

Abstract
Interferon Regulatory Factor 7 (IRF7) is a key component of the cellular response to virus infection that culminates in physiologically relevant IFNα production. We studied molecular mechanisms governing responses to respiratory viral infection that are characterized by transient induction and subsequent shut-off of interferon (IFN) gene expression. We asked whether alterations in IRF7 protein stability occurred during virus infection that might contribute to this regulation. To this end, we measured IRF7 half-life in various cell types and found it to be short-lived, in marked contrast to the pronounced stability of the related transcription factor, IRF3. Furthermore, virus infection accelerated IRF7 degradation in a proteosome-dependent manner in most cell types. However, plasmacytoid dendritic cells (pDC), which constitute the major circulating IFN producing cell type, displayed a distinct pattern of regulation. Infection of lymphoid tissues, where the majority of IRF7 is expressd in pDC, attenuated the normal proteosome-mediated degradation of IRF7, resulting in a long-lived protein. Stabilization was partially stimulated by autocrine IFN as a positive feedback mechanism, but was partially IFN independent. Thus, two distinct post-translational mechanisms regulate IRF7 activity in response to viral infection, with protein turnover attenuating responses post-infection in most cell types while infection-induced protein stabilization contributes to the heightened IFN production characteristic of pDC.
Keywords: Interferon, Posttranslational protein stabilization, Antiviral inane immunity, Proteosome, Plasmacytoid dendritic cells
Interferon regulatory factor 7 (IRF7) is a primary regulator of physiologic type I interferon (IFN) production in the local and systemic reaction to viral infection, and has been characterized as the “master regulator” of type I IFN production [1, 2]. Previous studies have demonstrated that IRF7 protein expression is necessary and sufficient for virus-induced IFN responses [1, 3, 4]. In most cell types, induction of IRF7 abundance through a positive feedback regulatory circuit is necessary for IFN production [5, 6], while the high IFN production capacity of plasmacytoid dendritic cells (pDC) is at least in part due to high constitutive levels of IRF7 [4, 7]. Absence of IRF7 [1] or failure to induce its expression [5, 6] abrogates IFNα gene expression and impairs antiviral innate immunity. Moreover, correct spatio-temporal regulation of IRF7 protein within subcellular compartments maintains the integrity of the signal transduction pathway in infected cells [3].
A second component of the IFN transcriptional machinery is the related protein IRF3. The activity of both IRF3 and IRF7 is dependent on virus-induced serine phosphorylation within a carboxyl-terminal regulatory domain, a modification that stimulates protein dimerization, nuclear retention, and interaction with transcriptional coactivators. However, activated IRF3 is capable of inducing transcription from only a limited subset of type I IFN genes due to its restricted DNA binding specificity, while IRF7 activates a robust response comprised of a broader spectrum on IFN isotypes (reviewed in ref. [8]). The IFN transcriptional response during viral infection is transient, displaying a rapid burst of gene expression that wanes over time. While the mechanisms involved in IFN gene induction through regulated phosphorylation of IRF3 and IRF7 have been intensely studied, mechanisms involved in limiting the extent and duration of the response remain incompletely understood.
One aspect regulating the kinetics of IFN gene induction could be the duration of IRF3 or IRF7 phosphorylation, either through controlling the length of kinase activity or by counteracting effects of phosphatases. No specific protein phosphatases have been described that dephosphorylate these proteins, but one of the IRF3 kinases, TBK1, is a client of the chaperone HSP90 [9], which may be involved in modulating its activity towards IRF3, thereby controlling the duration of phosphorylation. Another mechanism controlling transcription factor activity could be disappearance of the protein, and regulated degradation of IRF3 has been documented in infected cells [10, 11]. Virus infection-dependent IRF3 phosphorylation accelerates protein turnover through a proteosome-dependent process, thereby decreasing the levels of activated IRF3 at late times after infection and presumably contributing to the decrease in IFN gene expression. However, IRF7 level is the major determinant of total IFN gene transcription, so understanding its regulation is critical for a complete picture of IFN gene regulation.
