

Abstract
Multicomponent therapies, originating through deliberate mixing of drugs in a clinical setting, through happenstance, and through rational design, have a successful history in a number of areas of medicine, including cancer, infectious diseases, and CNS disorders. We have developed a high-throughput screening method for identifying effective combinations of therapeutic compounds. We report here that systematic screening of combinations of small molecules reveals unexpected interactions between compounds, presumably due to interactions between the pathways on which they act. Through systematic screening of ≈120,000 different two-component combinations of reference-listed drugs, we identified potential multicomponent therapeutics, including (i) fungistatic and analgesic agents that together generate fungicidal activity in drug-resistant Candida albicans, yet do not significantly affect human cells, (ii) glucocorticoid and antiplatelet agents that together suppress the production of tumor necrosis factor-α in human primary peripheral blood mononu-clear cells, and (iii) antipsychotic and antiprotozoal agents that do not exhibit significant antitumor activity alone, yet together prevent the growth of tumors in mice. Systematic combination screening may ultimately be useful for exploring the connectivity of biological pathways and, when performed with reference-listed drugs, may result in the discovery of new combination drug regimens.
Modern biological research and much of drug discovery is often driven by the search for new molecularly targeted therapeutics (1–3). In this approach, a specific protein is studied in vitro, in cells and in whole organisms, and evaluated as a drug target for a specific therapeutic indication (3, 4). The refinement of this approach has resulted in the ability to discover compounds with great selectivity for a chosen protein target. The recent success of Gleevec (imatinib mesylate), an inhibitor of the breakpoint cluster region-abelson (BCR-ABL) kinase, and of selective cyclooxygenase-2 (COX-2) inhibitors Vioxx (rofecoxib) and Celebrex (celecoxib) are evidence that the target-based approach can be successful (5, 6).
Systems biology, however, has revealed that human cells and tissues are composed of complex, networked systems with redundant, convergent and divergent signaling pathways (7–10). For example, the redundant function of proteins involved in cell-cycle regulation (11) has inspired efforts to intervene simultaneously at multiple points in these signaling pathways (12). A drug discovery approach consonant with this systems biology framework, and complementary to the target-based approach, entails identification of combinations of small molecules that perturb cellular signaling networks in a desired fashion.
Recognition of the potential for multipoint intervention in biology and medicine has a long history. As early as 1928, Loewe (13) observed and quantified effects of combinations of compounds that were different from, and not predicted by, the activities of the constituents. The concepts of synergy, additivism, and antagonism have been explored extensively, particularly in the fields of pharmacology and toxicology (14–17). Moreover, patients with infectious diseases and with cancer have benefited from combination chemotherapy, where combination drugs are in many cases the standard of care (18, 19). This clinical experience has led to the testing of combinations of drugs in patients as an explicit strategy for drug improvement by physicians (18, 20). However, this clinical mixing, and its in vitro surrogate, has generally been conducted with agents already known to be effective in the therapeutic area of interest, or where there is a clear rationale for the combination. Such limited combination testing samples only a tiny fraction of combination space and is unlikely to have resulted in the selection of optimal combinations among the very large number of possibilities. A comparatively small number of compounds will provide a very large number of combinations; a collection of 1,000 compounds yields ≈500,000 pairwise combinations, and many more higher-order combinations. Moreover, variations in molar ratio and timing of compound addition can be relevant and increase the size of the search space. Therefore, whereas combinatorial diversity is valuable in that it allows for the simultaneous use of multiple probe molecules, an efficient screening method is needed. We report herein the development and application of such a method.
Methods
Combination High-Throughput Screening (cHTS) Procedure. Compounds were stored as dry powders at the temperature specified by the supplier. Before screening, compounds were weighed into polypropylene vials and dissolved in DMSO when possible, and into distilled, deionized water when necessary. Compound solutions were transferred into polypropylene 384-well master plates containing an appropriate volume of diluent (generally DMSO) by using a Perkin–Elmer Life Sciences (Boston) Multiprobe. Compounds were then serially diluted left to right, or top to bottom by using a Perkin–Elmer MiniTrak. Compounds were transferred to a dilution daughter plate and mixed to create a diluted compound stock in the appropriate assay buffer. All dilution and assay plates [Nunc (Rochester, NY) polystyrene, untreated or tissue-culture treated, as required] were filled by using an Apogent (Hudson, NH) PlateMate 384-well pipettor. Aliquots from two or more compound dilution plates were then dispensed to the final combination assay plate by using custom protocols driving a MiniTrak. Cells were added to the assay plate postcompound addition by using a Thermo Labsystems Multidrop (Ventraa, Finland) in a biosafety cabinet. Cells and compounds were then incubated at the temperature and CO2 concentration specified for each assay. Postincubation processing of the assay plates included direct readout of cell number by using a BMG (Offenberg, Germany) NEPHELOstar and the addition of a detection reagent and further incubation before fluorescence detection by using a Wallac (Gaithersburg, MD) Victor V plate reader. In the case of immunoinflammatory screening, assay plates were spun and supernatants transferred to a protein-binding plate for incubation before ELISA processing and detection of time-resolved fluorescence by using a Victor V plate reader.
