

Abstract
Charge transport and catalysis in enzymes often rely on amino acid radicals as intermediates. The generation and transport of these radicals are synonymous with proton-coupled electron transfer (PCET), which intrinsically is a quantum mechanical effect as both the electron and proton tunnel. The caveat to PCET is that proton transfer (PT) is fundamentally limited to short distances relative to electron transfer (ET). This predicament is resolved in biology by the evolution of enzymes to control PT and ET coordinates on highly different length scales. In doing so, the enzyme imparts exquisite thermodynamic and kinetic controls over radical transport and radical-based catalysis at cofactor active sites. This discussion will present model systems containing orthogonal ET and PT pathways, thereby allowing the proton and electron tunnelling events to be disentangled. Against this mechanistic backdrop, PCET catalysis of oxygen–oxygen bond activation by mono-oxygenases is captured at biomimetic porphyrin redox platforms. The discussion concludes with the case study of radical-based quantum catalysis in a natural biological enzyme, class I Escherichia coli ribonucleotide reductase. Studies are presented that show the enzyme utilizes both collinear and orthogonal PCET to transport charge from an assembled diiron-tyrosyl radical cofactor to the active site over 35 Å away via an amino acid radical-hopping pathway spanning two protein subunits.
Keywords: proton-coupled electron transfer, amino acid radicals, tunnelling, tyrosine, catalysis, ribonucleotide reductase
1. Introduction
Enzymes often rely on the coupling of electrons and protons to affect primary metabolic steps involving charge transport and catalysis. Amino acid radical generation and transport are synonymous with proton-coupled electron transfer (PCET; Stubbe et al. 2003) as is the activation of most substrate bonds at enzyme active sites (Chang et al. 2004a). PCET is especially prevalent at metallo-cofactors that activate substrates at carbon, oxygen, nitrogen and sulphur atoms. PCET charge transport and activation events in enzymes embody ‘quantum catalysis’ inasmuch as PCET is intrinsically a quantum mechanical effect because both the electron and the proton tunnel. The caveat to PCET is that the transfer of the proton, as the heavier particle, is fundamentally limited to short distances, whereas the electron, as the lighter particle, may transfer over very long distances (Moser et al. 1992; Winkler & Gray 1992; Gray & Winkler 1996). When transport distances are short, the electron and proton may transfer together. When they are long, however, the predicament of the disparate transfer distances is resolved by the evolution of enzymes to control proton-transfer (PT) and electron-transfer (ET) coordinates on highly different length scales. Adding to the challenge of effecting PCET over long distances with appreciable rates are the requirements that charge transport occurs under mild physiological conditions, with minimal thermodynamic driving force, with low over-potentials and with specificity. To do so, enzymes impart exquisite thermodynamic and kinetic controls over the electron and proton during radical transport and catalysis.
Figure 1 presents the two basic scenarios for PCET in biology.
Figure 1.

Two basic scenarios for PCET in biology: collinear PCET, which may involve bond making/breaking; and orthogonal PCET, where PT to a base (B:) occurs along a separate coordinate than ET. This pathway is usually accompanied by bond breaking/making.
The electron and the proton may transfer along the same collinear path with or without X–H bond breaking. The former describes long-range ET in biology. Here, electron transport along pathways containing X–H⋯Y bonds is modulated by the hydrogen bond, typically via the electronic coupling matrix element (Beratan et al. 1991; Lin et al. 2005). The coupling between the proton and the electron is more pronounced when the X–H bond breaking is involved. This subclass of PCET includes hydrogen atom transfer (HAT), which is the specific case for an electron and a proton originating from the same atom (Cukier 2002). Amino acid radical generation often occurs by HAT (Stubbe & van der Donk 1998) as does the activation of the C–H bonds of substrates by oxidized cofactors such as those in lipoxygenase (Kohen & Klinman 1998; Liang & Klinman 2004), galactose oxidase (Maradufu et al. 1971; Ito et al. 1994; Whittaker 2005) and ribonucleotide reductase (RNR; Stubbe et al. 1983). In these cases, the thermodynamics for transport of the electron and the proton compels that they couple. This is most easily elucidated by the square scheme shown in figure 2. The thermochemistry of the diagonal PCET pathway is the sum of the thermochemistry of the constituent ET and PT stepwise pathways, which are indicated along the edges of the square scheme (Mayer 2004). For the case of amino acid radicals and for substrate activation at physiological pH values, the intermediate states of the stepwise pathways are energetically uphill. If the HAT pathway is energetically favoured with regard to these uphill steps for initial ET or PT, then the reaction may be directed along the diagonal.
Figure 2.

Square scheme describing the thermochemistry of different pathways of a HAT reaction. The overall reaction free energy is obtained by combining the relevant reduction potentials and pKa values corresponding to stepwise ET/PT (or PT/ET) reactions around the edges. Yet, in many cases, direct HAT along the diagonal is favoured in order to avoid energetic intermediates.
The other major category of PCET in biology (figure 1) is characterized by ET and PT pathways that are orthogonal to each other. Theoretical treatments of PCET confirm that proton motion can affect electron transport even when the electron and proton do not move along collinear coordinates (Cukier 1995, 1996, 1999, 2002; Cukier & Nocera 1998; Soudackov & Hammes-Schiffer 1999, 2000; Hammes-Schiffer 2001a,b). Furthermore, the same electron and proton do not have to be coupled throughout the entire transformation. As the electron moves, it may encounter different protons along a transport chain. All that is required for direct coupling is that the kinetics (and thermodynamics) of electron transport depends on the position of a specific proton or set of protons at any given time. It is direct coupling of the electron and proton that is the most elementary characteristic of a PCET event. The case of orthogonal PCET is more frequent than might be expected because the evolution of this pathway permits enzymes to manage the disparate electron and proton length scales. Electrons can transfer into and out of active sites over long distances in concert with protons that hop to or from the active site along amino acid side chains or along structured water channels. Mono-oxygenases such as cytochrome P450 (Poulos et al. 1985, 1987; Vidakovic et al. 1998; Meunier et al. 2004) and peroxidases (Miller et al. 1990; Newmyer & Ortiz de Montellano 1995; Tanaka et al. 1997) are exemplars of enzymes that operate by this type of PCET in biology. Redox activation of these enzymes to produce haem–oxo intermediates of compounds I and II occurs with the movement of protons along water channels or by amino acid side chains that connect the redox cofactor to the external aqueous environment (Pond et al. 2001).
Other oxidases also derive function from orthogonalized PCET pathways at the enzyme active site. The recent crystal structures of photosystem II (PSII; Ferreira et al. 2004; Loll et al. 2005) support suggestions that as the oxygen-evolving complex (OEC) steps through its various S-states (Vrettos & Brudvig 2002; Hoganson & Tommos 2004), substrate-derived protons are shuttled to the lumen via a proton exit channel, the headwater of which appears to be the D61 residue hydrogen bonded to Mn-bound water (Barber et al. 2004). As shown by the structure reproduced in figure 3, D61 is diametrically opposite to YZ, which has long been known (Barry & Babcock 1987; Debus et al. 1988a,b) to be the electron relay between the reaction centre and the OEC. The crystal structure of PSII also suggests that orthogonal PCET may be used to generate amino acid radicals. Functional schemes of PSII prior to the crystal structure suggested that YZ was linked to a water channel via H190 (Tommos & Babcock 1998, 2000) and hence was the nexus for the requisite electron and proton transport of OEC. However, the more recently obtained crystal structure shows the YZ–H190 pair to be relatively isolated by α-helices. Yet, for reasons described in figure 2, oxidation of YZ requires proton dissociation from the phenolic oxygen. The quandary is overcome by the intervention of H190, which is positioned in hydrogen-bonding contact to YZ. Redox activity from YZ may be supported by PT to and from H190 since electrons are transferred through the phenol ring of tyrosine. We emphasize that the crystal structure does not capture conformational dynamics that may expose the H190–YZ pair to the solvent. Indeed, the efficiency of YZ oxidation can be titrated with pH, in both the natural system and the H190 mutants (Debus 2001), suggesting some degree of proton access from the YZ–H190 pair to the bulk.
Figure 3.
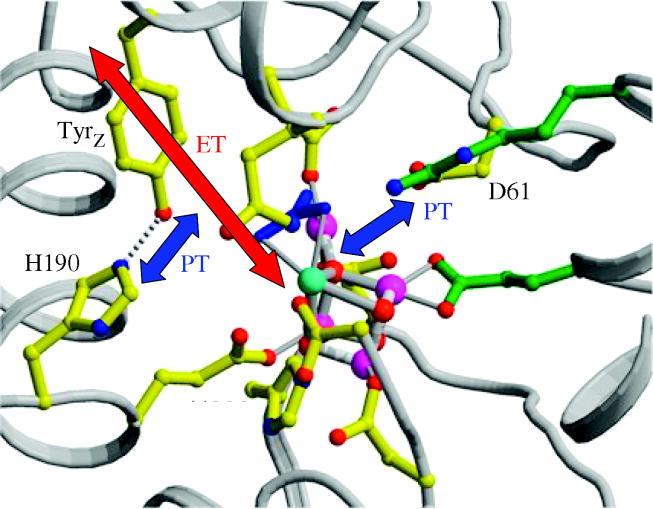
The 3.4 Å resolution structure of the oxygen evolving complex (OEC) and the immediate peptide environment adapted from Ferreira et al. (2004). The direction of proposed PT and ET pathways are indicated with arrows.