To date, two studies reporting discordant results have addressed IRF7 protein stability [12, 13], and no consistent picture of its regulation has emerged. Sato et al. examined the stability of recombinant murine IRF7 expressed in NIH3T3 cells and found it to be a relatively unstable protein with a half-life of 0.5-1h [12]. In contrast, Zhang et al. reported that the half-life of endogenous IRF7 in human lymphoma tumor cell lines was greater than 24 h, inicating a relatively stable protein [13]. The discordant results between these studies could be due to species, cell type, or tumor-specific differences in IRF7 or to differences in experimental approaches. In addition, neither study addressed the stablity of IRF7 in the context of IFN gene expression, that is, following induction by IFN-dependent positive feedback or after phosphorylation-dependent activation during viral infection. Coupled with the observation that IRF7 protein levels are widely different between fibroblasts and pDC [4, 7], we were prompted to examine endogenous IRF7 protein stability in normal cells of the mouse.
We found endogenously expressed IRF7 to be a short-lived protein in fibroblasts; and likewise in splenocyte and thymocyte populations where the majority of IRF7 is expressed by pDC [4]. IFN induction of IRF7 produced a moderately stabilizing effect, but virus infection decreased the stability of both IRF3 and IRF7 in fibroblasts, suggesting that virus infection promoted IRF protein degradation. Degradation was dependent on the 26S proteosome, suggesting that virus-induced phosphorylation targeted degradation. In contrast, virus infection of splenocytes led to increased IRF7 stability, converting it into a more long-lived protein. These results suggest that the pDC population that comprises the IFN producing cell type for systemic cytokine production retains high IRF7 levels by preventing virus-induced protein degradation, permitting prolonged IFNα production following infection.
Materials and Methods
Mice. Mice deficient for STAT1 (stat1-/-) and wild type controls on the same inbred background (Balb/c and C57BL6/J) were bred under specific pathogen-free conditions, in accordance with approved protocols at New York University School of Medicine. All mice were 8-12 weeks of age.
Cell isolation and culture. Stat1-/- and wild type immortalized embryo fibroblasts (MEFs) were grown in DMEM (Mediatech, Herndon, VA) containing 10% calf serum and subcultured using 0.05% trypsin (Mediatech). Human embryo kidney 293T cells were cultured on tissue culture plates coated with 0.1% gelatin in DMEM with 10% calf serum, and were transfected by the calcium phosphate procedure, as previously described [14]. Spleens harvested from euthanized animals were digested by injection of Liberase CI and DNase I (Roche Applied Science, Indianapolis, IN) and incubation for 30 min at 37°C. Single cell suspensions were prepared by passage through a cell strainer and red blood cells were removed by lysis in buffered ammonium chloride solution, as described [4]. Thymocyte cultures were prepared by crushing organs between frosted glass microscope slides.
Protein extracts were prepared in IP lysis buffer (1% Triton X-100, 300 mM NaCl, 50 mM HEPES pH 7.6, 1 mM Na3VO4, 1 mM DTT, 1 mM PMSF, and 5 μg/ml aprotinin and leupeptin).
Virus infections. Newcastle disease virus, Manhattan strain (NDV), was grown in 10-d embryonated chicken eggs. Monolayer cells were infected with NDV using 107 pfu/ml in DMEM containing 0.1% bovine serum albumin for 1 h at 37°C, followed by normal growth medium for 12 h. Splenocytes and thymocytes were infected by resuspending 5 x 106 cells in 50 μl of RPMI containing NDV at 108 pfu/ml and incubation at 37°C for 1 h with frequent agitation, followed by replating in normal growth medium.
Reagents and antibodies. Immunoblotting was performed using the following antibodies: rabbit anti-IRF7, anti-IRF3, anti-IRF4, and anti-IRF8 (Zymed, South San Francisco, CA); monoclonal anti-α-tubulin and anti-FLAG M2 (Sigma-Aldrich, St. Louis, MO); and monoclonal HA (Roche Applied Science). Where indicated, cells were treated with 500 IU/ml of mouse IFNα/β (Access Biomedical, San Diego, CA). Western blot films were quantified by using Quantity One software (Bio-Rad, Richmond, CA). Expression plasmids used for transfection have been previously described [15, 16]. Cycloheximide (CHX) was obtained from Sigma-Aldrich and used at 75 μg/ml; MG132 was obtained from Alexis Corporation (San Diego, CA) and used at 50 μM.
