

Abstract
Mitochondria are the primary site of skeletal muscle fuel metabolism and ATP production. Although insulin is a major regulator of fuel metabolism, its effect on mitochondrial ATP production is not known. Here we report increases in vastus lateralis muscle mitochondrial ATP production capacity (32–42%) in healthy humans (P < 0.01) i.v. infused with insulin (1.5 milliunits/kg of fat-free mass per min) while clamping glucose, amino acids, glucagon, and growth hormone. Increased ATP production occurred in association with increased mRNA levels from both mitochondrial (NADH dehydrogenase subunit IV) and nuclear [cytochrome c oxidase (COX) subunit IV] genes (164–180%) encoding mitochondrial proteins (P < 0.05). In addition, muscle mitochondrial protein synthesis, and COX and citrate synthase enzyme activities were increased by insulin (P < 0.05). Further studies demonstrated no effect of low to high insulin levels on muscle mitochondrial ATP production for people with type 2 diabetes mellitus, whereas matched nondiabetic controls increased 16–26% (P < 0.02) when four different substrate combinations were used. In conclusion, insulin stimulates mitochondrial oxidative phosphorylation in skeletal muscle along with synthesis of gene transcripts and mitochondrial protein in human subjects. Skeletal muscle of type 2 diabetic patients has a reduced capacity to increase ATP production with high insulin levels.
Keywords: cytochrome c oxidase, NADH dehydrogenase subunit IV, amino acids, citrate synthase
Insulin is the key postprandial hormone involved in fuel metabolism (1). Mitochondria are the major functional components of cellular fuel oxidation and ATP production (2). However, the effect of insulin on skeletal muscle mitochondrial function and oxidative capacity have not been defined. Previous studies have suggested a relationship between insulin action and the oxidative capacity of skeletal muscle (3–10). For example, the ratio between oxidative and glycolytic enzyme activities is decreased in skeletal muscle from type 2 diabetic subjects compared with nondiabetic subjects (6). A recent report by Boirie et al. (11) on miniature swine has also demonstrated that insulin infusion preferentially stimulates the fractional synthesis rate (FSR) of skeletal muscle mitochondrial proteins. In contrast, 2 wk of insulin treatment did not stimulate muscle mitochondrial protein synthesis and cytochrome c oxidase (COX) activity in people with type 2 diabetes mellitus (DM) (12). Skeletal muscle insulin resistance in people with type 2 DM is associated with a reduced proportion of fatigue resistant slow-oxidative (type I) muscle fibers compared with fatigable fast-glycolytic muscle fibers (type II) (7, 8).
Mitochondrial ATP production depends on many factors, including the availability of enzyme complexes, ADP, and fuel oxidation. Enzymes constitute major components of the mitochondrial protein complex, and protein synthesis depends on well coordinated transcriptional regulation of both nuclear and mitochondrial genes (13). The current study tested the hypothesis that insulin enhances skeletal muscle capacity for mitochondrial ATP production. Therefore, we examined healthy subjects during 8 h of euglycemic i.v. insulin to high physiological levels comparable to peak postprandial concentrations. Mitochondrial ATP production, protein synthesis, and mRNA transcripts encoding key mitochondrial proteins from both the nuclear and mitochondrial genomes were measured from vastus lateralis muscle biopsy samples to define underlying mechanisms. After demonstrating the effects of sustained high physiological insulin on muscle mitochondrial function, the study was extended to men and women with type 2 DM to determine whether the enhancement of ATP production by insulin is reduced compared with non-DM matched controls.
Materials and Methods
Subjects. All studies were performed at the Mayo Medical Center General Clinical Research Center. Initial studies involved healthy human volunteers. These individuals were physically active, being involved in moderate-intensity activities (e.g., daily walking), but none were highly trained, participating infrequently (<60 min/wk) in higher-intensity exercise or recreation (e.g., running). Healthy subject characteristics are shown in Table 1. These volunteers were without personal or family history (first-degree relative) of DM. Subjects with type 2 DM and matched controls without DM or a family history of DM were recruited for additional studies as described below. Fat mass and fat-free mass (FFM) were measured by using dual x-ray absorptiometry (Lunar DPX-IQ, Madison, WI).
Table 1. Subject characteristics.