Candida albicans Proliferation Assay. In these experiments, amphotericin B (Sigma) was used as a positive control for antifungal activity. Stock solutions of each compound were prepared and stored at -20°C. Before use, stock solutions were diluted into growth medium to produce 10× solutions. A clinical isolate (strain 17) of fluconazole-resistant C. albicans was seeded in 384-well plates at a density of 1,000 cells per ml in growth medium (RPMI medium 1640; 2% glucose without bicarbonate or phenol red, buffered with 0.165 M Mops to pH 7.0) with 11% Alamar blue (BioSource International, Camarillo, CA) at 35°C for 16 h. Compounds were tested in a 36-point dose matrix by using a six-point, 4-fold dilution series centered on the empirically derived concentration required for 50% effect (EC50) for each compound in this assay. Alamar blue, which is metabolically reduced by mitochondria to produce a fluorescent dye, was quantified fluorometrically by using a Fusion plate reader (Packard) to determine the percentage of viable cells remaining. Fluconazole (Interchem, Paramus, NJ) and phenazopyridine (Sigma) were additionally tested at the indicated concentrations for their effect on the proliferation of fluconazole-resistant C. albicans (strain 17; Seattle Biomedical Research Institute, Seattle). Results are the average of three measurements.
Colony-Forming Unit (cfu) Assay. Fungicidal activity was measured with a cfu assay. Fluconazole-sensitive C. albicans was seeded at a starting density of 500 cfu/ml in 10 ml of RPMI medium 1640 supplemented with 2% glucose. Cultures were treated as indicated above and grown for 24 h at 32°C with shaking at 250 rpm and the absorbance of each culture was measured. To remove drugs, the cultures were spun down and resuspended in medium lacking drugs. One thousand yeast-sized particles were then plated on sabouraud agar plates with no supplementary compounds and incubated overnight at 32°C. The number of colonies was counted in each case.
Dye Efflux Assay. Fluconazole-resistant C. albicans cultures were pretreated with compound(s) for 30 min at 32°C and then treated with the fluorescent dye rhodamine G for 1 h at 32°C to allow loading of the dye into cells. Cultures were spun down and resuspended in medium alone to remove both unloaded dye and the test compound(s). Cells were allowed to recover without compound(s) present for 2 hat 32°C (resulting in dye efflux in untreated cells) and then diluted into ice-cold PBS to stop dye efflux. Cells were spun down again and resuspended in ice-cold PBS for microscopic analysis. Fluorescence, indicating a loss of dye efflux capability remaining in cells, was detected by using an inverted fluorescence compound microscope (Nikon TS100-F) fitted with a tetramethylrhodamine B isothiocyanate (TRITC) filter set. Images were collected by using a cooled charge-coupled device camera (Spot; Diagnostic Instruments, Sterling Heights, MI) with a fixed set of acquisition parameters.
Tumor Necrosis Factor α (TNF-α) and IFN-γ ELISAs. Primary human white blood cells (purified from whole blood supplied by the Rhode Island Blood Center, Providence, RI) were stimulated with phorbol 12-myristate 13-acetate (PMA) (Sigma) and ionomycin (Sigma) and simultaneously treated with the indicated concentrations of dexamethasone and/or dipyridamole for 18 h. Supernatants were removed and subjected to an ELISA to determine the percent inhibition of cytokine production.
A549 Tumor Cell in Vitro BrdUrd Incorporation Assay. Cell cultures composed of A549 nonsmall cell lung carcinoma cells (21) were treated with the indicated concentrations of chlorpromazine and pentamidine for 90 h and viability was measured by BrdUrd incorporation.
A549 Tumor Cell in Vivo Mouse Xenograft Assay. A549 lung carcinoma cells were injected into severe combined immunodeficient (SCID) mice and allowed to form a tumor with an average volume of 400 mm3. Mice were treated over 14 days with saline alone, 20 mg/kg paclitaxel alone (Monday, Wednesday, and Friday), 20 mg/kg pentamidine alone (daily), 5 mg/kg chlorpromazine alone (daily), or 20 mg/kg pentamidine and 5 mg/kg chlorpromazine (daily). After treatment was discontinued, the mice were monitored until the tumors in the saline-treated mice reached 1,000 mm3 in size.