Results and discussion
Half-life analysis of exogenously expressed IRF7
To examine a role for proteosomes in IRF7 turnover, we measured IRF7 levels in infected cells treated with the proteosome inhibitor, MG132 (Fig. 1A). For these experiments, we expressed Flag-tagged IRF7 in 293T cells. Proteosome inhibition resulted in accumulation of modestly increased levels of phosphorylated IRF7, indicated by the slower migrating form, following NDV infection (compare lanes 2 and 4). The MG132-mediated stabilization of IRF7 in infected cells suggested that infection might target IRF7 degradation. To examine this effect in greater detail, we compared differential IRF7 protein degradation rates in uninfected and infected cells. To focus on protein degradation, we blocked further protein synthesis by CHX treatment. As shown in Fig. 1B, the stability of wild type IRF7 was diminished by virus infection (upper panel). To determine if virus-targeted degradation of IRF7 was dependent on phosphorylation, we performed similar experiments using a non-phosphorylatable, inactive IRF7 mutant, IRF7-SSAA [17]. Similar to wild type protein, the inactive IRF7-SSAA mutant was also destabilized in infected cells (Fig. 1B, middle panel), suggesting that virus-stimulated degradation is not wholly dependent on direct IRF7 phosphorylation. We also examined the stability of a constitutively active form of the protein, IRF7-M15, in which all regulatory serine residues were converted to aspartic acid residues [18]. This activated protein was also less stable than wild type protein, even in the absence of virus infection (Fig. 1B, lower panel), suggesting that phosphorylation in response to infection may also contribute to protein degradation.
Fig. 1.
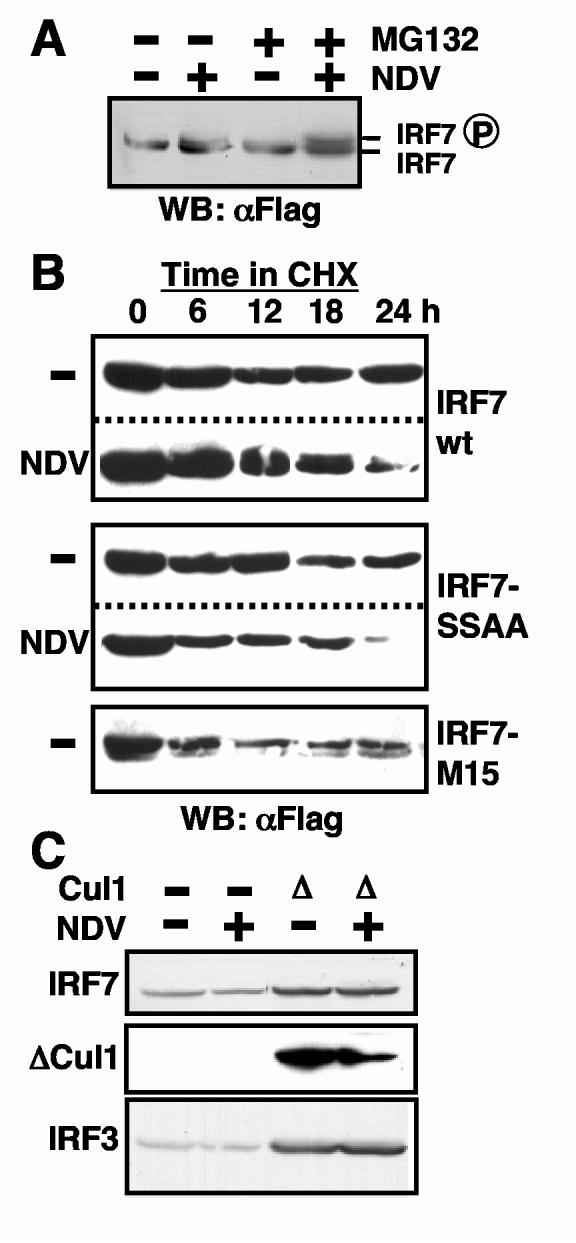
IRF7 turnover is proteosome-dependent. (A) IRF7 expressed by transfection in 293T cells was increased in abundance following proteosome inhibition by 50 μM MG132, particularly the slower migrating phosphorylated form observed following infection with NDV. (B) Virus infection (NDV) resulted in decreased IRF7 stability relative to uninfected cells (-), as measured by decreased abundance following CHX treatment. Non-phosphorylatable IRF7-SSAA mutant was destabilized by viral infection, while constitutively active IRF7-M15 was unstable even in uninfected cells. (C) Over-expression of dominant-negative Cul1 (Δ) inhibited IRF7 and IRF3 turnover. IRF7 was tagged with HA, ΔCul1 with Flag, and transfected IRF3 was detected by a specific antibody on western blots of whole cell protein extracts.