| Group
|
||||
|---|---|---|---|---|
| Characteristic | High insulin/saline | Low insulin | Type 2 DM | Matched control |
| Age, y | 31.5±3.8 | 31.0±9.3 | 58.9±9.5 | 57.1±10.1 |
| Weight, kg | 68.8±11.4 | 76.0±7.3 | 76.6±9.8 | 79.7±13.3 |
| BMI, kg/m2 | 23.1±2.3 | 25.0±1.3 | 27.3±4.0 | 26.2±2.7 |
| Fat, % | 30.4±7.1 | 33.4±7.8 | 34.3±10.9 | 37.7±9.9 |
| FFM, kg | 47.8±9.5 | 48.4±8.6 | 31.5±3.8 | 31.5±3.8 |
| Waist, cm | — | — | 97.6±10.5 | 92.6±12.2 |
| Citrate synthase, μmol/min per g | 18.0±3.7 | 21.8±7.8 | 17.3±4.7 | 16.8±3.1 |
| COX, μmol/min per g | 16.7±5.4 | 14.5±6.7 | 5.9±2.1 | 9.5±5.4 |
Values are means ± SD. There were no statistically significant differences between the subjects receiving the high physiological or low-dose insulin infusions (P > 0.05). There were no significant differences between patients with type 2 DM and matched control subjects (P > 0.05). BMI, body mass index. Enzyme specific activities are presented in terms of g of wet weight
Materials. l-[1,2-13C]Leucine (99 mol % enriched) was purchased from Isotec (Miamisburg, OH) and Mass Trace (Woburn, MA). The isotopic and chemical purity were checked by gas chromatography/MS. Tracer solutions were tested for sterility and pyrogenicity and were prepared in sterile water. Humulin R insulin (Lilly, Indianapolis) was used for insulin-glucose clamps. Human growth hormone (GH) was obtained from Genentech (South San Francisco, CA), glucagon was from Lilly Research Laboratories, and somatostatin was from Bachem (Torrance, CA). A crystalline amino acid solution of 10% Travasol was obtained from Baxter Healthcare (Deerfield, IL).
Study Protocol. The protocol was approved by the Mayo Clinic Institutional Review Board. Six subjects (three male and three female) participated with informed consent on two occasions in a randomized, placebo-controlled, cross-over design. They were admitted to the Mayo Medical Center General Clinical Research Center (GCRC) at 1700 hours on two separate occasions. Female participants were studied in the luteal phase of their menstrual cycle. On each admission, subjects ingested a standard meal at 1800 hours and snack at 2200 hours. Thereafter, they remained fasting until the end of the inpatient period. All subjects were on a standard weight-maintaining diet (carbohydrate/protein/fat, 55:15:30% by calories) provided from the Mayo Medical Center GCRC for 3 consecutive days before each inpatient study period. At 0400 hours of each inpatient period, a priming dose of l-[1,2-13C]leucine (1.8 mg/kg of FFM) was administered through a peripheral forearm vein. This was followed by a continuous infusion of l-[1,2-13C]leucine (1.8 mg/kg of FFM per h). Insulin [1.5 milliunits (mU)/kg of FFM per min] or normal saline infusion was randomized and began at 0700 hours. During the insulin infusion, somatostatin (7 μg/kg of FFM per h) was infused to suppress endogenous insulin production, and glucagon (1 ng/kg of FFM per min), and GH (5 ng/kg of FFM per min) were replaced. In addition, a standard amino acid solution (10% Travasol) was infused (0.6 μmol of leucine per kg of FFM per min) to maintain leucine concentrations near fasting levels during insulin infusion (14, 15). Arterialized blood glucose was measured every 10 min with a Beckman glucose analyzer (Fullerton, CA). Glucose (40% solution) infusion rate was adjusted to maintain euglycemia during high physiological insulin.
Another group of six healthy subjects (Table 1) was tested identically except under conditions of low-dose insulin infusion (0.15 mU/kg of FFM per min). Somatostatin, glucagon, GH, and amino acid tracer infusion rates were identical to above. Amino acid (10% Travasol) replacement was not required to maintain leucine concentrations.
Patients with Type 2 DM. Studies were extended to patients with type 2 DM (n = 9; six female and three male) and control subjects (n = 9) matched for age, sex, activity, body mass, and composition (Table 1). Type 2 DM was verified by clinical history, fasting glucose, and oral glucose tolerance testing. Each volunteer was admitted to the Mayo Medical Center General Clinical Research Center on two occasions after standardized diets as described above. All oral hypoglycemic, insulin-sensitizing drugs and long-acting insulins were discontinued 10 days before admission. Each subject was studied under conditions of low-dose (0.25 mU/kg of FFM per min) insulin and high-dose (1.5 mU/kg of FFM per min) insulin infusion for 7 h (0600–1300 hours) on separate days. Somatostatin was infused and GH was replaced as above. Euglycemia was maintained by glucose (40% solution) infusion when necessary. Replacement amino acids were not infused.
Muscle Biopsies. Vastus lateralis muscle samples (≈300 mg each) were obtained under local anesthesia (lidocaine, 2%), with a percutaneous needle as described (16). Baseline samples from the healthy subjects were obtained before insulin or saline infusion at 0700 hours. Samples were also obtained after 4 h (1100 hours) and from the contralateral thigh at 8 h (1500 hours). Muscle samples were immediately frozen in liquid nitrogen and kept at -80°C. A 50-mg fresh muscle sample was used to measure mitochondrial ATP production. A fresh 50-mg sample of vastus lateralis muscle was also obtained from DM patients and matched control subjects after 7 h (1300 hours) of insulin to measure mitochondrial ATP production.