Results
cHTS. To perform the large number of requisite experiments for combinatorial screening, and because no adequate commercial system exists, we have designed and implemented a custom robotic screening and informatics system. This technology, which we refer to as cHTS, can be applied to two-component or higher-order screening, and incorporates both an efficient experimental strategy and analytic methods to determine whether a beneficial combination interaction occurs between compounds.
Our systematic testing of all pairwise combinations for a compound set begins by defining the activity of each compound as a single agent in the assay system, and then by testing in two groups (active agents and inactive agents) all pairwise combinations of these compounds. Separating the testing of active and inactive compounds makes an efficient and complete search of all pairwise combinations tractable, when combined with an automated robotic screening and informatics system. Inactive compounds, showing no detectable activity as single agents are tested in pools initially (four compounds per pool) and active pools are then deconvoluted to identify the specific pairwise combination with activity of interest. Because these compounds are inactive on their own and because we have observed that active combinations comprising two inactive compounds are infrequent, we can take advantage of the higher efficiency allowed by pooling, without the confusion generated by overlapping activities. Compounds that show detectable activity on their own (active compounds) are more difficult to assess in pools at a single concentration and are best tested at a range of concentrations to identify potency shifts as well as increases in intrinsic activity. We test each active compound against all other compounds (both active and inactive) in dose matrices comprising six concentrations (including zero) for each compound. Thus, our standard screening matrix for active single agents takes place in 36 different wells of a microtiter plate.
We have implemented a number of algorithms for quantifying synergy in our screening experiments. For example, median effect and isobolographic analyses effectively identify combinations in which one drug enhances the potency of the other drug, but these models are not appropriate for combinations in which one drug enhances only the intrinsic activity of the other (15–17, 22, 23). Clinically and mechanistically useful combination interactions may occur through either a shift in potency or an increase in intrinsic activity. Moreover, additivism and synergy can both be useful therapeutically, and are readily distinguished from the vast majority of random combinations that are neither synergistic nor additive.
We employ three standard reference models of additivism to identify synergies. The highest single agent (HSA) model is the larger of the effects produced by each of the combination's single agents at the same concentrations as in the mixture. In contrast, the Bliss additivism model (23) predicts the combined response C for two single compounds with effects A and B is
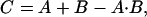 |
where each effect is expressed as fractional inhibition between 0 and 1. These effect-based synergy models make no assumptions about the functional form of the dose-response curves, and do not require dose-response information that lies outside the range sampled by each screening matrix. The third model, Loewe additivity (13), measured by the combination index (15), is dose-based, and applies only to activity levels achieved by the single agents. Compound combinations that show excess inhibition at a chosen significance level over a synergy model are selected for followup. Such candidates are confirmed by using repeat assays on 384-well plates at higher density concentration coverage, and by using other in vitro and in vivo assays.
Inhibiting Growth of Fluconazole-Resistant C. albicans. The identification of azole-enhancing agents and pathways is important for the treatment of fungal infections caused by C. albicans, in part because resistance to fluconazole and crossresistance with triazole antifungal drugs represents a growing clinical problem (24). We applied our cHTS method to screen all pairwise combinations of 30 compounds that inhibit C. albicans proliferation. In the first stage of this experiment, we empirically identified 30 reference-listed drugs (Fig. 1A) with moderate to high activity in a C. albicans antifungal assay by using the viability dye Alamar blue, which is reduced by mitochondrial reductases in live fungal cells. These 30 compounds were identified by testing ≈500 reference-listed drugs at 4 μg/ml for their effect on the viability of proliferating C. albicans in 384-well plates (data not shown). Of these 30 drugs, 9 are currently used as antifungal agents and 21 are used for other indications (Fig. 1A).
Fig. 1.

Multicomponent therapeutics that prevent proliferation of fluconazole-resistant C. albicans. All 435 possible two-component combinations of 30 compounds were tested in duplicate in 36-point dose matrices to identify synergistic combinations. C. albicans cells were treated with compounds in 384-well plates and the extent of inhibition of Alamar blue fluorescence was determined. (A) Overview of synergistic combinations in C. albicans Alamar blue proliferation assay. Yellow columns and rows indicate compounds that are commonly used as antifungal agents. Purple squares indicate combinations for which neither compound is used as an antifungal agent on its own and red indicates combinations for which one, but not both, of the compounds is used as an antifungal agent on its own. (B) A sample combination dose matrix, showing the combined effect of pentamidine and phenazopyridine at six concentrations (including a zero concentration point) each. The experimentally measured inhibition of Alamar blue signal is shown for each pair of combinations. The color of the squares also indicates the level of Alamar blue inhibition. (C) The calculated excess inhibition over the predicted Bliss additivism model. The predicted Bliss additive effect (see text) was subtracted from the experimentally observed inhibition at each pair of concentrations. (D) The calculated excess inhibition over the HSA.