Orthogonal PCETs are operable in the concerted function of the bacterial photosynthetic reaction centre and cytochrome bc1 complex. Excitation of the reaction centre initiates charge separation via ET, which results in the sequential two-electron reduction of quinone QB. The quinone reduction is coupled to two sequential proton uptakes to form the hydroquinone, the second of which is a strongly coupled PCET event (Okamura et al. 2000). Cytochrome bc1 couples the oxidation of hydroquinone at the Q0-binding site to translocation of the protons across the membrane. This coupling most probably proceeds via either concerted two-electron chemistry or conformationally gated one-electron chemistry involving the semi-quinone intermediate (Osyczka et al. 2004).
Orthogonal PCET is also prevalent in reductases. Crystal structures of hydrogenases (Peters et al. 1998; Nicolet et al. 1999; Volbeda & Fontecilla-Camps 2005) indicate that the mechanism for hydrogen production occurs by transporting protons into the active site along pathways distinct from those traversed by the electron equivalents. Electrons are injected putatively into the active site via a chain of [FeS] clusters, while proton channels and acidic/basic residues at the active site manage the substrate inventory.
This discussion will present our approach to developing new chemical and biological tools to unravel the mechanistic details of PCET. The discussion will begin by presenting model systems that permit the proton and electron tunnelling events to be disentangled for collinear and orthogonal PCETs. We will then show, by the lessons learned from these model systems, that PCET may be exploited in biomimetic mono-oxygenases, which display unprecedented multifunctional, catalytic activity. The discussion will conclude with the case study of radical-based quantum catalysis in a natural biological enzyme, class I Escherichia coli RNR. This enzyme utilizes both collinear and orthogonal PCETs to transport charge from an assembled diiron-tyrosyl radical cofactor to the active site over 35 Å away via an amino acid radical-hopping pathway spanning two protein subunits.
2. Proton-coupled electron transfer model systems
The need to account for the effect of proton motion on ET in the PCET problem requires the development of new methods in chemistry and biology. In devising these new approaches, lessons learnt from the study of ET should not be ignored. Paramount among these is the benefits garnered from mechanistic studies of unimolecular ET over a fixed distance. This is best seen from the large body of work in the 1970s on the bimolecular ET reactions between redox proteins (primarily cytochrome c) and untethered small-molecule reactants (Marcus & Sutin 1985). Diffusion of reactants and large Coulombic contributions obscured the factors governing ET events. Only with the advent of new methods that enabled ET to be examined over fixed distances were theories able to be tested and redox reactions developed with predictability. The same benefits are expected to be garnered from studying PCET at fixed distance, but the challenge is compounded by the requirement to control two distances—those of ET and PT. The task is a challenge to synthesis because it must deliver control over the primary coordination sphere for tuning the ET event, and control over the secondary coordination sphere for tuning the PT event. In biology, the secondary and tertiary structures about redox cofactors provide a fixed distance pathway for PT. Such principles need to be incorporated in models designed to probe the mechanistic details of PCET. Some of these strategies under development in our group are described.
(a) Collinear proton-coupled electron transfer networks
The first model systems designed to interrogate PCET reactions in a controlled manner are depicted in figure 4a. An electron donor (D) and acceptor (A) are assembled with a PT interface (–[H+]–), thus aligning ET and PT coordinates in a collinear D–[H+]–A fashion. PCET is triggered by laser excitation of the donor or acceptor and resolved kinetically by performing time-resolved spectroscopy. These systems have provided tangible kinetics benchmarks for PCET reactions (Turró et al. 1992; Kirby et al. 1995, 1997; Roberts et al. 1995, 1997; Deng et al. 1997; Yeh et al. 2001b; Damrauer et al. 2004) and stimulated the development of theories to describe PCET (Cukier & Nocera 1998; Hammes-Schiffer 2001a,b).
Figure 4.

(a) Model collinear PCET complexes assembled from a symmetric carboxylic acid dimer interface or an asymmetric amidinium–carboxylate interface. D is a photo-excitable donor; A is an electron acceptor. (b) An assembly used to study the role of the mediating proton in collinear PCET by the temperature dependence of the deuterium isotope effect (Damrauer et al. 2004; Hodgkiss et al. 2006).
The initial D–[H+]–A construct ([H+]=[(COOH)2], D=zinc(II) porphyrin, A=dinitrobenzene) exploited the propensity of carboxylic acids to form cyclic dimers in low polarity, non-hydrogen bonding solvents (Turró et al. 1992). Observation of a deuterium isotope effect for charge separation and recombination revealed the coupling between electron and proton. Within the –[(COOH)2]– interface, proton displacement on one side of the dicarboxylic acid interface is compensated by the concomitant displacement of a proton from the other side. Since charge redistribution within this interface is negligible, the only available mechanism for PCET arises from the dependence of the electronic coupling matrix element on the position of the protons within the interface (Cukier 1994; Zhao & Cukier 1995; Cukier et al. 2002). Similar results have been obtained for donors and acceptors separated by guanine–cytosine base pairs (Shafirovich et al. 1995; Sessler et al. 1996; Ward 1997) and related interfaces (Ghaddar et al. 2000; Chang et al. 2001), where net proton motion within the interface is minimal.
To induce proton motion along an ET pathway, collinear systems with asymmetric interfaces such as salt bridges between donor–acceptor pairs have been constructed (Kirby et al. 1995, 1997; Roberts et al. 1995, 1997; Deng et al. 1997). A prominent system of this type that we have exploited features an amidinium–carboxylate salt bridge (figure 4) as the asymmetric interface. The pronounced effect of the proton on the ET rate is immediately evident from a comparative kinetics study of a D–[amidinium–carboxylate]–A complex and its inverted interfacial D–[carboxylate-amidinium]–A counterpart (, A=dinitrobenzene; Roberts et al. 1995; Kirby et al. 1997). The rate of charge transfer between donor and acceptor along a linear D–(carboxylate–amidinium)–A pathway (kPCET=3.1×108 s−1) is attenuated ca 40-fold when the interface is switched, D–(amidinium–carboxylate)–A. From these experiments, we find that the driving force and reorganization energy depend on the charge distribution of the electron and the proton because the initial and the final charge values are dependent on whether the process corresponds to ET, PT or PCET. Therefore, the two parameters that determine the rate of a charge transfer reaction, the activation energy and the electronic coupling, depend on the reaction pathway. The coupling of the charge shift resulting from electron and proton motion to the polarization of the surrounding environment thus embodies the essential distinguishing characteristic of a PCET reaction.
The effects of tunnelling on PCET rates through asymmetric interfaces have been uncovered with assembly shown in figure 4b. Note that in figure 4b the - [H+] - interface is represented in its non-ionized tautomeric form, which has been shown to prevail for carboxylic acids of comparable pKαs in low-dielectric environments (Rosenthal et al. In press). Photoexcitation of the Zn(II) porphyrin photoreductant prompts ET to the naphthalene diimide electron acceptor via the amidinium–carboxylate acid interface (Damrauer et al. 2004). This reaction may be followed by monitoring the growth and decay of the porphyrin cation radical transient absorption (kPCET(fwd)=9×108 s−1 and kPCET(rev)=14×108 s−1). The forward rate constant is attenuated by nearly two orders of magnitude when compared with that measured for a dyad consisting of nearly identical donor and acceptor moieties positioned in a comparable geometry, but via covalent bonds rather than hydrogen bonds (Osuka et al. 1998). Since the thermodynamic driving force and solvent reorganization energies for ET in these two systems are comparable, this comparison implies that the bridging hydrogen bonds in our model system attenuate the electronic coupling between the electron donor and acceptor, consistent with earlier reports (Beratan et al. 1987, 1990; Onuchic & Beratan 1990). The nuclear and electronic contributions to the PCET reaction may be unravelled from temperature-dependent kinetics measurements (Hodgkiss et al. 2006). A small electronic coupling term (V=2.4 cm–1) supports the contention that the hydrogen-bonding interface is the bottleneck for electronic coupling in this system. Extending the temperature-dependent measurements to a deuterated salt bridge yields the surprising result shown in figure 5. The kinetic isotope effect switches from being normal at high temperature (kH/kD∼1.2, 300 K) to inverted at low temperature (kH/kD∼0.9, 120 K). This is interpreted in a model where fluctuations within the hydrogen-bonding bridge dynamically modulate electronic coupling for ET, and consequently the rate of charge-separation becomes sensitive to the nature of proton modes within the bridge (Pressé & Silbey 2006). Thermal population of vibrational states is the most probable cause for the reverse isotope effect in this system, where the low-frequency mode is a localized vibration in the hydrogen bond. At low enough temperatures, the thermally induced shift in the deuteron probability density contributes more to the PCET rate compared with the protiated form of the interface (due to the difference in zero-point energies). The normal isotope effect is recovered with increasing temperature as the lowest lying excited states of the hydrogen bond vibration of interest begin contributing to the PCET rate. This microscopic insight into the role of mediating protons is very pertinent to collinear PCET in biology, where asymmetrical hydrogen-bonding networks are frequently the bottlenecks for electron transport. However, this level of understanding can only be reached when the relevant dynamics are isolated in well-defined and spectroscopically accessible model systems such as that shown in figure 4b.