FSR of Muscle Proteins. Muscle biopsy samples were prepared for determining synthesis rates of muscle mitochondrial protein fractions as described (16–19). Isotopic enrichment of [13C]leucine from the muscle fractions was measured by using modified gas chromatography/on-line combustion/isotope ratio MS as described (17). Muscle tissue fluid leucine enrichment was also measured by using MS (20).
The FSRs for mitochondrial protein fraction were calculated by using the following equation (16, 21):
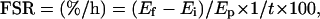 |
where Ef and Ei represent the enrichments as atom percent excess of 13C derived from the combustion of muscle fraction leucine obtained from the 8-h and 4-h muscle biopsies, respectively. Ep is the precursor pool (tissue fluid leucine) enrichment, and t represents the time between biopsies in hours.
RNA Isolation and cDNA Synthesis. Total RNA was extracted from skeletal muscle tissue (≈20 mg) by the guanidinium method (TriReagent, Molecular Research Center, Cincinnati). Total RNA (1 μg) was treated with DNase (Life Technologies, Gaithersburg, MD) and then reverse transcribed by using the TaqMan reverse transcription reagents (PE Biosystems, Foster City, CA) according to the manufacturer's instruction. Oligo(dT) primers were used.
Real-Time PCR. Primers and probes were selected by using the primer express software (PE Biosystems) and screened for mispriming of isoforms by using the OLIGO PRIMER ANALYSIS software version 5.0 (National Biosciences, Plymouth, MN). The internal probes and the 28S rRNA probe were constructed as described (16). The internal probes were labeled at the 5′-ends with the reporter dye 6′-carboxyfluorescein and at the 3′-end with the quencher dye 6′-carboxytetramethylrhodamine and were phosphate-blocked at the 3′-end to prevent extension. The 28S rRNA probe 5′-end was labeled with a fluorescent dye (PE Biosystems). Taq DNA polymerase allows for the separation of the reporter from the quencher. The resulting fluorescence was measured at each cycle of amplification by the ABI sequence detection system (Perkin–Elmer ABI Prism 7700 RT-PCR).
The probes for the nucleus-encoded COX IV were designed to span an exon. Because the mitochondrial genome does not contain introns, the mitochondrion-encoded NADH dehydrogenase subunit IV (ND IV) and COX III reverse primers were designed to target several final nucleotides specific to the target gene, including a poly(A)+ tail string present only in the RNA. The following primer and probe sequences were used for COX III, COX IV, and ND IV. COX III (GenBank accession no. NC_001807) forward primer: CGCCTGATACTGGCATTTTGT; reverse primer: TTTTTTTTTTTTTTTTTTTTTTTTTAAGACC; and probe TGGTTTGACTATTTCTGTATGTCTCCATCTATTG. COX IV (GenBank accession no. XM 008055) forward primer: CCTCCTGGAGCAGCCTCTC; reverse primer: TCAGCAAAGCTCTCCTTGAACTT; and probe TGCGATACAACTCGACTTTCTCATCCAT. ND IV (GenBank accession no. NC_001807) forward primer: CCCCATTCTCCTCCTATCCC; reverse primer: TTTTTTTTTTTTTTTTTTTTTTTTTTAAGAG; and probe CAACCCCGACATCATTACCGGGT.
The signal for the 28S rRNA was used to normalize against differences in RNA isolation and RNA degradation and in the efficiencies of the reverse transcription and PCRs. The final quantitation was achieved with a relative standard curve (16).
Hormones and Substrates. Plasma concentration of insulin was measured by using a two-site immunoenzymatic assay, and plasma glucagon levels were measured by a direct double-antibody RIA (Linco Research Immunoassay, St. Charles, MO). Human GH was measured with a two-site immunoenzymatic assay performed on the Access automated immunoassay system (Beckman Instruments, Chaska, MN).
Plasma levels of amino acids were measured by an HPLC system (HP 1090, 1046 fluorescence detector and cooling system) with precolumn o-phthalaldehyde derivatization (22). Glucose was analyzed on site by an analyzer by using an enzymatic technique (Beckman Instruments).
Mitochondrial ATP Production. A 50-mg portion of each muscle sample was kept fresh on ice in saline-soaked gauze for mitochondrial studies. Rapid separation of mitochondria by differential centrifugation was performed as described (23, 24). Aliquots of the final mitochondrial suspension were used for measuring mitochondrial ATP production rate with a bioluminescent technique (23, 24). The reaction mixture included a luciferin–luciferase ATP-monitoring reagent (formula SL, BioThema, Dalarö, Finland), substrates for oxidation, and 35 μM ADP. Substrates added (in mM final concentration) were as follows: 1 pyruvate plus 1 malate (PM), 0.05 palmitoyl-L-carnitine plus 1 malate (PCM), 10 glutamate plus 1 malate (GM), 20 succinate plus 0.1 rotenone (SR), 1 pyruvate plus 0.05 palmitoyl-L-carnitine plus 10 α-ketoglutarate plus 1 malate (PPKM), 10 α-ketoglutarate, or 1 N,N,N′,N′-tetramethyl-p-phenylenediamine plus 5 ascorbate (TA), with blank tubes used for measuring background. All reactions for a given sample were monitored simultaneously and calibrated with addition of an ATP standard (BioOrbit 1251 luminometer, Turku, Finland).
