infected with adenovirus at a moi of 200, and then treated 4 h later with LPS and Nω-nitro-l-arginine methyl ester (NAME). Cell lysates were immunoblotted for NOS2. (D) Viral E1A inhibits NO production from infected cells. RAW cells were infected with adenovirus for 24 h, and then treated with LPS for 16 h. Some cells received the NOS inhibitor NAME. The [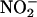 ] in the supernatant was measured by using the Griess assay (mean ± SD; n = 3).
] in the supernatant was measured by using the Griess assay (mean ± SD; n = 3).
We compared the amount of NO released from DETA-NONOate to the amount of NO produced by stimulated macrophages, by using the Griess assay. We found that 100 μM DETA-NONOate releases 25–50 μM NO over 24 h; furthermore, 1,000 μM DETA-NONOate releases 100–125 μM NO over 24 h. RAW cells stimulated with 100 ng/ml LPS released 10–50 μM NO over 24 h.
We then explored whether adenovirus infection induces NOS2 expression. We began by infecting the murine monocyte cell line RAW 264.7 with a WT adenovirus type 5 or a mutant adenovirus deficient in E1A (Ad/dl312) at a moi of 0, 20, or 200 for 40 h. Lysates of infected cells were immunoblotted with an antibody to NOS2. We found that the WT adenovirus does not induce NOS2 expression, but infection with a mutant adenovirus lacking the E1A region does induce NOS2 expression (Fig. 1B). These data suggest that adenovirus infection activates NOS2 expression, but E1A directly or indirectly inhibits NOS2 expression.
We then repeated this experiment with LPS-stimulated RAW cells. (These experiments were performed so that we could subsequently explore the role of E1A in cells that were expressing NOS2 but were not infected.) We infected RAW cells with a WT or mutant adenovirus deficient in E1A. The infected cells were then treated with LPS (100 ng/ml) to induce NOS2. As a control, some LPS-treated cells were also treated with the NO synthase (NOS) inhibitor Nω-nitro-L-arginine methyl ester (NAME). We measured NOS2 steady-state protein levels and NO production and found that LPS induces NOS2 protein expression in noninfected cells (Fig. 1C), whereas WT adenovirus infection blocks NOS2 expression. However, infection with a mutant adenovirus deficient in E1A does not block NOS2 expression (Fig. 1C). (NAME inhibition of NOS causes a slight increase in steady-state NOS levels.) We also measured the effect of adenovirus infection on NO synthesis and found that cells stimulated by LPS produce higher levels of NO after infection by an adenovirus deficient in E1A, as compared with cells infected by a WT adenovirus (Fig. 1D). (The immunoblot in Fig. 1B is exposed longer than the immunoblot in Fig. 1C to demonstrate that adenovirus infection alone can induce NOS2 expression.)
These data suggest that E1A produced by adenoviruses directly or indirectly inhibits NOS2 expression. However, E1A is required for other adenovirus genes to be expressed, and thus other adenoviral proteins may also play a role in suppressing NOS2 expression. We therefore expressed E1A and E1A mutants in cells by using plasmid vectors, to explore the specific role of E1A in the regulation of NOS2 expression, while eliminating the effects of other viral proteins.
E1A Expressed by Plasmid Vector Inhibits NOS2 Expression. To confirm that E1A is the adenoviral protein that inhibits NOS2 expression, we next examined NOS2 expression in cell lines transfected transiently or stably with an E1A expression vector. We first generated stably transfected RAW cell lines that express a fusion polypeptide consisting of E1A fused to the ligand binding domain of the estrogen receptor α subunit (E1A-ER). The ligand binding domain of the ER subunit normally remains in the cytoplasm, but when bound to estrogen or 4-HT translocates into the nucleus. The E1A-ER fusion polypeptide remains in the cytoplasm of stably transfected RAW cells (Fig. 2A Upper Left). However, exposure to 4-HT causes the E1A-ER fusion polypeptide to translocate to the nucleus (Fig. 2A Upper Center). Adenovirus infection of RAW cells also leads to E1A expression in the nucleus (Fig. 2A Upper Right). We also constructed RAW cells stably transfected with a vector expressing ER alone as negative controls. We used these stably transfected cells to explore the effect of ectopic expression of E1A on NOS2 expression.