Figure 5.

Temperature dependence of the rate of PCET in the porphyrin assembly shown in figure 4b for a protiated (solid circles) and deuterated (open circles) amidinium–carboxylate interface in the solvent 2-methyl tetrahydrofuran (2-MeTHF). Data are presented in a modified Arrhenius form with linear fits (Hodgkiss et al. 2006). Reproduced with permission from J. Phys. Chem. B. Copyright © 2006, Am. Chem. Soc.
(b) Orthogonal proton-coupled electron transfer networks
The collinear PCET networks of §2a impose an inherent limitation on negotiating the length-scale disparity between PT and ET. The network is assembled by the hydrogen bonds of the PT interface; hence, PT distances are confined to the hydrogen bond length scale. To overcome this limitation, we are constructing the PCET model systems shown in figure 6a in which ET and PT coordinates are orthogonalized. The PT distance is established by a rigid scaffold that poises an acid–base group above an ET conduit. In the ‘Hangman’ porphyrin architecture, a carboxylic acid or amidine is positioned over a PFeIII(OH) (P=porphyrin) redox platform via a xanthene or dibenzofuran spacer (Yeh et al. 2001a; Chang et al. 2003; Chng et al. 2003). By appending an electron acceptor to the porphyrin platform, the PCET reaction may generate the ferryl: PFeIII(OH)−e−−H+→PFeIV(O) by PT to the hanging group upon ET from the haem centre. Conversely, by appending an electron donor to the porphyrin platform, the PCET reaction PFeIII(OH)+e−+H+→PFeII(OH2) may be accessed, where PT occurs from the hanging group upon ET to the haem.
Figure 6.

(a) Schematic of model systems for studying fixed-distance orthogonal PCET. Excitation of the photo-oxidant triggers ET from the iron porphyrin, which donates a proton to the base (B) appended to the Hangman pillar. The key features of this design are that the ET and PT kinetics can be tuned through variation of the spacer and pillar, respectively, and driving forces for each of these processes can be tuned independently by varying the photo-oxidant and the base. Alternatively, if a photo-excitable donor is appended to the porphyrin, the PCET reaction can be run in the opposite direction (ET to the porphyrin along with protonation upon reduction). (b) A line drawing and crystal structure of the Hangman porphyrin showing the water channel adapted from (Yeh et al. 2001a).
The Hangman constructs allow us to investigate incisively the role of proton tunnelling in PCET because the PT distance is easily tuned with the length of the Hangman pillar. For example, we have structurally characterized the Hangman porphyrin xanthene (HPX; figure 6b) and shown that a water molecule (with a binding energy of 5.8 kcal mol−1; Chang et al. 2003) is suspended between the xanthene carboxylic acid hanging group and the hydroxide ligand (Yeh et al. 2001a). This is the first synthetic redox active site displaying an assembled water molecule as part of a structurally well-defined PT network. By interposing a water molecule near the Hangman xanthene spacer, we can switch from a configuration requiring a long-distance PT event (approx. 3.5 Å) coupled to ET, to one requiring two short-distance PT events (1.8 Å, proton hopping along a water chain). Alternatively, we have shown that the PT distance may be tuned with the length of the spacer (Chang et al. 2003). With our ability to control precisely the PT distance, theoretical predictions of how the isotope effect varies in a PCET reaction (Decornez & Hammes-Schiffer 2000) with PT distance may be tested.
(c) Proton-coupled electron transfer biocatalysis
A PT network disposed orthogonally to a haem redox cofactor is prevalent in structures of enzymes that derive their function from oxygen activation. The structure of cytochrome P450 is exemplary in this respect (Poulos et al. 1986). Figure 7a focuses on the haem and its hard-wired water channel along which PT is directed. The highly activated ferryl oxygen of the redox cofactor, FeIV=O (compound I-type intermediate in which a FeIV=O centre resides in a porphyrin cation radical, ), is produced by PT from the water channel to a ferric peroxy intermediate, as shown in figure 7b. Formation of the high-valent metal oxo fragment is thus accomplished by coupling PT to an internal 2e− redox event.
Figure 7.

(a) High-resolution structure of cytochrome P450, displaying a water channel above the haem adapted from Poulos et al. (1986). (b) The peroxo-shunt mechanism of mono-oxygenases produces compound I (()FeIV=O), which oxidizes substrates by their nucleophilic attack on the electrophilic oxo of the ()FeIV=O core. Reproduced with permission from Inorg. Chem. 44, 6879–6892. Copyright © 2006, Am. Chem. Soc.
The Hangman porphyrin is a simplified construct of cytochrome P450. The protonic pendant acid group of the Hangman porphyrin plays the same role as the water channel of the natural enzyme. Accordingly, the functional activity of cytochrome P450 is captured by the Hangman construct (Chang et al. 2003). PT from the acid–base hanging group in (HPX)FeIII peroxide complexes yields (HX)FeIV=O (Dempsey et al. 2005). In the presence of olefins, epoxidation occurs at high turnover (Chang et al. 2003). In the absence of substrate, the compound I-type intermediate reacts with peroxide to generate oxygen and water in a catalase-like reactivity, also at high turnover (Chng et al. 2003). Mono-oxygenase and catalase activities are lost when the scaffold is extended and the proton must transfer over long distance (Chang et al. 2003). Activity is also reduced severely when the pKa of the hanging acid–base group is increased (Chng et al. 2003).
The studies on these Hangman porphyrins and other macrocyclic redox platforms (Liu & Nocera 2005) clearly demonstrate that exceptional catalysis may be achieved when redox and PT properties of a cofactor are controlled independently. A key requirement is that the PT distance is kept short, which may be accomplished by orthogonalizing ET and PT coordinates. The benefits of incorporating PT functionality into redox catalysis can only be realized when a suitable geometry is established. Moreover, the Hangman platforms show that a multifunctional activity of a single metalloporphyrin-based scaffold is achieved by the addition of proton control to a redox platform. This observation is evocative of natural haem-dependent proteins that employ a conserved protoporphyrin IX cofactor to affect a myriad of chemical reactivities.
3. Proton-coupled electron transfer in enzymes: a study of ribonucleotide reductase
The study of PCET in natural systems provides the convenience that both the ET and the PT groups are held at fixed distance by the secondary and tertiary structures of the protein. Thus, the laborious synthesis attendant to constructing fixed-distance ET and PT pathways is circumvented. However, PCET investigations of the natural systems brings the new challenge that the PCET reaction is typically part of a more complicated cascade of events including, but not limited to, protein–protein interactions, binding/release of substrate, protons or redox equivalents, and intra-protein conformational dynamics, all of which may be necessary for enzymatic function. Thus, the PCET event must be isolated in the biological system, prompting us to develop new biochemical and biophysical methods for the study of PCET in biology. Paramount among these new methods are:
photo-active non-natural amino acids/redox platforms that enable PCET to be photo-triggered;
non-natural amino acids that permit the examination of ET and PT by control of pKa and redox potentials;
the implementation of new biochemical methods that permit the non-natural amino acids to be selectively introduced along putative PCET pathways of the enzyme.
We demonstrate the utility of these new methods with studies aimed at elaborating the PCET reactivity in RNR.