Mitochondrial integrity was monitored by measuring citrate synthase activity before and after freeze–thaw membrane disruption and Triton X-100 addition. Accordingly, mitochondria were 94 ± 1% intact with no differences between treatments. Muscle homogenate COX and citrate synthase enzyme activities were measured by using standard spectrophotometric techniques (16, 18).
Statistics. Multivariate repeated-measures ANOVA was used to detect differences between saline and insulin conditions when multiple measurements were compared. When a difference was observed, Duncan's multiple-range post hoc test was used. When single measurements between saline and high insulin conditions or between high and low insulin conditions were compared, paired two-tailed t tests were used. When single measurements between groups (low insulin vs. saline or high insulin, or DM vs. control) were compared, nonpaired two-tailed t tests were used. Statistical significance was set at P < 0.05.
Results
Insulin and Substrates. Subjects were studied during 8 h of insulin and saline infusion (Fig. 1A). During high-dose insulin infusion plasma insulin concentrations (395 ± 12 pmol/liter; mean ± SD) were ≈17-fold higher than during the saline infusion (23 ± 6 pmol/liter). In addition, plasma insulin concentrations (39 ± 3 pmol/liter) were higher in the subjects infused with low-dose insulin than in those infused with saline (P < 0.01). During high-dose insulin infusion plasma glucose was clamped to 5.0 ± 0.2 mmol/liter (mean ± SD), which was slightly higher than the mean glucose concentrations measured during the saline infusion 4.7 ± 0.1 mmol/liter (P < 0.01). Furthermore, low-dose insulin infusion was not adequate to maintain euglycemia, with plasma glucose rising to a peak value of 8.6 ± 0.7 mmol/liter at 3 h of infusion before decreasing to near baseline levels by 8 h (Fig. 1B). No attempts were made to reduce the glucose levels because our objective was to determine the effect of insulin. Moreover, these relatively high glucose levels provided an opportunity to determine whether elevated glucose per se had an effect on the outcome measures in this study. To prevent insulin-induced suppression of circulating amino acids, a standard amino acid replacement solution (10% Travasol) was infused during the high insulin conditions to maintain leucine concentrations near fasting levels. Consequently, there was no difference in plasma leucine levels between the high insulin and saline infusion conditions (Fig. 1C). However, with low-dose insulin infusion plasma leucine values were higher than with high-dose insulin and saline infusions (P < 0.01).
Fig. 1.

Plasma insulin (A), glucose (B), and leucine (C) concentrations during 8 h of high-dose insulin, low-dose insulin, or saline infusion. Values are means ± SD. Significant differences by ANOVA are indicated in text.
Skeletal Muscle Oxidative Capacity. The vastus lateralis muscle citrate synthase activity increased 28% (P < 0.01) after 8 h of high-dose insulin infusion, whereas no change was observed during saline or low-dose insulin (Fig. 2). The citrate synthase activity did not increase after 4 h of high-dose insulin. Likewise (Fig. 2), COX activity increased 29% (P < 0.01) after 8 h of high-dose insulin infusion compared with no change during the saline and low-dose insulin conditions. The changes in COX activity at 4 h of either low- or high-dose insulin infusion were not different from those during saline conditions.
Fig. 2.

Vastus lateralis muscle citrate synthase activity and COX activity after 8 h of saline, low-dose insulin, and high-dose insulin infusion. Values of these mitochondrial enzyme activities are expressed as a percentage of baseline (mean ± SEM). *, Statistically significant difference from the saline condition (P < 0.01); †, significantly different from the low insulin condition (P < 0.01).
Vastus lateralis muscle mitochondrial ATP production rates after 8 h of high-dose insulin infusion were increased 32–42% above baseline (P < 0.01). There was no increase observed during saline or low-dose insulin infusions regardless of the substrate used (Fig. 3). In addition, increases in ATP production observed after 4 h of high-dose insulin infusion (10–16%) were different from changes observed during saline infusion with TA, GM, and PCM substrate combinations and differed from changes observed during low-dose insulin infusion with TA, PM, GM, and PCM combinations (Fig. 3). Changes in mitochondrial ATP production rates during low-dose insulin or saline infusion after 4 or 8 h ranged from -10% to +6% (P > 0.05; Fig. 3).
Fig. 3.

Vastus lateralis muscle mitochondrial ATP production rates after 4 h (A) and 8 h (B) of saline, low-dose insulin, and high-dose insulin infusion. Values are expressed as a percentage of preinfusion baseline (mean ± SEM). Measurements were made in the presence of five different substrate combinations, TA, GM, PM, PCM, or SR. *, Significantly different (P < 0.05) from the saline values; †, significantly different from the low insulin values at 4 h; ‡, significant difference from the saline values and low insulin values at 8 h (P < 0.01).