All 435 possible two-component combinations of these 30 active compounds were tested in duplicate in 36-point dose matrices (six doses for each compound, representing 31,320 data points) for their effects on the proliferation of fluconazole-resistant C. albicans (e.g., Fig. 1B). The five test concentrations for each compound were chosen by first determining the EC50 of each compound as a single agent in this assay, and then selecting 4-fold and 16-fold higher and lower concentrations. Each combination was then scored to identify fungistatic or fungicidal effects that were greater than the effects of the individual components by using an Alamar blue proliferation assay. For each combination, we calculated the difference between the observed effect of each combination of doses and the predicted effect based on the three models of additivism described above (Fig. 1 C and D).
This combination screen of active compounds produced 22 pairwise combinations that showed an effect in excess of the HSA model, each for a specific range of compound concentrations, giving an overall HSA hit rate of ≈5%. Of the confirmed active pools, none comprised two known antifungal agents, six comprised antifungal and nonantifungal agents, and 16 comprised two nonantifungal agents (Fig. 1).
Enhancing the Activity of Fluconazole in Fluconazole-Resistant C. albicans. In a related screen, we sought to identify reference-listed compounds that could enhance the activity of fluconazole in fluconazole-resistant C. albicans. By using a similar experimental procedure to that used for the active compound screen (vide supra), we tested fluconazole in combination with 560 compounds, all in 36-point dose matrices, irrespective of their single-agent antifungal activity. This screen produced 20 confirmed pairwise combinations with combined activity in excess of the HSA model that were nonobvious (i.e., the second compound was not a known antifungal compound).
A notable example that emerged from this screen is the combination of fluconazole and phenazopyridine (PAP), which is a urinary tract analgesic (25). This combination of compounds synergistically inhibited the proliferation of otherwise fluconazole-resistant C. albicans (Fig. 2 A–C). The antifungal activity of PAP alone, which is modest, has not, to our knowledge, been previously reported. The combination of fluconazole and PAP was more effective than either compound alone (Fig. 2D). Moreover, a cfu assay determined that whereas fluconazole alone is fungistatic, the combination of PAP and fluconazole is fungicidal (Fig. 2E). The combination of fluconazole and PAP did not significantly affect the proliferation of primary human lung fibroblasts, A549 lung carcinoma cells, or mouse skin fibroblasts (data not shown), indicating that the combination is selective for C. albicans relative to mammalian cells.
Fig. 2.

The combination of fluconazole and phenazopyridine selectively inhibits proliferation of fluconazole-resistant C. albicans.(A) The percent inhibition of fluconazole-resistant C. albicans proliferation is shown for the indicated concentrations of fluconazole and phenazopyridine, determined by using an Alamar blue proliferation assay. The average of three measurements is shown. (B) The calculated excess inhibition over the Bliss additivism model. The predicted Bliss additive effect (see text) was subtracted from the experimentally observed inhibition at each pair of concentrations. (C) The calculated excess inhibition over the HSA. (D) Percent inhibition of fluconazole-resistant C. albicans proliferation at concentrations showing optimal synergy [250 μM fluconazole (flu) and/or 20 μM phenazolepyridine (PAP)]. (E) Fungicidal activity, determined by using a cfu assay. Fluconazole-resistant C. albicans were treated with 20 μM PAP and/or 250 μM flu, or 4 μM amphotericin B (amp B) as a fungicidal positive control, and in each case an equal number of yeast particles were plated in the absence of any compound. The number of colonies that grew after each treatment regime is indicated. (F) The combination of PAP and flu prevents dye efflux. Fluconazoleresistant C. albicans cells were treated with 20 μM PAP and/or 250 μM flu and the effect on efflux of rhodamine G was determined by using fluorescence microscopy. Phase-contrast images of the yeast cells are shown in each case to confirm that a large number of cells were present in each case, but that dye efflux was only effectively inhibited when both PAP and flu were present. For all numerical figures shown, the average of three measurements is represented. Error bars, 1 SD.
To test the possibility that this combination inhibits fungal cells' ability to eliminate fluconazole through multidrug resistance pumps or Candida drug resistance pumps, we performed dye efflux experiments. Fluconazole-resistant C. albicans cultures were pretreated with compound(s), treated with the dye rhodamine G, washed, and allowed to recover without compound(s) present. The ability of compound(s) to prevent rhodamine G efflux was determined by measuring the remaining fluorescence in fungal cells. Whereas fluconazole or PAP alone had little effect on dye efflux, the combination efficiently prevented dye efflux (Fig. 2F). This result demonstrates that the two compounds together affect membrane pump activity, either directly, or indirectly by disrupting mitochondrial ATP synthesis, even though neither agent on its own has such an effect.