(a) The putative proton-coupled electron transfer pathway in ribonucleotide reductase
Class I E. coli RNR is composed of two homodimeric subunits designated R1 and R2, and a complex between the two catalyses the reduction of nucleoside diphosphates to deoxynucleoside diphosphates (Jordan & Reichard 1998; Stubbe & van der Donk 1998; Stubbe et al. 2003). R2 harbours the diferric tyrosyl radical (·Y122) cofactor that initiates nucleotide reduction by generating a transient thiyl radical (·C439) in the enzyme active site located in R1 (Stubbe & Riggs-Gelasco 1998). The crystal structures of both R1 and R2 have been solved independently (Nordlund et al. 1990; Uhlin & Eklund 1994; Högbom et al. 2003) and a docking model, which places the Y· on R2 at a distance greater than 35 Å away from the C439 residue on R1, has been proposed (Uhlin & Eklund 1994). Radical transfer over this distance has been proposed to occur via a PCET radical-hopping pathway involving radical intermediates of the aromatic amino acid residues shown in figure 8 (Stubbe et al. 2003). Specifically, the radical is suggested to transfer along the pathway ·Y122→W48→Y356 in R2 to Y731→Y730→C439 in R1.
Figure 8.

Conserved residues of class I RNR that compose the putative PCET pathway for radical transport from ·Y122 in R2 to C439 in the R1 active site (Seyedsayamdost et al. 2005b). Distances are from the separate crystal structures of the R1 (Uhlin & Eklund 1994) and R2 (Högbom et al. 2003) subunit from the E. coli enzyme. Reproduced with permission from J. Am. Chem. Soc. (in press). Copyright © 2006, Am. Chem. Soc.
(b) Proton-coupled electron transfer in the R2 subunit of ribonucleotide reductase
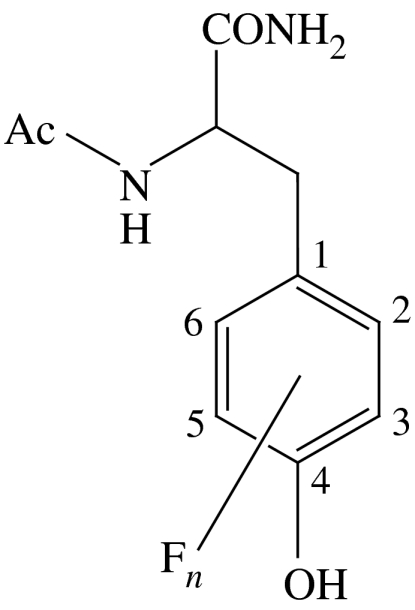
As the proposed gatekeeper for radical transport between R1 and R2, Y356 is pre-eminent to the PCET pathway of RNR. The limited repertoire of natural amino acids does not allow an informative perturbation to be made, and thus we have recently turned to site-specific insertions of unnatural amino acids into R2 at Y356 using protein ligation methods (Muir 2003). We have replaced Y356 with a series of fluorotyrosine (Yee et al. 2003a; Seyedsayamdost et al. 2005b), 3-nitrotyrosine (Yee et al. 2003b) and aniline (Chang et al. 2004b) unnatural amino acids, thus allowing us to systematically vary both the pKa and the reduction potential (Ep) of the radical at this position. The fluorotyrosines (FnY) listed in table 1 have permitted the most detailed investigation owing to a large range in Ep(FnY·/FnY−) values (from −50 to +270 mV, relative to Ep(Y·/Y) between 6<pH<9) and pKa values (from 5.6 to 9.9; Seyedsayamdost et al. 2006a). pH rate profiles of deoxynucleotide production by the FnY356-R2/R1 complexes suggest that the rate-determining step in RNR turnover can be changed from a physical (conformational) step to the radical propagation step, by altering the reduction potential with FnYs (Seyedsayamdost et al. 2006b). Figure 9 plots the activity of each FnY356-R2 relative to wild type versus Ep of FnY·, relative to Y·. For Ep>80 mV versus Y·, the activity of RNR is inhibited, and for Ep>200 mV nucleotide reduction is no longer detectable. These studies support the contention that Y356 is a redox active amino acid on the radical propagation pathway. Open and closed circles in figure 9 represent deprotonated (indicated by open circles) and protonated (filled circles) FnY-R2s, respectively, based on the pKa values in table 1. Both deprotonated and protonated forms have similar activities for Ep<80 mV versus Y·, establishing that the protonation state of Y356 does not affect the activity of the enzyme and hence that a proton at Y356 is not obligated to the pathway. These results lead us to propose that, upon oxidation of Y356, the proton is transferred ‘off-pathway’ in an orthogonal manner to bulk solution, either directly or via amino acid residues of the enzyme. This result becomes more profound when taken together with the suggestion that the oxygen of a water/hydroxo bound to Fe1 in the resting-state crystal structure of R2 (Högbom et al. 2003) is the probable PT partner of Y122 (Stubbe et al. 2003). With oxidation and reduction of the Y122 and Y356 termini coupled to an orthogonal PT over a short distance, radical transport through R2 may occur via long-range ET. However, we reiterate that Y356 is thermally labile in the crystal structure of R2 (Högbom et al. 2003), and hence its location between W48 of R2 and Y731 of R1 is unknown and may vary during turnover.
Table 1.
Physical properties of fluorotyrosine derivatives substituted for Y356 in R2 (data from Seyedsayamdost et al. 2005a).
 | ||
|---|---|---|
| fluorotyrosine | pKa | Ep(FnY·/FnY−) (mV) versus NHEa |
| Ac-Y-NH2 | 9.9 | 642 |
| Ac-3,5-F2Y-NH2 | 7.2 | 755 |
| Ac-2,3-F2Y-NH2 | 7.8 | 810 |
| Ac-2,3,5-F3Y-NH2 | 6.4 | 853 |
| Ac-2,3,6-F3Y-NH2 | 7.0 | 911 |
| Ac-F4Y-NH2 | 5.6 | 968 |
NHE, normal hydrogen electrode.
Figure 9.

Redox potential regimes of RNR activity (Seyedsayamdost et al. 2005b). Relative activities of FnY356-R2s versus Y-R2, plotted as a function of peak reduction potential difference between the corresponding Ac-FnY·-NH2 and Ac-Y·-NH2: (blue filled and open circles) 3,5-F2Y356-R2, (red filled and open circles) 2,3-F2Y356-R2, (magenta circles) 2,3,5-F3Y356-R2, (green filled and open circles) 2,3,6-F3Y356-R2, and (orange circles) F4Y356-R2. Filled circles represent data points where pH<pKa of the corresponding Ac-FnY-NH2; open circles represent data points where pH>pKa of the corresponding Ac-FnY-NH2. The three different regimes of RNR activity are highlighted as either gated by a physical/conformational change (regime 1), rate-limited by radical transport (regime 2), or reduced to background levels (regime 3) depending on the peak reduction potential difference between the corresponding Ac-FnY·-NH2 and Ac-Y·-NH2. Reproduced with permission from J. Am. Chem. Soc. (in press). Copyright © 2006, Am. Chem. Soc.
Against this backdrop of a long-distance ET in R2, a further insight into the role of PCET has come from transient kinetic studies of W–Y dipeptides. We have shown that the rate and the directionality of radical transfer between W and Y in dipeptides can be controlled by changing the pH of the bulk solution, thereby affecting the reduction potentials of the corresponding radicals (Reece et al. 2005). At physiological pH values, Y· has a lower reduction potential and the direction of ET is W·–Y→W–Y·; the direction changes at high pH, W–Y·→W·–Y−. These results dovetail with biochemical studies of RNR. Nordlund & Eklund (Nordlund et al. 1990; Nordlund & Eklund 1993) were the first to recognize the potential mechanistic importance of W48 and [·W48H]+ by proposing the Fe1→H118→D237→W48 pathway in cofactor assembly and nucleotide reduction, based on the structurally analogous and spectroscopically well-characterized Fe(haem)→H→D→W ET pathway in cytochrome c peroxidase. Subsequent studies have shown that, under certain conditions, [·W48H]+ plays a key role in cofactor assembly (Bollinger et al. 1991, 1994; Baldwin et al. 2000; Krebs et al. 2000). These results, taken with site-directed mutagenesis studies (Rova et al. 1995), have led us to support a model in which W48 is central to a PCET pathway in R2 that directs both the cofactor assembly and the initiation of nucleotide reduction (Stubbe et al. 2003). As for the latter, previous kinetic studies have led to the proposal that PCET occurs in the forward and reverse directions along the 35 Å pathway, each time nucleotide reduction occurs (Ge et al. 2003; Stubbe et al. 2003). The crystal structure R2 (Högbom et al. 2003) shows that the indole nitrogen of W48 is hydrogen bonded to the carboxylate oxygen of D237 located 2.9 Å away (figure 8). Based on the W–Y dipeptide results, D237 most probably provides a site to incorporate PCET by coupling ET along the radical transport pathway to an orthogonal PT to or from W48, thus providing a mechanism in R2 to control the direction of radical transport between Y122 and Y356.