Fig. 2.

Ectopic E1A decreases NOS2 expression. (A) 4-HT induces expression of an E1A fusion polypeptide in the nucleus. (Left and Center) RAW cells were stably transfected with a plasmid construct expressing a fusion polypeptide consisting of E1A and an estrogen receptor fragment (ER). Cells were treated with control or 4-HT for 16 h, and imaged by fluorescence microscopy for E1A or DNA (DNA stained with Hoechst 33342; Lower). The 4-HT treatment causes expression of E1A-fusion polypeptide in the nucleus (Center). This experiment was repeated in three different stably transfected cell clones with similar results. (Right) RAW cells were infected with WT adenovirus type 5 and imaged by fluorescence microscopy for E1A and DNA. (B) Ectopic E1A decreases NOS2 steady-state protein levels. RAW cells were stably transfected with a plasmid expressing ER alone or E1A-ER, treated for 6 h with 4-HT to induce ER or E1A-ER expression in the nucleus, and then treated for 16 h with LPS to induce NOS2 expression. Cell lysates were immunoblotted for NOS2 and nuclear extracts were immunoblotted for E1A. Total protein was stained with Coomassie. (This experiment was repeated three times with similar results.) (C) Ectopic E1A expression decreases steady-state NOS2 mRNA levels. RAW cells were stably transfected as above, and treated as above with medium, 4-HT, LPS, or 4-HT and LPS. Total RNA was analyzed by Northern blotting with a probe for NOS2 mRNA or for ribosomal phosphoprotein 36B4 mRNA as a control. (This experiment was repeated three times with similar results.) (D) Ectopic E1A expression decreases NOS2 transcription. RAW cells were stably transfected with a vector expressing ER (Upper) or E1A-ER (Lower), treated with 4-HT for 16 h, and then treated with LPS for 4 h. Transcription of NOS2 was analyzed by nuclear run-on assays (center). As controls, transcription of aldolase (left) and GAPDH (right) was also measured.
Our data show that LPS induces NOS2 expression in RAW cells stably transfected with a vector expressing ER alone (Fig. 2B, lanes 1–4). ER expression alone has no effect on NOS2 steady-state protein levels in RAW cells (Fig. 2B, lanes 3 and 4). However, expression of the fusion polypeptide E1A-ER decreases NOS2 steady-state protein levels in RAW cells (Fig. 2B, lanes 7 and 8).
We also examined the effect of E1A on NOS2 steady-state mRNA levels. By using the same cells, we found that LPS increases NOS2 expression in RAW cells expressing ER (Fig. 2C, lanes 3 and 4). However, E1A-ER fusion polypeptide decreases NOS2 steady-state mRNA levels (Fig. 2C, lanes 7 and 8).
To show that E1A inhibits NOS2 RNA transcription, we used a nuclear run-on assay. RAW cells expressing ER or E1A-ER were treated with LPS and 4-HT as above, nuclei were harvested from these cells, and radiolabeled new RNA transcripts were prepared from the nuclei for use as probes against NOS2 cDNA. We found that the E1A-ER fusion polypeptide inhibits NOS2 RNA synthesis, but not the synthesis of control RNA (Fig. 2D).
These data confirm that E1A directly inhibits NOS2 expression. No other adenoviral proteins are necessary to mediate the effects of E1A.