Cytokine Modulation in Primary Human Blood Cells. As part of our program to identify anti-inflammatory combination drugs that make use of multipoint intervention mechanisms, we created a phenotypic assay system that examines both intercellular and intracellular signaling networks and adapted this assay to cHTS. In the primary screen, we monitored the production of the immunostimulatory cytokine TNF-α in primary human blood cells in response to various methods of stimulation, including stimulation with lipopolysaccharide (LPS), and with PMA and ionomycin.
By using the described cHTS method (vide supra), we tested 20,000 combinations from a set of 600 approved drugs. Twenty-six of the most interesting combinations were confirmed by using higher-density 100-point dose matrices. One of the interesting combination effects we discovered was that the antiplatelet agent dipyridamole, when used in conjunction with the glucocorticoid dexamethasone, effectively prevents TNF-α production in response to PMA/ionomycin stimulation (Fig. 3 A and B). The TNF-α-suppressive activity of steroids is well documented (26–28) and it has been noted that dipyridamole exhibits TNF-α suppressive effects, possibly through potentiation of adenosine-mediated action at the adenosine A2a receptor (29–32). However, their combined action in vitro has not been described, nor would knowledge of their proposed modes of action have predicted that in combination they would yield the observed interaction effect (Fig. 3 A and B). In fact, many other combinations of singly active TNF-α-suppressing compounds were tested and few exhibited the steroid-sparing profile of dipyridamole (data not shown).
Fig. 3.

Corticosteroids combined with dipyridamole (DP) selectively inhibit cytokine production. (A) Percent inhibition of TNF-α production in stimulated human peripheral blood cells (average of two measurements) using dexamethasone and dipyridamole at the indicated concentrations. (B) Excess inhibition over HSA for dipyridamole and dexamethasone. (C and D) Dipyridamole and dexamethasone inhibit production of TNF-α (C), but not IFN-γ (D). As a comparison, two steroids, fludrocortisone and prednisolone (E), were tested in combination for their effect on TNF-α production. In each case, the dilution factor is calculated based on the highest concentration tested [3 μM DP, 0.25 μM dexamethasone, 1 μM fludrocortisone (FLU), and 1 μM prednisolone (PRED)] for each agent, selected based on the empirically determined dose curves.
An important feature of dipyridamole's action is that it enhances the glucocorticoid-mediated inhibition of TNF-α, but not of all cytokines (such as IFN-γ or IL-2; Fig. 3 C and D), nor does it significantly reduce cell viability (data not shown). Moreover, combinations of steroids alone do not result in TNF-α inhibition beyond a plateau of maximal effect (Fig. 3E), illustrating that some compounds are limited by a maximum achievable inhibition. A combination that increases TNF-α inhibition beyond the maximum level achieved by a glucocorticoid may be mechanistically and therapeutically valuable.
Inhibiting Proliferation of Cancer Cells. To determine whether multicomponent therapeutics discovered in vitro through cHTS could be effective in vivo, we first performed an antiproliferative screen to identify compound combinations that selectively kill, or prevent the proliferation, of human tumor cells in vitro. Using our cHTS method (vide supra), we tested 100,000 combinations from a set of ≈600 approved drugs. We identified and confirmed 13 synergistic combinations, after eliminating 28 synergistic combinations in which both compounds were known antineoplastics drugs.
One of these combinations, comprising the antipsychotic agent chlorpromazine and the antiprotozoal agent pentamidine, was selected for further testing in vivo. Both of these compounds have moderate antiproliferative activities on their own in vitro in A549 lung carcinoma cells (Fig. 4 A–C), but neither demonstrates substantial activity at concentrations that would be considered clinically relevant in vitro or in vivo, and neither is currently used as an anticancer drug (Fig. 4 A–C). In combination, however, these compounds prevented the growth of A549 lung carcinoma cells in vitro (Fig. 4 A–C) and in vivo in a human tumor xenograft assay in mice (Fig. 4D). The combination was even more effective than paclitaxel, a clinically used anticancer drug, at concentrations of the two drugs that correspond to clinically achievable plasma concentrations (Fig. 4D). This growth inhibition occurred without loss of body weight or other visible behavior changes characteristic of mice treated with standard chemotherapeutic drugs (data not shown). Additional in vitro testing in a normal lung fibroblast cell line (Fig. 4E) has demonstrated that this combination exhibits greater antiproliferative activity in A549 tumor cells than in cells derived from normal lung tissue. Together, these data indicate that the combination may possess a therapeutically useful tumor-selective activity.
Fig. 4.