Sensitivity of IRF7 protein turnover to the 26S proteosome inhibitor MG132 (Fig. 1A) suggested the possible involvement of ubiquitin-dependent degradation. To investigate this possibility further, we examined IRF protein abundance in the presence of a dominant-negative form of the SCF ubiquitin ligase complex protein, Cul1 [19]. Cells transfected with IRF7 and ΔCul1, a truncated form that inhibits SCF function, displayed higher levels of IRF7 (Fig. 1C). The modest reduction in IRF7 observed following virus infection was also reversed by ΔCul1 expression. Furthermore, IRF3 protein abundance was similarly increased when SCF was inhibited by ΔCul1 expression (Fig. 1C, bottom panel), suggesting ubiquitin-dependent protein turnover normally regulates IRF protein levels.
Half-life of endogenous IRF7
To examine IRF protein stability under less artificial conditions, we measured endogenous protein turnover. IRF7 is not expressed at detectable levels in resting fibroblasts [17]; therefore, we first induced the expression of the protein by treatment of MEFs with IFNα/β. Following IFNα/β treatment, cells were exposed to CHX to block protein synthesis, and levels of IRF7 and IRF3 were assayed at various time points by western blotting using specific antisera. As shown in Fig. 2A, IRF7 protein half-life was quite short and considerably shorter than that of IRF3. Quantitative densitometry (Fig. 2A, lower panel) suggested half-lives of 5 h for IRF7 and greater than 60 h for IRF3.
Fig. 2.

Endogenous IRF7 is a short-lived protein relative to long-lived IRF3. (A) MEFs were left untreated (No IFN) or were treated overnight with IFNα/β prior to incubation with CHX for the indicated times (h). Protein extracts were assayed for IRF7 (left panel) or IRF3 (right panel) by western blotting and densitometric quantitation (graphs) for calculation of half-lives. Note that no IRF7 was detectable in cells without IFN treatment. IRF7 and IRF3 half-lives were calculated for mouse thymocytes (B) and splenocytes (C) as described in (A), except that it was not necessary to pretreat cells with IFN.
Since we had previously found that resting lymphoid cell populations expressed constitutively high levels of IRF7 relative to other cell types, due primarily to high levels of IRF7 in pDC [4], we asked whether increased protein stability contributed to IRF7 abundance in a cell type-specific manner. Therefore, we examined IRF7 protein stability in quiescent splenocyte and thymocyte cultures obtained from naïve mice. Cell populations were treated with CHX in vitro, and IRF3 and IRF7 protein levels were quantified over time (Fig. 2B, C). In contrast to expectation, splenic and thymic IRF7 protein stability was considerably reduced relative to MEFs, with a predicted half-life of 1-2 h. Reduced IRF7 stability was not a general characteristic of protein stability in these cells, since IRF3 protein remained long-lived in both splenocyte and thymocyte cultures, displaying a half-life similar to that measured in MEFs (Fig. 2B, C, right panels). Therefore, the increased IRF7 protein levels observed in lymphoid organs relative to other mouse tissues is not due to prolonged stability of the protein in pDC.