Protein FSRs. Fig. 4 shows that the FSR of mitochondrial proteins was increased between 4 and 8 h of high-dose insulin infusion compared with saline infusion (20%) measured in the same subjects (P < 0.05). All six subjects showed an increase, although the increase was modest in two of the six. In addition, high-dose insulin infusion resulted in a higher mitochondrial protein FSR (57%) compared with a similar group of subjects during low-dose insulin infusion (Fig. 4). Fig. 4 FSR values use tissue fluid leucine as the precursor pool; however, similar changes were observed by using plasma α-ketoisocaproic acid or plasma leucine (data not shown).
Fig. 4.

Mitochondrial protein FSR for high-dose insulin and saline conditions in the same subjects, and low-dose insulin infusion in a separate group of subjects. Values are mean ± SEM. *, Significantly different from the saline value (P < 0.05); †, significantly different from the low insulin value (P < 0.05).
mRNA Transcript Levels. Fig. 5 shows that ND IV and COX IV mRNA transcript levels normalized for 28S rRNA were increased (164–180%) from baseline (time 0 h) after 8 h of high-dose insulin infusion compared with changes during saline infusion (from -19% to +4%). The increase in COX III mRNA (61%) above baseline was short of statistical significance (P < 0.08) compared with the saline condition. ND IV, COX III, and COX IV mRNA transcripts did not change after 4 h of high-dose insulin infusion, or after 4 or 8 h of low-dose insulin infusion.
Fig. 5.

ND IV (A), COX III (B), and COX IV (C) mRNA transcript levels normalized to 28S rRNA at baseline, 4 and 8 h of high-dose insulin, low-dose insulin, or saline. Values are mean ± SEM. *, Significant increase above baseline for the insulin condition compared with the saline condition (P < 0.05).
Type 2 DM Effects on Mitochondrial ATP Production. Fig. 6A shows the percentage changes in vastus lateralis mitochondrial ATP production from low-dose to high-dose insulin in type 2 DM patients and non-DM controls. Mitochondrial ATP production was increased from low-dose to high-dose insulin infusion in non-DM controls when GM, PPKM, α-ketoglutarate, and PCM were used as substrates (P < 0.02). Mitochondrial ATP production did not change in type 2 DM patients with these substrates. Neither group showed an increase when PM and SR were used as substrates. There were no differences in low- or high-dose insulin levels between type 2 DM patients and non-DM control subjects (Fig. 6B).
Fig. 6.

(A) Vastus lateralis muscle mitochondrial ATP production rates after 7 h of high-dose insulin infusion. Values (mean ± SEM) are expressed as a percentage change from the low-dose insulin condition. Measurements were made in the presence of six different substrate combinations (KG, 10 mMα-ketoglutarate). (B) Insulin concentrations sustained during 7 h of insulin infusion (mean ± SD). *, Significant difference between the low-dose and high-dose insulin conditions (P < 0.02).
Discussion
The current study demonstrates that insulin infusion increases the mitochondrial capacity for oxidative phosphorylation in skeletal muscle. This study also demonstrates in human muscle that mitochondrial protein synthesis is increased by insulin infusion. This increased mitochondrial protein synthesis was associated with increased activities of mitochondrial oxidative enzymes: citrate synthase and COX. In addition, these changes occurred in association with an increase in the muscle transcript levels encoding mitochondrial proteins. Collectively, these findings advance an emerging paradigm that establishes insulin not only as the predominant postprandial anabolic hormone but also as an important regulator of skeletal muscle mitochondrial oxidative phosphorylation. Extending the investigation to type 2 DM patients showed that insulin did not increase muscle mitochondrial ATP production, unlike the significant increase observed in matched control subjects.
Evidence linking insulin action and skeletal muscle oxidative capacity has been previously described but inconclusive. For example, insulin-responsive glucose transporter proteins (GLUT-4) and oxidative enzyme activities increase in parallel with exercise training in rat (25, 26) and human (27) skeletal muscle. In addition, direct analysis of human skeletal muscle by using immunohistochemistry and stereologic techniques show that type I fibers, which by definition have relatively high oxidative enzyme capacities and are fatigue resistant, have more GLUT-4 transporters than do type II fibers (28). Furthermore, insulin-resistant muscles from type 2 DM patients have lower percentages of type I fibers and lower density of GLUT-4 protein in type I fibers (29). Moreover, increased capillaries around all muscle fibers, especially type 2 fibers, are reported to predict progression to DM in people with impaired glucose tolerance (30). Lower oxidative capacities, as measured by enzyme activities (6, 10, 31) and rates of fatty acid oxidation (32) have been observed in insulin-resistant skeletal muscle. Expression of mRNA transcripts encoding mitochondrial proteins has also been predictive of skeletal muscle insulin sensitivity. Huang et al. (33) found decreased levels of ND subunit I mRNA expression in muscle from patients with type 2 DM compared with control subjects. In this study (33), ND subunit I mRNA expression was significantly correlated to insulin sensitivity. In contrast, Antonetti et al. (34) demonstrated increases in COX subunit I, COX subunit III, and ND IV mRNA expression in muscle from type 2 DM patients. However, it is unclear whether the muscle specimens obtained from amputated limbs in that study (34) were from patients under suboptimal glucose control and/or with peripheral vascular disease. Recent studies performed in people with type 2 DM demonstrated that 2 wk of insulin treatment did not stimulate COX activity and mitochondrial protein synthesis (12).