A Critical Region of the NOS Promoter Mediates the Effects of E1A. We next explored the effect of E1A on transactivation of the NOS2 5′-flanking region. We transiently cotransfected RAW cells with both an E1A expression vector and a reporter vector containing the NOS2 5′-flanking region driving expression of Photinus luciferase (41). (The total amount of DNA transfected into cells was kept constant.) Cotransfected cells were then treated with LPS, and the amount of luciferase activity was measured in cell lysates. Our data show that increasing amounts of E1A expression inhibited NOS2 promoter transactivation (Fig. 3A).
Fig. 3.

E1A inhibits NOS2 promoter transactivation. (A) E1A inhibits transactivation of the entire NOS2 promoter. RAW cells were transiently cotransfected with a vector expressing E1A and with a NOS2 reporter construct consisting of the NOS2 5′-flanking region driving expression of luciferase. Cells were treated with LPS for 16 h, and the amount of luciferase was measured in a luminometer (Upper; mean ± SD; n = 3). (Lower) E1A immunoblot and total protein stain of the cell lysates. (B) E1A inhibits transactivation of a minimal NOS2 promoter. RAW cells were transiently cotransfected with the full-length E1A expression vector and various deletion mutants of the NOS2 5′-flanking region reporter construct diagrammed Left. Cotransfected cells were then treated with medium alone or with LPS, and luciferase activity was measured (Right). LPS increases NOS2 promoter transactivation in RAW cells in the absence of E1A. However, LPS fails to increase NOS2 promoter activity in RAW cells also expressing E1A. E1A inhibits LPS activation of the NOS2 promoter, even if only 262 bp of the NOS2 promoter region remain (mean ± SD; n = 3).
We then determined the region of the NOS2 5′-flanking region that is a target of E1A. We cotransfected RAW cells with an E1A expression vector, and with various deletion mutants of the NOS2 5′-flanking region driving expression of luciferase. We then measured the effect of LPS on NOS2 promoter transactivation in these cotransfected RAW cells. We found that the NOS2 promoter fragment has a basal level of transactivation (Fig. 3B), and that LPS increases transactivation of the NOS2 5′ flanking region (Fig. 3B). LPS is able to transactivate the NOS2 promoter as successive portions of the 5′ region are deleted (Fig. 3B), though such LPS transactivation of the NOS2 promoter decreases somewhat as the IFN response element (IRE) at -925 to -915 is deleted. However, deletion of the NOS2 promoter region from -262 to -42 abrogates baseline expression and LPS induction (Fig. 3B, bottom line). This fragment contains from -86 to -77 the κB element binding site for NF-κB. These data suggest that the region of the NOS2 promoter between -262 and -42 is critical for NOS2 expression, as we and others have shown (40, 41).
E1A alone inhibits baseline transactivation of NOS2 (Fig. 3B). Furthermore, E1A inhibits LPS induction of NOS2 (Fig. 3B). Successive deletions of the NOS2 promoter have no effect on E1A inhibition of the NOS2 promoter. Because E1A inhibits NOS2 promoter transactivation of a minimal NOS2 promoter (262 bp of the NOS2 5′-flanking region), these data indirectly suggest that the region of the NOS2 promoter between -262 and -42 mediates the inhibitory effects of E1A.
E1A Inhibits NF-κB Binding to a NOS2 κB Site. The region of the NOS2 promoter -262 to -42 that mediates the inhibitory effects of E1A contains an NF-κB binding element from -86 to -77. NF-κB regulates NOS2 transcription (45, 50, 51, 61). We hypothesized that E1A decreases NF-κB binding to the NOS2 promoter. To test this hypothesis, we measured NF-κB binding activity in nuclear extracts of stably transfected RAW cells. Our results show that LPS increases κB binding activity in nuclear extracts from RAW cells (Fig. 4 Left, lanes 1 and 2 vs. 3 and 4). The ER polypeptide alone has no effect on κB binding activity (Fig. 4 Left, lanes 3 vs. 4). However, the E1A-ER fusion polypeptide reduces the κB binding activity in RAW cells stimulated with LPS (Fig. 4 Left, lanes 7 vs. 8). This κB binding activity is caused by NF-κB, because a competitor κB oligonucleotide blocks binding, and because an antibody to the NF-κB subunit p50 produces a supershift, and an antibody to subunit p65 decreases the intensity of the original band (Fig. 4 Right).