(c) Proton-coupled electron transfer in R1 subunit of ribonucleotide reductase
The Y731↔Y730↔C439 triad in R1 connects Y356 of R2 to the active site. As shown by the distances in figure 8, the triad is in hydrogen-bonding contact. We have developed the methods summarized in figure 10 to investigate this pathway. Briefly, the 20-mer C-terminal peptide tail (R2C20, NH2–YLVGQIDSEVDTDDLSNFQL–COOH) of the R2 subunit has been retained because this peptide contains the critical Y356 and the binding determinant of R2 to R1 (Climent et al. 1991, 1992). Using solid-phase peptide synthesis, the C-terminal peptide tail of R2 is produced with a photo-oxidant appended proximal to Y356 on the peptide (figure 10, red circle). Laser excitation of the modified peptide provides a method to generate ·Y356, bypassing hole generation at the metallo-cofactor therein and allowing us to instantaneously ‘turn on’ the PCET pathway in RNR. The design shown in figure 10 is predicated on using a photo-generated ·Y356 to initiate radical transport into R1, produce the thiyl radical at the active site, and consequently induce RNR activity. We established the validity of this construct using tryptophan as the photo-oxidant (Chang et al. 2004c). Photoionization of W with UV light (λ<290 nm) irreversibly generates W·, which in turn oxidizes Y within the pH range relevant to RNR (Reece et al. 2005). The excitation wavelength required for W photoionization (and consequently W· formation) falls within the protein envelope. The drawbacks attendant to the protein acting as an ‘inner-filter’ of the excitation light led us to explore the viability of unnatural benzophenone-containing amino acids (BPA) as an excited-state oxidant of tyrosine (Reece et al. in preparation). Excitation of the BPA-modified peptide with UV light (λexc>325 nm) yields the benzophenone-ketyl radical and tyrosyl radical on the sub-nanosecond timescale; the photo-generated radicals recombine with a 500 ns time constant. Figure 11 shows that, under single turnover conditions with the peptide bound to R1 and in the presence of CDP substrate and ATP effector, the enzyme is active when excited by light and inactive in the absence of excitation. Mutation of Y730 to F breaks the hydrogen bond network in R1 and increases the electron and proton tunnelling distance, as now the radical must tunnel directly from C439 to reduce the hole on Y731·. As seen in figure 11, this mutant is effectively inactive towards photo-initiated nucleotide reduction. This result provides strong evidence that radical transport is indeed pathway specific in R1 and suggests a collinear PCET pathway.
Figure 10.

Experimental design for studying the kinetics of radical transport along ·Y356→Y731→Y730→C439 pathway. ·Y356 is generated photochemically by a proximal photo-oxidant (red circle) on the R2C19 peptide. NDP, nucleoside diphosphate substrate; dNDP, deoxynucleoside diphosphate product; R2C19, 19-mer C-terminal peptide tail of R2.
Figure 11.

Light-initiated single turnover experiments in R1-peptide complexes. Samples were irradiated using 299 nm long pass filters at room temperature for 2 min. Light (cyan) and dark (grey) bars correspond to light reactions and dark controls, respectively. R1 was purified and dC product quantitated as previously described (Chang et al. 2004c).
(d) A model for proton-coupled electron transfer in ribonucleotide reductase
Figure 12 presents the present model for PCET in RNR. Beginning at the cofactor, an orthogonal PT between Y122 and the diiron oxo/hydroxo cofactor establishes the need only for the transfer of an electron through the span of R2. Oxidation of Y356, the redox terminus of the R2 pathway, demands a PCET reaction; however, this also appears to involve a PT orthogonal to the ET pathway. By moving the protons at Y122 and Y356 off pathway, the radical transport in R2 involves a long-distance ET coupled to short PT hops at the tyrosine endpoints. In setting up the radical transport pathway in this fashion, the very different PT and ET length scales are managed in RNR. The direction of ET along the pathway may logically be ascribed to control by an orthogonal PT between W48 and D236. Dipeptide W–Y studies are consistent with this contention. Within R1, the activity studies of the Ac-(W/BPA)-R2C20 peptide and R1, together with those of the Y730/731F R1 mutant, suggest a collinear PCET pathway through R1 in which both the electron and the proton may be transferred between Y731–Y730–C439. Such a transfer is unusual inasmuch as radical transport occurs by committing both the proton and electron to the pathway. For instance, in photolyase, radical transport also occurs among a triad of amino acid radicals (WWW) on the sub-nanosecond timescale to form the [·W306H]+ radical on the protein surface, which then deprotonates to bulk solution (Aubert et al. 2000). In this case, ET is used for radical transport rather than the PCET, as observed in R1 of RNR. In contrast to most systems studied to date in biology, RNR appears to incorporate all the variances of PCET mechanisms in its transport of a radical across two subunits and over 35 Å.
Figure 12.

Proposed model for radical transport in RNR. The mode of transport at the interface (between Y356 and 731) is undefined.
(e) Future prospects
As of now, we have no direct information on the radical transport mechanism between subunits, i.e. Y356 and Y731. To resolve the ·Y356→Y731 radical hop from the peptide into R1, new tools need to be developed to distinguish the two Y·s spectrally. The fluorotyrosines prove useful in this regard. Figure 13 shows the transient absorption spectra of benzophenone–fluorotyrosine dipeptides (BPA-FnY-OMe) following nanosecond excitation (Seyedsayamdost et al. 2005a). The feature at 547 nm is constant for each BPA-FnY-OMe and corresponds to the benzophenone ketyl radical; the absorption to the blue is attributed to FnY·. Shifts in the maximum are observed from 395 to 415 nm depending on the extent and location of fluorine substitution. In most cases, the FnY· spectra are clearly distinguished from that of Y·. Thus, incorporation of fluorotyrosines at position 356 on the R2 peptide should allow us the direct measurement of the time-course of ·Y356→Y731 by time-resolved UV–vis spectroscopy. These studies will be facilitated by photo-oxidants that can be excited well to the red of the RNR's absorption profile. We have addressed this issue as well by delineating the details of Y oxidation directly by developing the photo-oxidation of Y by BPA, which can be excited at λexc>300 nm (Reece et al. in preparation). Even further shifts into the red spectral region can be achieved by exploiting the spectroscopy of metal-based photo-oxidants. We have shown that the metal-to-ligand charge transfer excited states of ReI polypyridyl complexes (Reece & Nocera 2005) can be engineered to efficiently oxidize Y with λexc>400 nm. Therefore, choice of the appropriate photo-oxidant and a comprehensive study of the FnY-R2C19 peptides should allow direct characterization of the amino acid radical intermediates in R1 and the rate for radical injection into R1. The PCET contribution to these kinetics can be de-convolved by exploiting the Ep and pKa differences of the FnY as we have done in the studies described in §3b.
Figure 13.

The transient absorption spectrum of (black) BPA-Y-OMe, (cyan) BPA-3-FY-OMe, (blue) BPA-3,5-F2Y-OMe, (red) BPA-2,3-F2Y-OMe, (green) BPA-2,3,6-F3Y-OMe, (violet) BPA-2,3,5-F3Y-OMe and (orange) BPA-F4Y-OMe obtained 100 ns after excitation of ca 500 μM solutions of each dipeptide buffered to pH 4.0 with 20 mM succinic acid and normalized to the ketyl radical absorption peak at 547 nm (Seyedsayamdost et al. 2006a). Reproduced with permission from J. Am. Chem. Soc. Copyright © 2006, Am. Chem. Soc.
Acknowledgements
The National Institutes of Health supported this work with grants GM47274 (D.G.N.) and GM29595 (J.S.).
Footnotes
One contribution of 16 to a Discussion Meeting Issue ‘Quantum catalysis in enzymes—beyond the transition state theory paradigm’.