Effect of virus infection on IRF7 protein stability
We wanted to determine if the destabilizing effect of virus on IRF7 observed in 293T cells also occurred with endogenous protein. Therefore, we measured protein half-lives following NDV infection. In this experiment, it was not necessary to pre-induce IRF7 levels, since the protein is transcriptionally up-regulated by positive feedback during infection [5, 20]. MEFs were infected with NDV for 12 h, and IRF7 protein levels were measured a various times after CHX addition. IRF7 protein levels decreased significantly more rapidly in infected cells (Fig. 3A) compared with uninfected cells (Fig. 2A). IRF7 protein half-life calculated from densitometric measurements was approximately 1 h in infected cells, compared to 5 h in uninfected cells. Similarly, IRF3 stability was decreased in infected cells (Fig. 3A, right panel), consistent with previous studies [10, 11]. However, IRF3 half-life (6 h) remained significantly longer than that of IRF7, even in infected cells.
Fig. 3.

Virus infection accelerates IRF7 degradation in MEFs, but stabilizes IRF7 in thymocytes and splenocytes. IRF7 and IRF3 protein half-lives were measured as described in Fig. 2, except that cells were first infected with NDV for 12 h prior to CHX addition. No IFN pretreatment was used in these experiments, since autocrine IFN produced during infection resulted in detectable levels of IRF7 protein in MEFs.
We next examined the effect of virus infection on IRF protein stability in infected thymocytes and splenocytes. Surprisingly, IRF7 was significantly stabilized by virus infection in these cells (Fig. 3B, C). Stability measurements of IRF7 in infected thymocytes and splenocytes revealed that it was converted to a relatively stable protein, increasing its half-life from approximately 1-2 h in the absence of infection to 8 h following infection. In contrast, the half-life of IRF3 was largely unaffected by virus infection, remaining a relatively stable protein with a half-life of greater than 12 h. Therefore, in contrast to MEFs, the cells from lymphoid organs expressing IRF7 (largely pDC) do not destabilize IRF7 in response to infection but rather display heightened protein stability. This finding may reflect a protective effect that contributes to the ability of pDC to produce high levels of IFN following viral infection.
Role of type I IFN in stabilizing IRF7 following infection
In order to determine what role IFN feedback played in the altered stability of the IRF7 protein during infection, we tested IRF7 stability in thymocytes and splenocytes pretreated with IFN. IFN-treated thymocytes (Fig. 4A) and splenocytes (Fig. 4B) showed increased IRF7 stability. However, this incease in stablity was not as great as that observed following virus infection, with a predicted half-life of approximately 5 h, which was intermediate between that observed in uninfected and virus-infected lymphoid cells.
Fig. 4.

Partial stabilization of IRF7 by IFN treatment of thymocytes and splenocytes. IRF7 and IRF3 half-lives were measured in thymocytes (A) and splenocytes (B) as described in Fig. 2, except cells were pretreated overnight with IFN prior to CHX treatment.
These results suggested that the increase in IRF7 stability and the resulting increase in protein levels following viral infection of lymphoid cells was due to at least two distinct mechanisms. One mechanism was dependent on autocrine IFN produced during positive feedback, but a second additive effect was virus dependent but could not be replaced by IFN treatment. To investigate this possibility further, we measured IRF protein stability in IFN-resistant stat1-/- cells. Splenocytes were isolated from stat1-/- mice and IRF7 half-life was measured in uninfected and NDV-infected cultures (Fig. 5). Mock-infected cultures showed a short half-life (2 h), equivalent to that observed in wild type splenocytes (Fig. 2C). Similar to wild type cells, virus infection resulted in prolonged stability, with a half-life of 7 h (Fig. 5, lower panel). Interestingly, however, the enhanced stablity produced by viral infecton of stat1-/- splenocytes was intermediate between that of uninfected cells and virus-infected wild type splenocytes. These results reinforce the notion that virus-induced IRF7 protein stabilization is due to two mechanisms. IFN produced following infection increases protein stability, as a component of positive feedback, and combines with a second virus-induced but IFN-independent mechanism that results in further augmentation of IRF7 stability. These virus-induced stabilization mechanisms are specific to lymphoid organs, where IRF7 is highly expressed in pDC.