The increases in mitochondrial function observed during insulin infusion in the current study could be due to increases in mitochondrial protein and therefore an increase in effective ATP-producing protein units. Alternatively, existing mitochondrial proteins could become more efficient in producing ATP per unit of mitochondrial protein, or both. It is well recognized that increased availability of mitochondrial protein is associated with increased respiration (13, 35). By increasing mitochondrial protein synthesis and possibly decreasing protein breakdown (36–38), insulin may increase mitochondrial protein content and could thus enhance mitochondrial respiration. Previous studies have meticulously revealed many of the subcellular mechanisms by which insulin stimulates muscle protein synthesis, most involving the up-regulation of translational steps (39, 40). However, Boirie et al. (11) showed that this up-regulation may not be a global effect on all proteins, but that specific protein pools (i.e., mitochondrial proteins) may be targeted. It may be that specific proteins having greater numbers of mRNA transcript messages available for translation would be favored in such a scenario. Presumably, increases in mitochondrial function would depend on all mitochondrial protein components being in sufficient supply. In the present study, we observed increases in transcript levels for key mitochondrial enzymes encoded by both the mitochondrial genome (ND IV) and the nuclear genome (COX IV). The present study confirms that mitochondrial protein synthesis is increased in the presence of insulin by ≈20–25%.
Increased activity or efficiency of existing tricarboxylic acid cycle and oxidative phosphorylation components could also increase ATP production. In the present study, we find increases in citrate synthase and COX activities in skeletal muscle after high physiological insulin. Because these enzymes are measured in excess reagents, it is assumed that the increase in maximal reaction velocity is not affected by substrate kinetics. However, allosteric activators or inhibitors could remain in effect under these conditions. Modulators of COX have been identified and are thought to be an important regulatory mechanism of this enzyme complex (41, 42). In addition, evidence that acute exercise increases skeletal muscle citrate synthase activity well before mRNA transcription and protein synthesis increases has been reported (43, 44). Skeletal muscle citrate synthase activity did not increase after 5 h of insulin infusion in one study (45), which is consistent with the current results showing no significant insulin effect on enzyme activity after 4 h. Nevertheless, changes in allosteric activators or inhibitors of these oxidative enzymes must be considered.
The high circulating insulin concentrations attained during high-dose infusion are similar to those observed postprandially, except of course that the elevation is extended and unchanged for 8 h. Similar insulin levels also occur in people with type 2 DM on insulin treatment (46). It is unknown whether pathways leading to increased muscle oxidative capacity would be stimulated between 4 and 8 h after a shorter or lesser exposure to insulin. Similarly, the persistence of the changes evoked in the current study are unknown. It also remains possible that increases in substrate flux and oxidation rather than direct insulin effects per se may have signaled skeletal muscle cells to up-regulate genes favoring oxidative metabolism. Changes in levels of free fatty acids and other secondary events that occur after insulin administration may also affect the mitochondrial functions.
In conclusion, the current data indicate that the expression of mRNA transcripts encoding key mitochondrial proteins from both the nuclear (COX IV) and mitochondrial (ND IV) genomes in human skeletal muscle is enhanced by high physiological insulin sustained for 8 h. In addition, insulin stimulated skeletal muscle mitochondrial protein synthesis and oxidative capacity, as measured by enzyme activities and mitochondrial ATP production rate. The largest increase in ATP production capacity after 8 h of high physiological insulin was associated with a period of at least 4 h of increased mitochondrial protein synthesis. Finally, the studies performed in type 2 DM patients demonstrate a diminished stimulation of muscle mitochondrial ATP production by insulin. This observation has potential implications in the pathogenesis of type 2 DM.
Acknowledgments
We acknowledge the valuable support of the General Clinical Research Center staff and nursing staff (Novo-Nordisk supported) and the skillful technical support of Jane Kahl, Rebecca Kurup, Mai Persson, Carole Berg, Charles Ford, and Jaime Gransee. This study was supported by the National Institutes of Health Grants R01 DK41973 and MO1 RR00585 and the Mayo Foundation. C.S.S. was supported by the Clinical Investigator Program of the Mayo Clinic. K.R.S. was supported by National Institutes of Health Training Grant T32 DK 07352 and a Thompson–Mayo Fellowship from the Mayo Foundation.