References
- Aubert C, Vos M.H, Mathis P, Eker A.P.M, Brettel K. Intraprotein radical transfer during photoactivation of DNA photolyase. Nature. 2000;405:586–590. doi: 10.1038/35014644. doi:10.1038/35014644 [DOI] [PubMed] [Google Scholar]
- Baldwin J, Krebs C, Ley B.A, Edmondson D.E, Huynh B.H, Bollinger J.M., Jr Mechanism of rapid electron transfer during oxygen activation in the R2 subunit of Escherichia coli ribonucleotide reductase. 1. Evidence for a transient tryptophan radical. J. Am. Chem. Soc. 2000;122:12 195–12 206. doi:10.1021/ja001278u [Google Scholar]
- Barber J, Ferreira K, Maghlaoui K, Iwata S. Structural model of the oxygen-evolving center of photosystem II with mechanistic implications. Phys. Chem. Chem. Phys. 2004;6:4737–4742. doi:10.1039/b407981g [Google Scholar]
- Barry B, Babcock G.T. Tyrosine radicals are involved in the photosynthetic oxygen-evolving system. Proc. Natl Acad. Sci. USA. 1987;84:7099–7103. doi: 10.1073/pnas.84.20.7099. doi:10.1073/pnas.84.20.7099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beratan D.N, Onuchic J.N, Hopfield J.J. Electron tunneling through covalent and noncovalent pathways in proteins. J. Chem. Phys. 1987;86:4488–4498. doi:10.1063/1.452723 [Google Scholar]
- Beratan D.N, Onuchic J.N, Betts J.N, Bowler B.E, Gray H.B. Electron-tunneling pathways in ruthenated proteins. J. Am. Chem. Soc. 1990;112:7915–7921. doi:10.1021/ja00178a011 [Google Scholar]
- Beratan D.N, Betts J.N, Onuchic J.N. Protein ET rates set by the bridging secondary and tertiary structure. Science. 1991;252:1285–1288. doi: 10.1126/science.1656523. [DOI] [PubMed] [Google Scholar]
- Bollinger J.M, Jr, Edmonson D.E, Huynh B.H, Filley J, Norton J.R, Stubbe J. Mechanism of assembly of the tyrosyl radical-dinuclear iron cluster cofactor of ribonucleotide reductase. Science. 1991;253:292–298. doi: 10.1126/science.1650033. [DOI] [PubMed] [Google Scholar]
- Bollinger J.M, Jr, Tong W.H, Ravi N, Huynh B.H, Edmondson D.E, Stubbe J. Mechanism of assembly of the tyrosyl radical-diiron(III) cofactor of E. coli ribonucleotide reductase. 3. Kinetics of the limiting Fe2+ reaction by optical, EPR, and Mössbauer spectroscopies. J. Am. Chem. Soc. 1994;116:8024–8032. doi:10.1021/ja00097a009 [Google Scholar]
- Chang C.J, Brown J.D.K, Chang M.C.Y, Baker E.A, Nocera D.G. Electron transfer in hydrogen-bonded donor–acceptor supramolecules. In: Balzani V, editor. Electron transfer in chemistry. vol. 3.2.4. Wiley-VCH; Weinheim, Germany: 2001. pp. 409–461. [Google Scholar]
- Chang C.J, Chng L.L, Nocera D.G. Proton-coupled O–O activation on a redox platform bearing a hydrogen-bonding scaffold. J. Am. Chem. Soc. 2003;125:1866–1876. doi: 10.1021/ja028548o. doi:10.1021/ja028548o [DOI] [PubMed] [Google Scholar]
- Chang C.J, Chang M.C.Y, Damrauer N.H, Nocera D.G. Proton-coupled electron transfer: a unifying mechanism for biological charge transport, amino acid radical initiation and propagation, and bond making/breaking reactions of water and oxygen. Biochim. Biophys. Acta. 2004a;1655:13–28. doi: 10.1016/j.bbabio.2003.08.010. doi:10.1016/j.bbabio.2003.08.010 [DOI] [PubMed] [Google Scholar]
- Chang M.C.Y, Yee C.S, Nocera D.G, Stubbe J. Site-specific replacement of a conserved tyrosine in ribonucleotide reductase with an aniline amino acid: a mechanistic probe for a redox-active tyrosine. J. Am. Chem. Soc. 2004b;126:16 702–16 703. doi: 10.1021/ja044124d. doi:10.1021/ja044124d [DOI] [PubMed] [Google Scholar]
- Chang M.C.Y, Yee C.S, Stubbe J, Nocera D.G. Turning on ribonucleotide reductase by light-initiated amino acid radical generation. Proc. Natl Acad. Sci. USA. 2004;101:6882–6887. doi: 10.1073/pnas.0401718101. doi:10.1073/pnas.0401718101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chng L.L, Chang C.J, Nocera D.G. Catalytic O–O activation chemistry mediated by iron Hangman porphyrins with a wide range of proton-donating abilities. Org. Lett. 2003;5:2421–2424. doi: 10.1021/ol034581j. doi:10.1021/ol034581j [DOI] [PubMed] [Google Scholar]
- Climent I, Sjöberg B.-M, Huang C.Y. Carboxyl-terminal peptides as probes for Escherichia coli ribonucleotide reductase subunit interaction: kinetic analysis of inhibition studies. Biochemistry. 1991;30:5164–5171. doi: 10.1021/bi00235a008. doi:10.1021/bi00235a008 [DOI] [PubMed] [Google Scholar]
- Climent I, Sjöberg B.-M, Huang C.Y. Site-directed mutagenesis and deletion of the carboxyl terminus of Escherichia coli ribonucleotide reductase protein R2. Effects on catalytic activity and subunit interaction. Biochemistry. 1992;31:4801–4807. doi: 10.1021/bi00135a009. doi:10.1021/bi00135a009 [DOI] [PubMed] [Google Scholar]
- Cukier R.I. Mechanism for proton-coupled electron-transfer reactions. J. Phys. Chem. 1994;98:2377–2381. doi:10.1021/j100060a027 [Google Scholar]
- Cukier R.I. Proton-coupled electron transfer through an asymmetric hydrogen-bonded interface. J. Phys. Chem. 1995;99:16 101–16 115. doi:10.1021/j100043a060 [Google Scholar]
- Cukier R.I. Proton-coupled electron transfer reactions: evaluation of rate constants. J. Phys. Chem. 1996;100:15 428–15 443. doi:10.1021/jp961025g [Google Scholar]
- Cukier R.I. A theory for the rate constant of a dissociative proton-coupled electron-transfer reaction. J. Phys. Chem. A. 1999;103:5989–5995. doi:10.1021/jp990329a [Google Scholar]
- Cukier R.I. A theory that connects proton-coupled electron-transfer and hydrogen-atom transfer reactions. J. Phys. Chem. B. 2002;106:1746–1757. doi:10.1021/jp012396m [Google Scholar]
- Cukier R.I, Nocera D.G. Proton-coupled electron transfer. Annu. Rev. Phys. Chem. 1998;49:337–369. doi: 10.1146/annurev.physchem.49.1.337. doi:10.1146/annurev.physchem.49.1.337 [DOI] [PubMed] [Google Scholar]
- Cukier R.I, Daniels S, Vinson E, Cave R.J. Are hydrogen bonds unique among weak interactions in their ability to mediate electronic coupling? J. Phys. Chem. A. 2002;106:11 240–11 247. doi:10.1021/jp026230c [Google Scholar]
- Damrauer N.H, Hodgkiss J.M, Rosenthal J, Nocera D.G. Observation of proton-coupled electron transfer by transient absorption spectroscopy in a hydrogen-bonded, porphyrin donor–acceptor assembly. J. Phys. Chem. B. 2004;108:6315–6321. doi: 10.1021/jp049296b. doi:10.1021/jp049296b [DOI] [PubMed] [Google Scholar]
- Debus R.J. Amino acid residues that modulate the properties of tyrosine YZ and the manganese cluster in the water oxidizing complex of photosystem II. Biochim. Biophys. Acta. 2001;1503:164–186. doi: 10.1016/s0005-2728(00)00221-8. doi:10.1016/S0005-2728(00)00221-8 [DOI] [PubMed] [Google Scholar]
- Debus R.J, Barry B.A, Sithole I, Babcock G.T, McIntosh L. Directed mutagenesis indicates that the donor to P680+ in photosystem II is tyrosine-161 of the D1 polypeptide. Biochemistry. 1988a;27:9071–9074. doi: 10.1021/bi00426a001. doi:10.1021/bi00426a001 [DOI] [PubMed] [Google Scholar]
- Debus R.J, Barry B.A, Babcock G.T, McIntosh L. Site-directed mutagenesis identifies a tyrosine radical involved in the photosynthetic oxygen-evolving system. Proc. Natl Acad. Sci. USA. 1988b;85:427–430. doi: 10.1073/pnas.85.2.427. doi:10.1073/pnas.85.2.427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decornez H, Hammes-Schiffer S. Model proton-coupled electron transfer reactions in solution: predictions of rates, mechanisms, and kinetic isotope effects. J. Phys. Chem. A. 2000;104:9370–9384. doi:10.1021/jp001967s [Google Scholar]