Fig. 4.

E1A inhibits NF-κB binding activity. RAW cells were stably transfected with constructs expressing the estrogen receptor alone (ER), or expressing the E1A-ER fusion polypeptide. Transfected cells were then treated with medium, LPS, 4-HT, or both. Nuclear extracts were analyzed for binding activity to a radiolabeled oligonucleotide derived from the NOS2 5′-flanking region containing a κB element. (Left) 4-HT induction of E1A inhibits κB binding activity. (Right) Antibody to p50 or p65 decreases the intensity of the κB DNA–protein complex, and antibody to p50 supershifts the complex, suggesting that E1A regulates NF-κB binding to the κB element. (These experiments were repeated three times with similar results.)
Amino Terminus of E1A Inhibits NOS2 Transcription. To determine which domains of the E1A protein mediate the inhibition of NOS2 expression, we cotransfected RAW cells with the NOS2 promoter–luciferase reporter vector and with different vectors expressing deletion mutants of E1A (20). Transfected cells were treated with LPS, and NOS2 promoter transactivation was measured by the luciferase assay. Our data show that full-length E1A completely inhibits NOS2 promoter transactivation (Fig. 5), whereas deletion of the carboxyl-terminal region of E1A (amino acid residues 139–289) has little effect on NOS2 promoter inhibition. However, deletions of the amino terminus of E1A (residues 1–120) decreased the inhibitory effect of E1A. Deletion of the amino-terminal domain (residues 1–25) has a modest effect on NOS2 promoter inhibition. However, deletion of the CR1 and CR2 domains (residues 25–132) permits full expression of NOS2. These data suggest that the CR1 and CR2 domains are the regions of E1A that most inhibit NOS2 expression.
Fig. 5.

Amino-terminal domains of E1A inhibit NOS2 promoter transactivation. RAW cells were cotransfected with the full-length NOS2 promoter-reporter construct and with vectors expressing deletion mutants of E1A. Transfected cells were stimulated with medium alone or with LPS, and the amount of luciferase in cell lysates was measured. WT E1A blocks transactivation of the NOS2 promoter. However, E1A lacking the amino-terminal domains (N, CR1, and CR2) fails to block transactivation of the NOS2 promoter.
Discussion
The major finding of this study is that adenovirus inhibits production of the antiviral effector NO by blocking expression of NOS2. Adenoviral inhibition of NOS2 is mediated by the amino terminus, CR1, and CR2 domains of E1A. E1A inhibits NOS2 directly or indirectly by suppressing NOS2 transcription. E1A acts on the 5′-flanking region immediately upstream of the NOS2 gene, in part by decreasing NF-κB DNA binding activity. Because the CR1 and CR2 domains of E1A interact with CBP/p300, E1A may also inhibit NOS2 expression by interfering with acetyltransferase activity. Viral inhibition of NO production is a mechanism that may enable viruses to evade the host innate immune system.
E1A Domain Structure and Function. Adenoviruses evade the host immune system by several distinct mechanisms. Our data suggest that one such mechanism is E1A inhibition of NOS2 expression and NO production, critical components of the innate immune system. The two alternatively spliced transcripts of E1A share domains that regulate viral and host DNA replication and suppress the host immune system. The domains of E1A that inhibit NOS2 transcription, the amino-terminal domain, CR1, and CR2, interact with host proteins to modulate specific host functions. The amino-terminal domain of E1A regulates host gene transcription by interacting with a variety of host proteins, including transcription factors, coactivators and repressors, and nucleosome-remodeling factors. This amino-terminal region of E1A has a modest inhibitory effect on NOS2 transcription. The CR1 and CR2 domains of E1A interact with several host proteins, including the pocket proteins Rb, p107, and p130. E1A interaction with Rb family members frees E2F transcription factors previously bound to Rb family members which can stimulate transcription of selected genes. By interacting with Rb, the CR1 and CR2 domains modulate the activity of other proteins that interact with Rb, including histone deacetylase, which regulates gene transcription, and hBRM and BRG-1, which regulate nucleosomal remodeling. Our data show that these CR1 and CR2 domains play a major role in E1A inhibition of NOS2 expression (Fig. 5).