Abbreviations: FSR, fractional synthesis rate; DM, diabetes mellitus; COX, cytochrome c oxidase; ND IV, NADH dehydrogenase subunit IV; GH, growth hormone; FFM, fat-free mass; mU, milliunit; PM, pyruvate plus malate; GM, glutamate plus malate; PCM, palmitoyl-l-carnitine plus malate; SR, succinate plus rotenone; PPKM, pyruvate plus palmitoyl-l-carnitine plus α-ketoglutarate plus malate; TA, N,N,N′,N′-tetramethyl-p-phenylenediamine plus ascorbate.
References
- 1.Cheng, A., Dube, N., Gu, F. & Tremblay, M. L. (2002) Eur. J. Biochem. 269, 1050-1059. [DOI] [PubMed] [Google Scholar]
- 2.Sherratt, H. S. & Turnbull, D. M. (1990) Baillieres Clin. Endocrinol. Metab. 4, 523-560. [DOI] [PubMed] [Google Scholar]
- 3.Ballinger, S. W. (1992) Nat. Genet. 1, 11-15. [DOI] [PubMed] [Google Scholar]
- 4.Eriksson, K.-F. & Lindgarde, F. (1996) Diabetologia 39, 573-579. [DOI] [PubMed] [Google Scholar]
- 5.Regensteiner, J. G., Sippel, J. M., McFarling, E. T., Wolfel, E. E. & Hiatt, W. R. (1995) Med. Sci. Sports Exercise 27, 875-881. [PubMed] [Google Scholar]
- 6.Simoneau, J. A. & Kelly, D. E. (1997) J. Appl. Physiol. 83, 166-171. [DOI] [PubMed] [Google Scholar]
- 7.Lillioja, S., Young, A. A., Culter, C. L., Ivy, J. L., Abbott, W. G. H., Zawadzki, J. K., Yki-Järvinen, H., Christin, L., Secomb, T. W. & Bogardus, C. (1987) J. Clin. Invest. 80, 415-424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marin, P., Andersson, B., Krotkiewski, M. & Bjorntorp, P. (1994) Diabetes Care 17, 382-386. [DOI] [PubMed] [Google Scholar]
- 9.van den Ouweland, J. M., Lemkes, H. H., Ruitenbeek, W., Sandkuijl, L. A., de Vijlder, M. F., Struyvenberg, P. A., van de Kamp, J. J. & Maassen, J. A. (1992) Nat. Genet. 1, 368-371. [DOI] [PubMed] [Google Scholar]
- 10.Kelley, D. E., He, J., Menshikova, E. V. & Ritov, V. B. (2002) Diabetes 51, 2944-2950. [DOI] [PubMed] [Google Scholar]
- 11.Boirie, Y., Short, K. R., Ahlman, B., Charlton, M. & Nair, K. S. (2001) Diabetes 50, 2652-2658. [DOI] [PubMed] [Google Scholar]
- 12.Halvatsiotis, P., Short, K. R., Bigelow, M. L. & Nair, K. D. (2002) Diabetes 51, 2395-2404. [DOI] [PubMed] [Google Scholar]
- 13.Hood, D. A., Takahashi, M., Connor, M. K. & Freyssenet, D. (2000) Exercise Sport Sci. Rev. 28, 68-73. [PubMed] [Google Scholar]
- 14.Flakoll, P. J., Kulaylat, M., Frexes-Steed, M., Hourani, H., Brown, L. L., Hill, J. O. & Abumrad, N. N. (1989) Am. J. Physiol. 257, E839-E847. [DOI] [PubMed] [Google Scholar]
- 15.Frexes-Steed, M., Warner, M. L., Bulus, N., Flakoll, P. & Abumrad, N. N. (1990) Am. J. Physiol. 258, E907-E917. [DOI] [PubMed] [Google Scholar]
- 16.Balagopal, P., Schimke, J. C., Ades, P. A., Adey, D. & Nair, K. S. (2001) Am. J. Physiol. 280, E203-E208. [DOI] [PubMed] [Google Scholar]
- 17.Balagopal, P., Ljungqvist, O. & Nair, K. S. (1997) Am. J. Physiol. 272, E45-E50. [DOI] [PubMed] [Google Scholar]
- 18.Rooyackers, O. E., Adey, D. B., Ades, P. A. & Nair, K. S. (1996) Proc. Natl. Acad. Sci. USA 93, 15364-15369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rooyackers, O. E., Balagopal, P. & Nair, K. S. (1997) Muscle Nerve Suppl. 5, S93-S96. [PubMed] [Google Scholar]
- 20.Ljungqvist, O., Persson, M., Ford, G. C. & Nair, K. S. (1997) Am. J. Physiol. 273, E564-E570. [DOI] [PubMed] [Google Scholar]
- 21.Halliday, D. & McKeran, R. O. (1975) Clin. Sci. Mol. Med. 49, 581-590. [DOI] [PubMed] [Google Scholar]