- Dempsey J.L, Esswein A.J, Manke D.R, Rosenthal J, Soper J.D, Nocera D.G. Molecular chemistry of consequence to renewable energy. Inorg. Chem. 2005;44:6879–6892. doi: 10.1021/ic0509276. doi:10.1021/ic0509276 [DOI] [PubMed] [Google Scholar]
- Deng Y, Roberts J.A, Peng S.M, Chang C.K, Nocera D.G. The amidinium–carboxylate salt bridge as a proton-coupled interface to electron transfer pathways. Angew. Chem. Int. Ed. Engl. 1997;36:2124–2127. doi:10.1002/anie.199721241 [Google Scholar]
- Ferreira K.N, Iverson T.M, Maghlaoui K, Barber J, Iwata S. Architecture of the photosynthetic oxygen-evolving center. Science. 2004;303:1831–1838. doi: 10.1126/science.1093087. doi:10.1126/science.1093087 [DOI] [PubMed] [Google Scholar]
- Ge J, Yu G, Ator M.A, Stubbe J. Pre-steady-state and steady-state kinetic analysis of E. coli class I ribonucleotide reductase. Biochemistry. 2003;42:10 071–10 083. doi: 10.1021/bi034374r. doi:10.1021/bi034374r [DOI] [PubMed] [Google Scholar]
- Ghaddar T.H, Castner E.W, Isied S.S. Molecular recognition and electron transfer across a hydrogen bonding interface. J. Am. Chem. Soc. 2000;122:1233–1234. doi:10.1021/ja9930465 [Google Scholar]
- Gray H.B, Winkler J.R. Electron transfer in proteins. Annu. Rev. Biochem. 1996;65:537–561. doi: 10.1146/annurev.bi.65.070196.002541. doi:10.1146/annurev.bi.65.070196.002541 [DOI] [PubMed] [Google Scholar]
- Hammes-Schiffer S. Theoretical perspectives on proton-coupled electron transfer reactions. Acc. Chem. Res. 2001a;34:273–281. doi: 10.1021/ar9901117. doi:10.1021/ar9901117 [DOI] [PubMed] [Google Scholar]
- Hammes-Schiffer S. Proton-coupled electron transfer. In: Balzani V, editor. Electron transfer in chemistry. vol. 1.1.5. Wiley-VCH; Weinheim, Germany: 2001. pp. 189–237. [Google Scholar]
- Hodgkiss, J. M., Damrauer, N. H., Pressé, S., Rosenthal, J. & Nocera, D. G. 2006. Temperature–isotope dependence reveals electron-transfer driven by proton-fluctuations in a hydrogen-bonded donor–acceptor assembly. J. Phys. Chem. B [DOI] [PubMed]
- Hoganson C.W, Tommos C. The function and characteristics of tyrosyl radical cofactors. Biochim. Biophys. Acta. 2004;1655:116–122. doi: 10.1016/j.bbabio.2003.10.017. doi:10.1016/j.bbabio.2003.10.017 [DOI] [PubMed] [Google Scholar]
- Högbom M, Galander M, Andersson M, Kolberg M, Hofbauer W, Lassmann G, Nordlund P, Lendzian F. Displacement of the tyrosyl radical cofactor in ribonucleotide reductase obtained by single-crystal high-field EPR and 1.4-Å X-ray data. Proc. Natl Acad. Sci. USA. 2003;100:3209–3214. doi: 10.1073/pnas.0536684100. doi:10.1073/pnas.0536684100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito N, Phillips S.E.V, Yadav K.D.S, Knowles P.F. Crystal structure of a free radical enzyme, galactose oxidase. J. Mol. Biol. 1994;238:794–814. doi: 10.1006/jmbi.1994.1335. doi:10.1006/jmbi.1994.1335 [DOI] [PubMed] [Google Scholar]
- Jordan A, Reichard P. Ribonucleotide reductases. Annu. Rev. Biochem. 1998;67:71–98. doi: 10.1146/annurev.biochem.67.1.71. doi:10.1146/annurev.biochem.67.1.71 [DOI] [PubMed] [Google Scholar]
- Kirby J.P, van Dantzig N.A, Chang C.K, Nocera D.G. Formation of porphyrin donor–acceptor complexes via an amidinium–carboxylate salt bridge. Tetrahedron Lett. 1995;36:3477–3480. doi:10.1016/0040-4039(95)00569-X [Google Scholar]
- Kirby J.P, Roberts J.A, Nocera D.G. Significant effect of salt bridges on electron transfer rate. J. Am. Chem. Soc. 1997;119:9230–9236. doi:10.1021/ja970176+ [Google Scholar]
- Kohen A, Klinman J.P. Enzyme catalysis: beyond classical paradigms. Acc. Chem. Res. 1998;31:397–404. doi:10.1021/ar9701225 [Google Scholar]
- Krebs C, Chen S, Baldwin J, Ley B.A, Patel U, Edmondson D.E, Huynh B.H, Bollinger J.M., Jr Mechanism of rapid electron transfer during oxygen activation in the R2 subunit of Escherichia coli ribonucleotide reductase. 2. Evidence for and consequences of blocked electron transfer in the W48F variant. J. Am. Chem. Soc. 2000;122:12 207–12 219. doi:10.1021/ja001279m [Google Scholar]
- Liang Z.-X, Klinman J.P. Structural bases of hydrogen tunneling in enzymes: progress and puzzles. Curr. Opin. Struct. Biol. 2004;14:648–655. doi: 10.1016/j.sbi.2004.10.008. doi:10.1016/j.sbi.2004.10.008 [DOI] [PubMed] [Google Scholar]
- Lin J, Balabin I.A, Beratan D.N. The nature of aqueous tunneling pathways between electron-transfer proteins. Science. 2005;310:1311–1313. doi: 10.1126/science.1118316. doi:10.1126/science.1118316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.-Y, Nocera D.G. Hangman salophens. J. Am. Chem. Soc. 2005;127:5278–5279. doi: 10.1021/ja042849b. doi:10.1021/ja042849b [DOI] [PubMed] [Google Scholar]
- Loll B, Kern J, Saenger W, Zouni A, Biesiadka J. Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature. 2005;438:1040–1044. doi: 10.1038/nature04224. doi:10.1038/nature04224 [DOI] [PubMed] [Google Scholar]
- Maradufu A, Cree G.M, Perlin A.S. Stereochemistry of dehydrogenation by d-galactose oxidase. Can. J. Chem. 1971;49:3429–3437. doi:10.1139/v71-575 [Google Scholar]
- Marcus R.A, Sutin N. Electron transfers in chemistry and biology. Biochim. Biophys. Acta. 1985;811:265–322. [Google Scholar]
- Mayer J.M. Proton-coupled electron transfer: a reaction chemist's view. Annu. Rev. Phys. Chem. 2004;55:363–390. doi: 10.1146/annurev.physchem.55.091602.094446. doi:10.1146/annurev.physchem.55.091602.094446 [DOI] [PubMed] [Google Scholar]
- Meunier B, de Visser S.P, Shaik S. Mechanism of oxidation reactions catalyzed by cytochrome P450 enzymes. Chem. Rev. 2004;104:3947–3980. doi: 10.1021/cr020443g. doi:10.1021/cr020443g [DOI] [PubMed] [Google Scholar]
- Miller M.A, Coletta M, Mauro J.M, Putnam L.D, Farnum M.F, Kraut J, Traylor T.G. Carbon monoxide recombination in cytochrome c peroxidase: effect of the local heme environment on carbon monoxide binding explored through site-directed mutagenesis. Biochemistry. 1990;29:1777–1791. doi: 10.1021/bi00459a017. doi:10.1021/bi00459a017 [DOI] [PubMed] [Google Scholar]
- Moser C.C, Keske J.M, Warncke K, Farid R.S, Dutton P.L. Nature of biological electron transfer. Nature. 1992;355:796–802. doi: 10.1038/355796a0. doi:10.1038/355796a0 [DOI] [PubMed] [Google Scholar]
- Muir T.W. Semisynthesis of proteins by expressed protein ligation. Annu. Rev. Biochem. 2003;72:249–289. doi: 10.1146/annurev.biochem.72.121801.161900. doi:10.1146/annurev.biochem.72.121801.161900 [DOI] [PubMed] [Google Scholar]
- Newmyer S.L, Ortiz de Montellano P.R. Horseradish peroxidase His-42→Ala, His-42→Val, and Phe-41→Ala mutants. Histidine catalysis and control of substrate access to the heme iron. J. Biol. Chem. 1995;270:19 430–19 438. doi: 10.1074/jbc.270.33.19430. doi:10.1074/jbc.270.33.19430 [DOI] [PubMed] [Google Scholar]
- Nicolet Y, Piras C, Legrand P, Hatchikian C.E, Fontecilla-Camps J.C. Desulfovibrio desulfuricans iron hydrogenase: the structure shows unusual coordination to an active site Fe binuclear center. Structure. 1999;7:13–23. doi: 10.1016/s0969-2126(99)80005-7. doi:10.1016/S0969-2126(99)80005-7 [DOI] [PubMed] [Google Scholar]