Fig. 5.
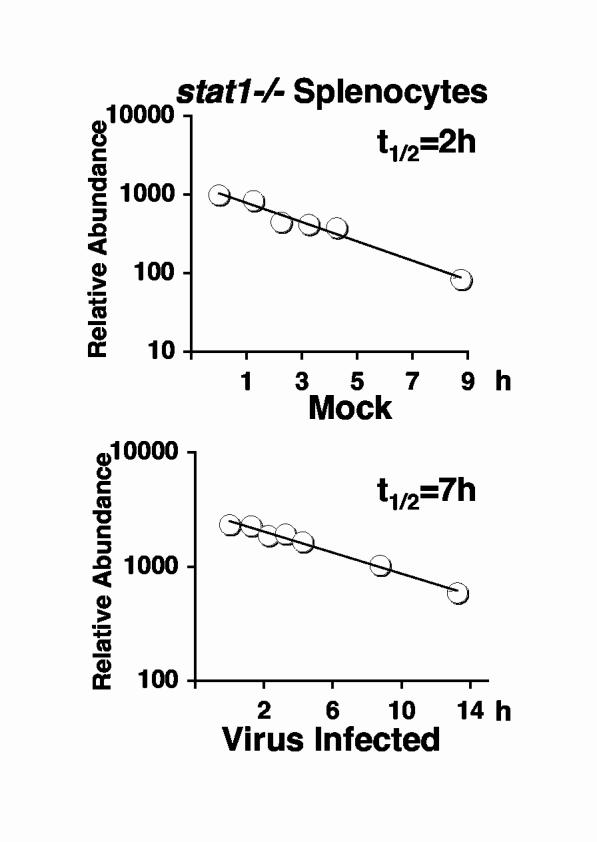
IRF7 Stabilization in response to virus infection is only partially dependent on STAT1. IRF7 half-lives were measured in splenocytes from stat1-/- mice before (upper panel) or after infection with NDV (lower panel). Calculated half-lives are indicated.
Data on endogenous IRF protein half-life are summarized in Fig. 6. In all cell types, IRF3 was a long lived protein in the absence of viral infection, with a half-life ranging from 20-60 h. In fibroblasts, its turnover was dramatically stimulated by NDV infection, as previously reported for infections with both Sendai virus and human papillomavirus [10, 11], suggesting that this is a common response to infection. Presumably, infection-mediated IRF3 protein turnover contributes to the ability of viruses to circumvent the innate antiviral response by preventing the early production of type I IFN [21]. However, virus-induced turnover of IRF3 was not observed in lymphoid organs, the major producers of systemic IFN.
Fig. 6.
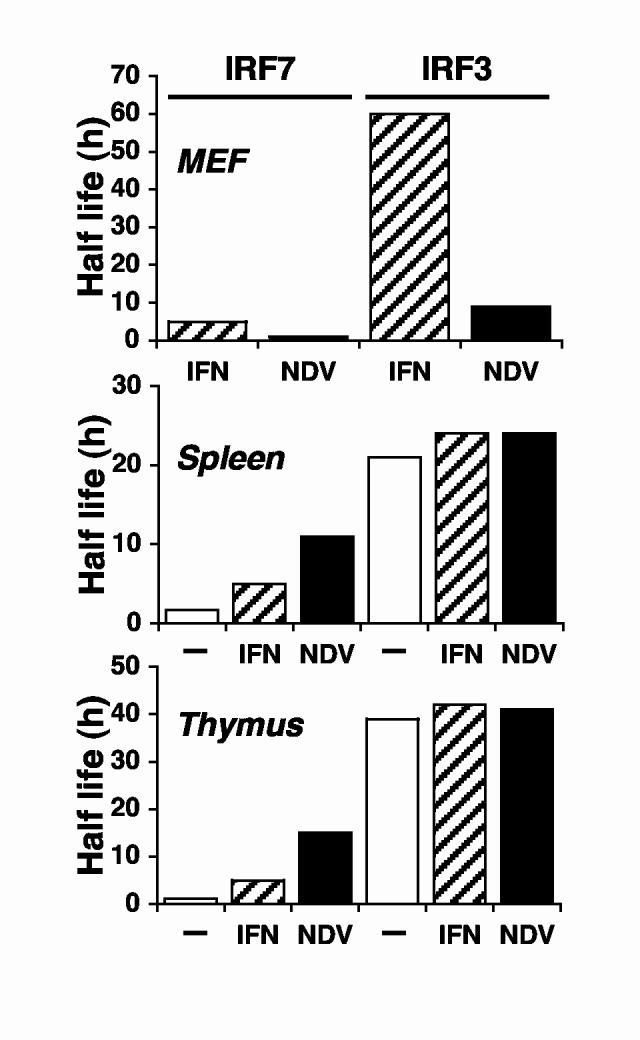
Comparison of IRF7 and IRF3 protein half-lives in different cells under different conditions. Calculated half-lives of IRF7 and IRF3 are shown for wild type MEFs (upper panel), splenocytes (middle panel) and thymocytes (lower panel) after the indicated treatments.