E1A and Mechanisms of NOS2 Inhibition. The mechanisms by which E1A inhibit NOS2 expression may provide insight into the regulation of NOS2 transcription. For example, the amino-terminal domain of E1A can interfere with the interaction between signal transducer and activator of transcription-1α and IFN regulatory factor-1, an interaction that is required for NOS2 transcription. However, the major influence of E1A on NOS2 transcription derives from the CR1 and CR2 regions, which interact with and regulate pocket proteins and the histone acetyltransferase (HAT) proteins CBP/p300 and pCAF. Because these CR1 and CR2 domains are the regions of E1A that most inhibit NOS2 transcription, these data suggest that acetylation and deacetylation play a major role in regulating NOS2 transcription. Transactivation of an analogous promoter, the IFN-β 5′ flanking region, requires not only NF-κB, IFN regulatory factor-1, and the high mobility group protein HMG-I(Y), but also the HAT activity of CBP/p300 (25, 26, 62, 63). The structure of the IFN-β 5′ flanking region is similar to the structure of the NOS2 5′ flanking region (23, 24). The concept that HAT activity is necessary for NOS2 transcription is supported by studies showing that sodium butyrate, an inhibitor of histone deacetylase, regulates NOS2 transcription (64). Because the CR1 and CR2 domains of E1A also regulate the cell cycle via interactions with Rb, it is possible that cell cycle regulators affect NOS2 expression as well. However, our data suggest that acetylation and deacetylation regulate NOS2 transcription, and that adenoviruses interfere with this pathway.
Viral Evasion of the Immune System. Viruses have evolved a variety of mechanisms to evade the immune system. Many RNA viruses have developed rapid replication, so that their life cycle is completed before the innate immune system can fully respond. Many DNA viruses replicate more slowly, and have developed strategies to deactivate the host immune system to survive. Our data show that adenovirus can suppress production of NO, an antiviral effector, by inhibiting NOS2 expression. Other viruses may also target production of radical effectors of the innate immune system as well. Viral inhibition of NO production is a mechanism that may enable viruses to evade the host innate immune system.
Acknowledgments
This research was supported in part by American Heart Association Established Investigator Grants EIG 0140210N (to C.J.L.), P01 HL56091 (to C.J.L.), P01 HL65608 (to C.J.L.), NIH R01 HL53615 (to C.J.L.), and R01 HL63706 (to C.J.L.), the Ciccarone Center for the Prevention of Heart Disease (C.J.L.), and the Cora and John H. Davis Foundation (C.J.L.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: NOS, NO synthase; NOS2, inducible NOS; E3, early region 3; LPS, lipopolysaccharide; 4-HT, 4-hydroxytamoxifen; Rb, retinoblastoma; moi, multiplicity of infection; ER, estrogen receptor; DETA-NONOate, diethylenetriamine NONOate.