Chlorpromazine, an antipsychotic agent, and pentamidine, an antiprotozoal agent, together selectively prevent tumor cell growth in vitro and in vivo. (A) Percent inhibition of A549 proliferation, measured by using a BrdUrd incorporation assay (average of two measurements). (B and C) Excess antiproliferative activity over the Bliss additivism model (B) and the HSA (C). The predicted Bliss additive (B) or HSA (C) level of inhibition was subtracted from the level observed in A and plotted to indicate concentration regimes in which synergy is observed. (D) Effect of chlorpromazine and pentamidine on the growth of A549 lung carcinoma cell tumors in SCID mice. Tumor size as a function of time is plotted for each cohort and is treated as indicated. (E) The antiproliferative effect of the combination of chlorpromazine (chlor) and pentamidine (pent) was measured in human lung carcinoma cells (A549; ref. 21), in colon carcinoma cells (HCT116), and in normal human lung fibroblasts by using Alamar blue (46) to illustrate that higher concentrations are required for inhibition of normal fibroblasts relative to carcinoma cells.
We sought to determine whether any of the previously described mechanisms and targets for pentamidine or chlorpromazine were relevant to the activity of this combination in the A549 proliferation assay. For chlorpromazine, the primary mechanism of action is dopamine receptor antagonism, but other ascribed mechanisms include α-adrenergic receptor antagonism, histamine receptor antagonism, and calmodulin inhibition (33, 34). Our experiments (data not shown) confirmed the previously reported observation that at high concentrations, the phenothiazines (including chlorpromazine) have antiproliferative activity as single agents, and that they can enhance certain existing chemotherapeutic agents, including radiomimetic drugs (35–40). However, as described below, the mechanism by which chlorpromazine acts to enhance pentamidine in A549 cells is distinct from its reported behavior with bleomycin, consistent with the idea that understanding the actions of single compounds is insufficient to predict fully their behavior in combination. Compounds that antagonize calmodulin (calmidazolium chloride and pimozide, but not trifluoperazine) mimic the effect of chlorpromazine in terms of single-agent activity and cooperativity with pentamidine in our assay system (data not shown). In contrast, compounds that antagonize dopamine receptors (pergolide, SKF-83566, and risperidone), adrenergic receptors (atenolol, dihydroergotamine, and imipramine) or histamine receptors (cimetidine, diphenhydramine, meclizine, and loratadine) had no activity as single agents or in combination with pentamidine in our assay system, suggesting that these receptors play no role in the antiproliferative activity of chlorpromazine.
For pentamidine, previously described mechanisms include DNA binding and consequent inhibition of DNA synthesis (41, 42), modulation of polyamine levels (43), and calmodulin antagonism (44). Whereas pentamidine is known to possess DNA minor groove-binding activity (42, 45), our studies indicate that it does not behave as a classical DNA-damaging agent in this A549 proliferation assay, nor simply as an inhibitor of DNA repair. To ascertain whether these known mechanisms are relevant to the ability of pentamidine to inhibit tumor cell proliferation in combination with chlorpromazine, we tested the effects of other compounds that operate by each of these mechanisms on tumor cell proliferation in the presence and absence of chlorpromazine (data not shown). DNA-binding agents (DAPI and WP631, but not netropsin) were active as single agents and cooperated with chlorpromazine in our assay system. Calmodulin inhibitors (calmidazolium chloride, pimozide, and trifluoperazine) were also active as single agents and cooperated with chlorpromazine in our assay system. Somewhat surprisingly, a modulator of polyamine levels (N,N-diethylspermine) cooperated with both pentamidine and chlorpromazine in our assay system.
From these results we conclude that (i) DNA binding and inhibition of calmodulin have individual and cooperative antiproliferative effects in A549 cells, (ii) dopamine, histamine, and adrenergic receptor antagonism do not have antiproliferative effects in A549 cells, and (iii) the mechanism underlying the antiproliferative effect of the combination of chlorpromazine and pentamidine is likely to involve in part, calmodulin inhibition, DNA binding, modulation of polyamine levels, and possibly other mechanisms.
Discussion
Using our cHTS technology to screen two-component combinations of reference-listed drugs in cell-based assays relevant to disease, we observed unexpected synergistic interactions that may be attributable to the interconnected signaling networks existing within and between cells. Our screening approach is complementary to other methods for exploring systems biology and protein networks and may be useful for determining connections between pathways that are functionally important. Because reference-listed drugs are well studied in terms of their molecular mechanisms and targets, these compounds may aid in understanding the underlying biological pathways that may be affected by combination treatment. The combinations identified may also serve as multicomponent drugs that are appropriate for advancement to clinical development.