- 22.Jones, B. N. & Gilligan, J. P. (1983) J. Chromatogr. 266, 471-482. [DOI] [PubMed] [Google Scholar]
- 23.Short, K. R., Nygren, J., Barazzoni, R., Levine, J. & Nair, K. S. (2001) Am. J. Physiol. 280, E761-E769. [DOI] [PubMed] [Google Scholar]
- 24.Wibom, R. & Hultman, E. (1990) Am. J. Physiol. 259, E204-E209. [DOI] [PubMed] [Google Scholar]
- 25.Henriksen, E. J. & Halseth, A. E. (1995) Am. J. Physiol. 268, R130-R134. [DOI] [PubMed] [Google Scholar]
- 26.Rodnick, K. J., Henriksen, E. J., James, D. E. & Holloszy, J. O. (1992) Am. J. Physiol. 262, C9-C14. [DOI] [PubMed] [Google Scholar]
- 27.Houmard, J. S., Shinebarger, M. H., Dolan, P. L., Legget-Frazier, N., Bruner, R. K., McCammon, M. R., Israel, R. G. & Dohm, G. L. (1993) Am. J. Physiol. 264, E896-E901. [DOI] [PubMed] [Google Scholar]
- 28.Gaster, M., Poulsen, P., Handberg, A., Schroder, H. D. & Beck-Nielsen, H. (2000) Am. J. Physiol. 278, E910-E916. [DOI] [PubMed] [Google Scholar]
- 29.Gaster, M., Staehr, P., Beck-Nielsen, H., Schroder, H. D. & Handberg, A. (2001) Diabetes 50, 1324-1329. [DOI] [PubMed] [Google Scholar]
- 30.Ericksson, K. F., Saltin, B. & Lindgarde, F. (1994) Diabetes 43, 805-808. [DOI] [PubMed] [Google Scholar]
- 31.He, J., Watkins, S. & Kelley, D. E. (2001) Diabetes 50, 817-823. [DOI] [PubMed] [Google Scholar]
- 32.Simoneau, J. A., Veerkamp, J. H., Turcotte, L. P. & Kelley, D. E. (1999) FASEB J. 13, 2051-2060. [DOI] [PubMed] [Google Scholar]
- 33.Huang, X., Eriksson, K.-F., Vaag, A., Lehtovirta, M., Hansson, M., Laurila, E., Kanninen, T., Thuesen Olesen, B., Kurucz, I., Koranyi, L., et al. (1999) Diabetes 48, 1508-1514. [DOI] [PubMed] [Google Scholar]
- 34.Antonetti, D. A., Reynet, C. & Kahn, C. R. (1995) J. Clin. Invest. 95, 1383-1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takahashi, M. & Hood, D. A. (1996) J. Biol. Chem. 271, 27285-27291. [DOI] [PubMed] [Google Scholar]
- 36.Meek, S. E., Persson, M., Ford, G. C. & Nair, K. S. (1998) Diabetes 47, 1824-1835. [DOI] [PubMed] [Google Scholar]
- 37.Gelfand, R. A. & Barrett, E. J. (1987) J. Clin. Invest. 80, 1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mitch, W. E. & Goldberg, A. L. (1996) N. Engl. J. Med. 335, 1897-1905. [DOI] [PubMed] [Google Scholar]
- 39.Kimball, S. R., Horetsky, R. L. & Jefferson, L. S. (1998) Am. J. Physiol. 274, C221-C228. [DOI] [PubMed] [Google Scholar]
- 40.Kimball, S. R., Jefferson, L. S., Fadden, P., Haystead, T. A. J. & Lawrence, J. C., Jr. (1996) Am. J. Physiol. 270, C705-C709. [DOI] [PubMed] [Google Scholar]
- 41.Bender, E. & Kadenbach, B. (2000) FEBS Lett. 466, 130-134. [DOI] [PubMed] [Google Scholar]
- 42.Kadenbach, B., Frank, V., Rieger, T. & Napiwotzki, J. (1997) Mol. Cell. Biochem. 174, 131-135. [PubMed] [Google Scholar]
- 43.Leek, B. T., Mudaliar, S. R., Henry, R., Mathieu-Costello, O. & Richardson, R. S. (2001) Am. J. Physiol. 280, R441-R447. [DOI] [PubMed] [Google Scholar]
- 44.Tonkonogi, M., Harris, B. & Sahlin, K. (1997) Acta Physiol. Scand. 161, 435-436. [DOI] [PubMed] [Google Scholar]
- 45.Kruszynska, Y. T., Mulford, M. I., Baloga, J., Yu, J. G. & Olefsky, J. M. (1998) Diabetes 47, 1107-1113. [DOI] [PubMed] [Google Scholar]
- 46.McMahon, M. M., Marsh, H. M. & Rizza, R. A. (1989) Diabetes 38, 291-303. [DOI] [PubMed] [Google Scholar]