References
- 1.Shenk, T. (1996) in Fields Virology, eds. Fields, B., Knipe, D. & Howley, P. (Lippincott–Raven, Philadelphia), Vol. 2, pp. 2111-2148. [Google Scholar]
- 2.Mahr, J. A. & Gooding, L. R. (1999) Immunol. Rev. 168, 121-130. [DOI] [PubMed] [Google Scholar]
- 3.Wold, W. S., Doronin, K., Toth, K., Kuppuswamy, M., Lichtenstein, D. L. & Tollefson, A. E. (1999) Curr. Opin. Immunol. 11, 380-386. [DOI] [PubMed] [Google Scholar]
- 4.Lukashok, S. A. & Horwitz, M. S. (1998) Curr. Clin. Top. Infect. Dis. 18, 286-305. [PubMed] [Google Scholar]
- 5.Thimmappaya, B., Weinberger, C., Schneider, R. J. & Shenk, T. (1982) Cell 31, 543-551. [DOI] [PubMed] [Google Scholar]
- 6.Schneider, R. J., Weinberger, C. & Shenk, T. (1984) Cell 37, 291-298. [DOI] [PubMed] [Google Scholar]
- 7.Reichel, P. A., Merrick, W. C., Siekierka, J. & Mathews, M. B. (1985) Nature 313, 196-200. [DOI] [PubMed] [Google Scholar]
- 8.Horwitz, M. S. (2001) Virology 279, 1-8. [DOI] [PubMed] [Google Scholar]
- 9.Wold, W. S. & Gooding, L. R. (1991) Virology 184, 1-8. [DOI] [PubMed] [Google Scholar]
- 10.Andersson, M., McMichael, A. & Peterson, P. A. (1987) J. Immunol. 138, 3960-3966. [PubMed] [Google Scholar]
- 11.Burgert, H. G. & Kvist, S. (1987) EMBO J. 6, 2019-2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kvist, S., Ostberg, L., Persson, H., Philipson, L. & Peterson, P. A. (1978) Proc. Natl. Acad. Sci. USA 75, 5674-5678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krajcsi, P., Dimitrov, T., Hermiston, T. W., Tollefson, A. E., Ranheim, T. S., Vande Pol, S. B., Stephenson, A. H. & Wold, W. S. (1996) J. Virol. 70, 4904-4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elsing, A. & Burgert, H. G. (1998) Proc. Natl. Acad. Sci. USA 95, 10072-10077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shisler, J., Yang, C., Walter, B., Ware, C. F. & Gooding, L. R. (1997) J. Virol. 71, 8299-8306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tollefson, A. E., Hermiston, T. W., Lichtenstein, D. L., Colle, C. F., Tripp, R. A., Dimitrov, T., Toth, K., Wells, C. E., Doherty, P. C. & Wold, W. S. (1998) Nature 392, 726-730. [DOI] [PubMed] [Google Scholar]
- 17.Gallimore, P. H. & Turnell, A. S. (2001) Oncogene 20, 7824-7835. [DOI] [PubMed] [Google Scholar]
- 18.Moran, E. & Mathews, M. B. (1987) Cell 48, 177-178. [DOI] [PubMed] [Google Scholar]
- 19.Frisch, S. M. & Mymryk, J. S. (2002) Nat. Rev. Mol. Cell Biol. 3, 441-452. [DOI] [PubMed] [Google Scholar]
- 20.Chakravarti, D., Ogryzko, V., Kao, H. Y., Nash, A., Chen, H., Nakatani, Y. & Evans, R. M. (1999) Cell 96, 393-403. [DOI] [PubMed] [Google Scholar]
- 21.Fry, C. J. & Peterson, C. L. (2002) Science 295, 1847-1848. [DOI] [PubMed] [Google Scholar]
- 22.Fry, C. J. & Peterson, C. L. (2001) Curr. Biol. 11, R185-R197. [DOI] [PubMed] [Google Scholar]
- 23.Maniatis, T., Falvo, J. V., Kim, T. H., Kim, T. K., Lin, C. H., Parekh, B. S. & Wathelet, M. G. (1998) Cold Spring Harbor Symp. Quant. Biol. 63, 609-620. [DOI] [PubMed] [Google Scholar]