In some instances, a synergistic combination that we, to our knowledge, discovered empirically will have relevant citations in the literature such that it may appear to have been almost predictable with hindsight. However, many other equivalent predictions would also be made from the literature and these usually do not result in synergy when tested experimentally. In this sense, biological literature contains many anticipatory observations and speculations regarding connections between pathways (and compounds), most of which do not ultimately prove to be synergistic when tested directly.
This approach might be expanded beyond small molecules to other methods of perturbation such as cDNA overexpression or RNA interference-based messenger RNA knockdown. Such systematic combination perturbations may ultimately help define the interactions among human genes and proteins, and the reagents that act on them, that elicit nonlinear and nonadditive phenotypic outcomes. A practical application of this cHTS methodology is the creation of combination drugs through systematic screening of compounds in disease-relevant phenotypic assays. Mining the combinatorics of multicomponent effects promises to result in rapid production of medicines and will define an approach to the systematic discovery of therapeutic agents.
Acknowledgments
We thank Joshua Lederberg and Jacob Goldfield for valuable advice.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: cHTS, combination high-throughput screening; cfu, colony-forming unit; HSA, highest single agent; PAP, phenazopyridine; TNF-α, tumor necrosis factor α.
References
- 1.Mokbel, K. & Hassanally, D. (2001) Curr. Med. Res. Opin. 17, 51-59. [DOI] [PubMed] [Google Scholar]
- 2.Shawver, L. K., Slamon, D. & Ullrich, A. (2002) Cancer Cell 1, 117-123. [DOI] [PubMed] [Google Scholar]
- 3.Gibbs, J. B. (2000) Science 287, 1969-1973. [DOI] [PubMed] [Google Scholar]
- 4.Lenz, G. R., Nash, H. M. & Jindal, S. (2000) Drug Discovery Today 5, 145-156. [DOI] [PubMed] [Google Scholar]
- 5.Druker, B. J. (2002) Trends Mol. Med. 8, S14-S18. [DOI] [PubMed] [Google Scholar]
- 6.Kalgutkar, A. S., Crews, B. C., Rowlinson, S. W., Marnett, A. B., Kozak, K. R., Remmel, R. P. & Marnett, L. J. (2000) Proc. Natl. Acad. Sci. USA 97, 925-930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitano, H. (2002) Science 295, 1662-1664. [DOI] [PubMed] [Google Scholar]
- 8.Kanehisa, M., Goto, S., Kawashima, S. & Nakaya, A. (2002) Nucleic Acids Res. 30, 42-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blume-Jensen, P. & Hunter, T. (2001) Nature 411, 355-365. [DOI] [PubMed] [Google Scholar]
- 10.Brent, R. (2000) Cell 100, 169-183. [DOI] [PubMed] [Google Scholar]
- 11.Zhang, P. (1999) Curr. Opin. Cell Biol. 11, 655-662. [DOI] [PubMed] [Google Scholar]
- 12.Shaheen, R. M., Tseng, W. W., Davis, D. W., Liu, W., Reinmuth, N., Vellagas, R., Wieczorek, A. A., Ogura, Y., McConkey, D. J., Drazan, K. E., et al. (2001) Cancer Res. 61, 1464-1468. [PubMed] [Google Scholar]
- 13.Loewe, S. (1928) Ergen. Physiol. 27, 47-187. [Google Scholar]
- 14.Chou, T.-C. & Talalay, P. (1983) Trends Pharmacol. Sci. 4, 450-454. [Google Scholar]
- 15.Chou, T.-C. & Talalay, P. (1984) Adv. Enzyme Regul. 22, 27-55. [DOI] [PubMed] [Google Scholar]
- 16.Berenbaum, M. C. (1989) Pharmacol. Rev. 2, 93-141. [PubMed] [Google Scholar]
- 17.Loewe, S. (1953) Arzneim.-Forsch. 3, 285-290. [PubMed] [Google Scholar]
- 18.DeVita, V. T., Jr. (1997) in Principles of Cancer Management: Chemotherapy, eds. DeVita, V. T., Jr., Hellman, S. & Rosenberg, S. A. (Lippincott, Philadelphia), pp. 333-347.
- 19.Gilbert, D. N. (2002) Sanford Guide to Antimicrobial Therapy (Antimicrobial Therapy, Vienna).