Virus-mediated protein turnover was also observed for IRF7 in fibroblasts, although the protein half-life in the absence of infection was much shorter than that of IRF3. Protein turnover, at least in 293T cells, appears to be ubiquitin and proteosome-dependent, since it was inhibited by MG132 and dominant-negative Cul1 (Fig. 1). It remains to be determined how virus infection stimulates the ubiquitinylation and degradation of IRF proteins, although phosphorylation-dependent recognition of proteins by the SCF complex is a common mechanism of targeting proteins for destruction [19]. A role for IRF7 phosphorylation was implicated by the decreased stability of the constitutively active IRF7-M15 mutant (Fig. 1B). However, phosphorylation-independent mechanisms are probably involved as well, since the inactive IRF7-SSAA mutant was also targeted for degradation in infected cells. IRF7 can also be directly targeted for degradation by virus-encoded ubiquitin ligases, as has been demonstrated for the HHV8-encoded Rta protein [22], but no similar activity is present in NDV.
A different picture of IRF7 protein stability emerged from studies of lymphoid tissues. In spite of the increased abundance of IRF7 in these tissues relative to others [4], IRF7 displayed a shorter half-life in both splenocytes and thymocytes compared with fibroblasts. Therefore, the increased protein abundance in these tissues is likely due largely to transcriptional or mRNA stability effects. However, virus-mediated protein turnover was not stimulated in these tissues for either IRF7 or IRF3, suggesting a very different mode of regulation. In fact, virus infection led to increased stability of IRF7, an effect that was in part dependent on IFN positive feedback through a STAT1-mediated pathway and in part due to effects of the virus not contributed by IFN. Most of the IRF7 protein in these lymphoid organs is expressed by pDC, the major IFN producing cells [4]. Both IFN treatment and virus infection drive pDC maturation from immature circulating cells to differentiated cells that contribute to adaptive immunity [23]. Therefore, it is possible that increased IRF7 stability could be a characteristic of DC differentiation. In any case, enhanced IRF7 stability in virus-infected lymphoid tissues likely contributes to the ability of pDC to produce high levels of type I IFN during innate immune responses.
Acknowledgments
We thank Valerio Dorrello and Michele Pagano for advice with half-life and proteosome studies and the gift of plasmids, and members of the laboratory for assistance, advice, and comments on the manuscript. This work was supported in part by NIH grants AI46503 and AI57158 and by a Grant-In-Aid from the American Heart Association.