- 24.Thanos, D. & Maniatis, T. (1995) Cell 83, 1091-1100. [DOI] [PubMed] [Google Scholar]
- 25.Parekh, B. S. & Maniatis, T. (1999) Mol. Cell 3, 125-129. [DOI] [PubMed] [Google Scholar]
- 26.Agalioti, T., Lomvardas, S., Parekh, B., Yie, J., Maniatis, T. & Thanos, D. (2000) Cell 103, 667-678. [DOI] [PubMed] [Google Scholar]
- 27.Look, D. C., Roswit, W. T., Frick, A. G., Gris-Alevy, Y., Dickhaus, D. M., Walter, M. J. & Holtzman, M. J. (1998) Immunity 9, 871-880. [DOI] [PubMed] [Google Scholar]
- 28.Leonard, G. T. & Sen, G. C. (1996) Virology 224, 25-33. [DOI] [PubMed] [Google Scholar]
- 29.Perkins, N. D., Felzien, L. K., Betts, J. C., Leung, K., Beach, D. H. & Nabel, G. J. (1997) Science 275, 523-527. [DOI] [PubMed] [Google Scholar]
- 30.Shao, R., Karunagaran, D., Zhou, B. P., Li, K., Lo, S. S., Deng, J., Chiao, P. & Hung, M. C. (1997) J. Biol. Chem. 272, 32739-32742. [DOI] [PubMed] [Google Scholar]
- 31.Shao, R., Hu, M. C., Zhou, B. P., Lin, S. Y., Chiao, P. J., von Lindern, R. H., Spohn, B. & Hung, M. C. (1999) J. Biol. Chem. 274, 21495-21498. [DOI] [PubMed] [Google Scholar]
- 32.Shurman, L., Sen, R. & Bergman, Y. (1989) J. Immunol. 143, 3806-3812. [PubMed] [Google Scholar]
- 33.Keicho, N., Higashimoto, Y., Bondy, G. P., Elliott, W. M., Hogg, J. C. & Hayashi, S. (1999) Am. J. Physiol. 277, L523-L532. [DOI] [PubMed] [Google Scholar]
- 34.MacMicking, J., Xie, Q. W. & Nathan, C. (1997) Annu. Rev. Immunol. 15, 323-350. [DOI] [PubMed] [Google Scholar]
- 35.De Groote, M. A. & Fang, F. C. (1995) Clin. Infect. Dis. 21, Suppl. 2, S162-S165. [DOI] [PubMed] [Google Scholar]
- 36.Mannick, J. B. (1995) Res. Immunol. 146, 693-697. [DOI] [PubMed] [Google Scholar]
- 37.Fang, F. C. (1997) J. Clin. Invest. 99, 2818-2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reiss, C. S. & Komatsu, T. (1998) J. Virol. 72, 4547-4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zsengeller, Z. K., Ross, G. F., Trapnell, B. C., Szabo, C. & Whitsett, J. A. (2001) Am. J. Physiol. 280, L503-L511. [DOI] [PubMed] [Google Scholar]
- 40.Xie, Q. W., Whisnant, R. & Nathan, C. (1993) J. Exp. Med. 177, 1779-1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lowenstein, C. J., Alley, E. W., Raval, P., Snowman, A. M., Snyder, S. H., Russell, S. W. & Murphy, W. J. (1993) Proc. Natl. Acad. Sci. USA 90, 9730-9734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nathan, C. & Xie, Q. W. (1994) Cell 78, 915-918. [DOI] [PubMed] [Google Scholar]
- 43.de Vera, M. E., Shapiro, R. A., Nussler, A. K., Mudgett, J. S., Simmons, R. L., Morris, S. M., Jr., Billiar, T. R. & Geller, D. A. (1996) Proc. Natl. Acad. Sci. USA 93, 1054-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taylor, B. S., Alarcon, L. H. & Billiar, T. R. (1998) Biochemistry (Moscow) 63, 766-781. [PubMed] [Google Scholar]