- 20.Di Leo, A. & Piccart, M. J. (1999) Semin. Oncol. 26, 27-32. [PubMed] [Google Scholar]
- 21.Lieber, M., Smith, B., Szakal, A., Nelson-Rees, W. & Todaro, G. (1976) Int. J. Cancer 17, 62-70. [DOI] [PubMed] [Google Scholar]
- 22.Fraser, T. R. (1871) Proc. Roy. Soc. Edinburgh 7, 506-511. [Google Scholar]
- 23.Berenbaum, M. C. (1981) Adv. Cancer Res. 35, 269-335. [DOI] [PubMed] [Google Scholar]
- 24.Rex, J. H., Rinaldi, M. G. & Pfaller, M. A. (1995) Antimicrob. Agents Chemother. 39, 1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zelenitsky, S. A. & Zhanel, G. G. (1996) Ann. Pharmacother. 30, 866-868. [DOI] [PubMed] [Google Scholar]
- 26.Joyce, D. A., Steer, J. H. & Abraham, L. J. (1997) Inflamm. Res. 46, 447-451. [DOI] [PubMed] [Google Scholar]
- 27.Wershil, B. K., Furuta, G. T., Lavigne, J. A., Choudhury, A. R., Wang, Z. S. & Galli, S. J. (1995) Int. Arch. Allergy Immunol. 107, 323-324. [DOI] [PubMed] [Google Scholar]
- 28.Joyce, D. A., Gimblett, G. & Steer, J. H. (2001) Inflamm. Res. 50, 337-340. [DOI] [PubMed] [Google Scholar]
- 29.Le Vraux, V., Chen, Y. L., Masson, I., De Sousa, M., Giroud, J. P., Florentin, I. & Chauvelot-Moachon, L. (1993) Life Sci. 52, 1917-1924. [DOI] [PubMed] [Google Scholar]
- 30.Eigler, A., Greten, T. F., Sinha, B., Haslberger, C., Sullivan, G. W. & Endres, S. (1997) Scand. J. Immunol. 45, 132-139. [DOI] [PubMed] [Google Scholar]
- 31.Hasko, G., Kuhel, D. G., Chen, J. F., Schwarzschild, M. A., Deitch, E. A., Mabley, J. G., Marton, A. & Szabo, C. (2000) FASEB J. 14, 2065-2074. [DOI] [PubMed] [Google Scholar]
- 32.Konstantinov, K., Galabov, A. & Mastikova, M. (1989) Br. J. Dermatol. 121, 59-63. [DOI] [PubMed] [Google Scholar]
- 33.Hardman, J. G. & Limbird, L. E. (2001) Goodman and Gilman's The Pharmacological Basis of Therapeutics (McGraw–Hill, New York), 10th Ed.
- 34.Marshak, D. R., Lukas, T. J. & Watterson, D. M. (1985) Biochemistry 24, 144-150. [DOI] [PubMed] [Google Scholar]
- 35.Nordenberg, J., Fenig, E., Landau, M., Weizman, R. & Weizman, A. (1999) Biochem. Pharmacol. 58, 1229-1236. [DOI] [PubMed] [Google Scholar]
- 36.Darkin, S., McQuillan, J. & Ralph, R. K. (1984) Biochem. Biophys. Res. Commun. 125, 184-191. [DOI] [PubMed] [Google Scholar]
- 37.Lialiaris, T., Pantazaki, A., Sivridis, E. & Mourelatos, D. (1992) Mutat. Res. 265, 155-163. [DOI] [PubMed] [Google Scholar]
- 38.Lehnert, S. (1986) Cancer Chemother. Pharmacol. 16, 269-272. [DOI] [PubMed] [Google Scholar]
- 39.Lazo, J. S., Chen, D. L., Gallicchio, V. S. & Hait, W. N. (1986) Cancer Res. 46, 2236-2240. [PubMed] [Google Scholar]
- 40.Hait, W. N., Lazo, J. S., Chen, D. L., Gallichio, V. S. & Filderman, A. E. (1988) J. Natl. Cancer Inst. 80, 246-250. [DOI] [PubMed] [Google Scholar]
- 41.Lowe, P. R., Sansom, C. E., Schwalbe, C. H. & Stevens, M. F. G. (1989) J. Chem. Soc. Chem. Commun. 16, 1164-1165. [Google Scholar]
- 42.Cory, M., Tidwell, R. R. & Fairley, T. A. (1992) J. Med. Chem. 35, 431-438. [DOI] [PubMed] [Google Scholar]
- 43.Basselin, M., Badet-Denisot, M. A., Lawrence, F. & Robert-Gero, M. (1997) Exp. Parasitol. 85, 274-282. [DOI] [PubMed] [Google Scholar]
- 44.Kitamura, Y., Arima, T., Imaizumi, R., Sato, T. & Nomura, Y. (1995) Eur. J. Pharmacol. 289, 299-304. [DOI] [PubMed] [Google Scholar]
- 45.Perez, J. M., Requena, J. M., Craciunescu, D., Doadrio, J. C. & Alonso, C. (1993) Chem. Biol. Interact. 89, 61-72. [DOI] [PubMed] [Google Scholar]
- 46.Jacobs, J. P., Garrett, A. J. & Merton, R. (1979) J. Biol. Stand. 7, 113-122. [DOI] [PubMed] [Google Scholar]