References
- [1].Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- [2].Honda K, Yanai H, Takaoka A, Taniguchi T. Regulation of the type I IFN induction: a current view. Int. Immunol. 2005;17:1367–1378. doi: 10.1093/intimm/dxh318. [DOI] [PubMed] [Google Scholar]
- [3].Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, Taniguchi T. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434:1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- [4].Prakash A, Smith E, Lee CK, Levy DE. Tissue-specific positive feedback requirements for production of type I interferon following virus infection. J. Biol. Chem. 2005;280:18651–18657. doi: 10.1074/jbc.M501289200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Marié I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998;441:106–110. doi: 10.1016/s0014-5793(98)01514-2. [DOI] [PubMed] [Google Scholar]
- [7].Izaguirre A, Barnes BJ, Amrute S, Yeow WS, Megjugorac N, Dai J, Feng D, Chung E, Pitha PM, Fitzgerald-Bocarsly P. Comparative analysis of IRF and IFN-alpha expression in human plasmacytoid and monocyte-derived dendritic cells. J. Leukoc. Biol. 2003;74:1125–1138. doi: 10.1189/jlb.0603255. [DOI] [PubMed] [Google Scholar]
- [8].Levy DE, Marié I, Prakash A. Ringing the interferon alarm: differential regulation of gene expression at the interface between innate and adaptive immunity. Curr. Opin. Immunol. 2003;15:52–58. doi: 10.1016/s0952-7915(02)00011-0. [DOI] [PubMed] [Google Scholar]
- [9].Yang K, Shi H, Qi R, Sun S, Tang Y, Zhang B, Wang C. Hsp90 Regulates Activation of IRF-3 and TBK-1 Stabilization in Sendai Virus-infected Cells. Mol. Biol. Cell. 2006 doi: 10.1091/mbc.E05-09-0853. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Ronco LV, Karpova AY, Vidal M, Howley PM. Human papillomavirus 16 E6 oncoprotein binds to interferon regulatory factor-3 and inhibits its transcriptional activity. Genes Dev. 1998;12:2061–2072. doi: 10.1101/gad.12.13.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lin R, Heylbroeck C, Pitha PM, Hiscott J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell. Biol. 1998;18:2986–2996. doi: 10.1128/mcb.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-α/β gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- [13].Zhang L, Wu L, Hong K, Pagano JS. Intracellular signaling molecules activated by Epstein-Barr virus for induction of interferon regulatory factor 7. J. Virol. 2001;75:12393–12401. doi: 10.1128/JVI.75.24.12393-12401.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Graham FL, van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90341-3. [DOI] [PubMed] [Google Scholar]
- [15].Marié I, Smith E, Prakash A, Levy DE. Phosphorylation-induced dimerization of interferon regulatory factor 7 unmasks DNA binding and a bipartite transactivation domain. Mol. Cell. Biol. 2000;20:8803–8814. doi: 10.1128/mcb.20.23.8803-8814.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Latres E, Chiaur DS, Pagano M. The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene. 1999;18:849–854. doi: 10.1038/sj.onc.1202653. [DOI] [PubMed] [Google Scholar]
- [17].Marié I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Caillaud A, Hovanessian AG, Levy DE, Marié IJ. Regulatory Serine Residues Mediate Phosphorylation-dependent and Phosphorylation-independent Activation of Interferon Regulatory Factor 7. J. Biol. Chem. 2005;280:17671–17677. doi: 10.1074/jbc.M411389200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jackson PK, Eldridge AG. The SCF ubiquitin ligase: an extended look. Mol. Cell. 2002;9:923–925. doi: 10.1016/s1097-2765(02)00538-5. [DOI] [PubMed] [Google Scholar]
- [20].Sato M, Hata N, Asagiri M, Nakaya T, Taniguchi T, Tanaka N. Positive feedback regulation of type I IFN genes by the IFN-inducible transcription factor IRF-7. FEBS Lett. 1998;441:106–110. doi: 10.1016/s0014-5793(98)01514-2. [DOI] [PubMed] [Google Scholar]
- [21].Levy DE, García-Sastre A. The virus battles: IFN induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev. 2001;12:143–156. doi: 10.1016/s1359-6101(00)00027-7. [DOI] [PubMed] [Google Scholar]
- [22].Yu Y, Wang SE, Hayward GS. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity. 2005;22:59–70. doi: 10.1016/j.immuni.2004.11.011. [DOI] [PubMed] [Google Scholar]
- [23].Becker Y. Immunological and regulatory functions of uninfected and virus infected immature and mature subtypes of dendritic cells--a review. Virus Genes. 2003;26:119–130. doi: 10.1023/a:1023427228024. [DOI] [PubMed] [Google Scholar]