- 45.Xie, Q. W., Kashiwabara, Y. & Nathan, C. (1994) J. Biol. Chem. 269, 4705-4708. [PubMed] [Google Scholar]
- 46.Martin, E., Nathan, C. & Xie, Q. W. (1994) J. Exp. Med. 180, 977-984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamijo, R., Harada, H., Matsuyama, T., Bosland, M., Gerecitano, J., Shapiro, D., Le, J., Koh, S. I., Kimura, T., Green, S. J., et al. (1994) Science 263, 1612-1615. [DOI] [PubMed] [Google Scholar]
- 48.Gao, J., Morrison, D. C., Parmely, T. J., Russell, S. W. & Murphy, W. J. (1997) J. Biol. Chem. 272, 1226-1230. [DOI] [PubMed] [Google Scholar]
- 49.Gupta, A. K. & Kone, B. C. (1999) Am. J. Physiol. 276, F599-F605. [DOI] [PubMed] [Google Scholar]
- 50.Ganster, R. W., Taylor, B. S., Shao, L. & Geller, D. A. (2001) Proc. Natl. Acad. Sci. USA 98, 8638-8643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Taylor, B. S., de Vera, M. E., Ganster, R. W., Wang, Q., Shapiro, R. A., Morris, S. M., Jr., Billiar, T. R. & Geller, D. A. (1998) J. Biol. Chem. 273, 15148-15156. [DOI] [PubMed] [Google Scholar]
- 52.Perrella, M. A., Pellacani, A., Wiesel, P., Chin, M. T., Foster, L. C., Ibanez, M., Hsieh, C. M., Reeves, R., Yet, S. F. & Lee, M. E. (1999) J. Biol. Chem. 274, 9045-9052. [DOI] [PubMed] [Google Scholar]
- 53.Pellacani, A., Chin, M. T., Wiesel, P., Ibanez, M., Patel, A., Yet, S. F., Hsieh, C. M., Paulauskis, J. D., Reeves, R., Lee, M. E. & Perrella, M. A. (1999) J. Biol. Chem. 274, 1525-1532. [DOI] [PubMed] [Google Scholar]
- 54.Li, M., Pascual, G. & Glass, C. K. (2000) Mol. Cell. Biol. 20, 4699-4707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bogdan, C. (2001) Nat. Immun. 2, 907-916. [DOI] [PubMed] [Google Scholar]
- 56.Laborda, J. (1991) Nucleic Acids Res. 19, 3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mustafa, S. B. & Olson, M. S. (1998) J. Biol. Chem. 273, 5073-5080. [DOI] [PubMed] [Google Scholar]
- 58.Horton, M. R., Olman, M. A., Bao, C., White, K. E., Choi, A. M., Chin, B. Y., Noble, P. W. & Lowenstein, C. J. (2000) Am. J. Physiol. 279, L707-L715. [DOI] [PubMed] [Google Scholar]
- 59.Cao, W., Britos-Bray, M., Claxton, D. F., Kelley, C. A., Speck, N. A., Liu, P. P. & Friedman, A. D. (1997) Oncogene 15, 1315-1327. [DOI] [PubMed] [Google Scholar]
- 60.Goldring, C. E., Reveneau, S., Algarte, M. & Jeannin, J. F. (1996) Nucleic Acids Res. 24, 1682-1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Saura, M., Zaragoza, C., Bao, C., McMillan, A. & Lowenstein, C. J. (1999) J. Mol. Biol. 289, 459-471. [DOI] [PubMed] [Google Scholar]
- 62.Kim, T. K., Kim, T. H. & Maniatis, T. (1998) Proc. Natl. Acad. Sci. USA 95, 12191-12196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wathelet, M. G., Lin, C. H., Parekh, B. S., Ronco, L. V., Howley, P. M. & Maniatis, T. (1998) Mol. Cell 1, 507-518. [DOI] [PubMed] [Google Scholar]
- 64.Sasahara, Y., Mutoh, M., Takahashi, M., Fukuda, K., Tanaka, N., Sugimura, T. & Wakabayashi, K. (2002) Cancer Lett. 177, 155-161. [DOI] [PubMed] [Google Scholar]