- Nordlund P, Eklund H. Structure and function of the Escherichia coli ribonucleotide reductase protein R2. J. Mol. Biol. 1993;232:123–164. doi: 10.1006/jmbi.1993.1374. doi:10.1006/jmbi.1993.1374 [DOI] [PubMed] [Google Scholar]
- Nordlund P, Sjöberg B.-M, Eklund H. Three-dimensional structure of the free radical protein of ribonucleotide reductase. Nature. 1990;345:593–598. doi: 10.1038/345593a0. doi:10.1038/345593a0 [DOI] [PubMed] [Google Scholar]
- Okamura M.Y, Paddock M.L, Graige M.S, Feher G. Proton and electron transfer in bacterial reaction centers. Biochim. Biophys. Acta. 2000;1458:148–163. doi: 10.1016/s0005-2728(00)00065-7. doi:10.1016/S0005-2728(00)00065-7 [DOI] [PubMed] [Google Scholar]
- Onuchic J.N, Beratan D.N. A predictive theoretical model for electron tunneling pathways in proteins. J. Chem. Phys. 1990;92:722–733. doi:10.1063/1.458426 [Google Scholar]
- Osuka A, Yoneshima R, Shiratori H, Okada S, Taniguchi S, Mataga N. Electron transfer in a hydrogen-bonded assembly consisting of porphyrin–diimide. Chem. Commun. 1998:1567–1568. doi:10.1039/a803541e [Google Scholar]
- Osyczka A, Moser C.C, Daldal F, Dutton P.L. Reversible redox energy coupling in electron transfer chains. Nature. 2004;427:607–612. doi: 10.1038/nature02242. doi:10.1038/nature02242 [DOI] [PubMed] [Google Scholar]
- Peters J.W, Lanzilotta W.N, Lemon B.J, Seefeldt L.C. X-ray crystal structure of the Fe-only hydrogenase (CpI) from Clostridium pasteurianum to 1.8 angstrom resolution. Science. 1998;282:1853–1858. doi: 10.1126/science.282.5395.1853. doi:10.1126/science.282.5395.1853 [DOI] [PubMed] [Google Scholar]
- Pond A.E, Ledbetter A.P, Sono M, Goodin D.B, Dawson J.H. Redox enzymes: correlation of three-dimensional structure and mechanism for heme-containing oxygenases and peroxidases. In: Balzani V, editor. Electron transfer in chemistry. vol. 3.1.4. Wiley-VCH; Weinheim, Germany: 2001. pp. 56–104. [Google Scholar]
- Poulos T.L, Finzel B.C, Gunsalus I.C, Wagner G.C, Kraut J. The 2.6-Å crystal structure of Pseudomonas putida cytochrome P-450. J. Biol. Chem. 1985;260:16 122–16 130. [PubMed] [Google Scholar]
- Poulos T.L, Finzel B.C, Howard A.J. Crystal structure of substrate-free Pseudomonas putida cytochrome P-450. Biochemistry. 1986;25:5314–5322. doi: 10.1021/bi00366a049. doi:10.1021/bi00366a049 [DOI] [PubMed] [Google Scholar]
- Poulos T.L, Finzel B.C, Howard A.J. High-resolution crystal structure of cytochrome P450cam. J. Mol. Biol. 1987;195:687–700. doi: 10.1016/0022-2836(87)90190-2. doi:10.1016/0022-2836(87)90190-2 [DOI] [PubMed] [Google Scholar]
- Pressé S, Silbey R.J. Anomalous temperature–isotope dependence in proton-coupled electron transfer. J. Chem. Phys. 2006;124:164504/1–7. doi: 10.1063/1.2188395. doi:10.1063/1.2188395 [DOI] [PubMed] [Google Scholar]
- Reece S.Y, Nocera D.G. Direct tyrosine oxidation using the MLCT excited states of rhenium polypyridyl complexes. J. Am. Chem. Soc. 2005;127:9448–9458. doi: 10.1021/ja0510360. doi:10.1021/ja0510360 [DOI] [PubMed] [Google Scholar]
- Reece S.Y, Stubbe J, Nocera D.G. pH dependence of charge transfer between tryptophan and tyrosine in dipeptides. Biochim. Biophys. Acta. 2005;1706:232–238. doi: 10.1016/j.bbabio.2004.11.011. doi:10.1016/j.bbabio.2004.11.011 [DOI] [PubMed] [Google Scholar]
- Reece, S. Y., Seyedsayamdost, M. R., Stubbe, J. & Nocera, D. G. In preparation.
- Roberts J.A, Kirby J.P, Nocera D.G. Photoinduced electron transfer within a donor–acceptor pair juxtaposed by a salt bridge. J. Am. Chem. Soc. 1995;117:8051–8052. doi:10.1021/ja00135a038 [Google Scholar]
- Roberts J.A, Kirby J.P, Wall S.T, Nocera D.G. Electron transfer within ruthenium(II) polypyridyl-(salt bridge)-dimethylaniline acceptor–donor complexes. Inorg. Chim. Acta. 1997;263:395–405. doi:10.1016/S0020-1693(97)05668-5 [Google Scholar]
- Rosenthal J, Hodgkiss J.M, Young E.R, Nocera D.G. Spectroscopic determination of proton position in PCET pathways of donor–acceptor supramolecular assemblies. J. Am. Chem. Soc. In press doi: 10.1021/ja062430g. [DOI] [PubMed] [Google Scholar]
- Rova U, Goodtzova K, Ingemarson R, Behravan G, Gräslund A, Thelander L. Evidence by site-directed mutagenesis supports long-range electron transfer in mouse ribonucleotide reductase. Biochemistry. 1995;34:4267–4275. doi: 10.1021/bi00013a016. doi:10.1021/bi00013a016 [DOI] [PubMed] [Google Scholar]
- Sessler J.L, Wang B, Springs S.L, Brown C.T. Comprehensive supramolecular chemistry. vol. 4. Pergamon Press; Oxford, UK: 1996. Electron- and energy-transfer reactions in noncovalently linked supramolecular model systems; pp. 311–336. [Google Scholar]
- Seyedsayamdost M.R, Reece S.Y, Nocera D.G, Stubbe J. Mono-, di-, tri-, and tetra-substituted fluorotyrosines: new probes for enzymes that use tyrosyl radicals in catalysis. J. Am. Chem. Soc. 2006a;128:1569–1579. doi: 10.1021/ja055926r. doi:10.1021/ja055926r [DOI] [PubMed] [Google Scholar]
- Seyedsayamdost M.R, Yee C.S, Reece S.Y, Nocera D.G, Stubbe J. pH rate profiles of FnY356-R2s (n=2, 3, 4) in E. coli ribonucleotide reductase: evidence that Y356 is a redox active amino acid along the radical propagation pathway. J. Am. Chem. Soc. 2006b;128:1562–1568. doi: 10.1021/ja055927j. doi:10.1021/ja055927j [DOI] [PubMed] [Google Scholar]
- Shafirovich V.Y, Courtney S.H, Ya N, Geacintov N.E. Proton-coupled photoinduced electron transfer, deuterium isotope effects, and fluorescence quenching in noncovalent benzo[a]pyrenetetraol-nucleoside complexes in aqueous solutions. J. Am. Chem. Soc. 1995;117:4920–4929. doi:10.1021/ja00122a024 [Google Scholar]
- Soudackov A, Hammes-Schiffer S. Multistate continuum theory for multiple charge transfer reactions in solution. J. Chem. Phys. 1999;111:4672–4687. doi:10.1063/1.479229 [Google Scholar]
- Soudackov A, Hammes-Schiffer S. Derivation of rate expressions for nonadiabatic proton-coupled electron transfer reactions in solution. J. Chem. Phys. 2000;113:2385–2396. doi:10.1063/1.482053 [Google Scholar]
- Stubbe J, Riggs-Gelasco P. Harnessing free radicals: formation and function of the tyrosyl radical in ribonucleotide reductase. Trends Biochem. Sci. 1998;23:438–443. doi: 10.1016/s0968-0004(98)01296-1. doi:10.1016/S0968-0004(98)01296-1 [DOI] [PubMed] [Google Scholar]
- Stubbe J, van der Donk W.A. Protein radicals in enzyme catalysis. Chem. Rev. 1998;98:705–762. doi: 10.1021/cr9400875. doi:10.1021/cr9400875 [DOI] [PubMed] [Google Scholar]
- Stubbe J, Ator M, Krenitsky T. Mechanism of ribonucleoside diphosphate reductase from Escherichia coli. Evidence for 3′-C–H bond cleavage. J. Biol. Chem. 1983;258:1625–1631. [PubMed] [Google Scholar]
- Stubbe J, Nocera D.G, Yee C.S, Chang M.C.Y. Radical initiation in the class I ribonucleotide reductase: long-range proton-coupled electron transfer? Chem. Rev. 2003;103:2167–2202. doi: 10.1021/cr020421u. doi:10.1021/cr020421u [DOI] [PubMed] [Google Scholar]
- Tanaka M, Ishimori K, Mukai M, Kitagawa T, Morishima I. Catalytic activities and structural properties of horseradish peroxidase distal His42→Glu or Gln mutant. Biochemistry. 1997;36:9889–9898. doi: 10.1021/bi970906q. doi:10.1021/bi970906q [DOI] [PubMed] [Google Scholar]
- Tommos C, Babcock G.T. Oxygen production in nature: a light-driven metalloradical enzyme process. Acc. Chem. Res. 1998;31:18–25. doi:10.1021/ar9600188 [Google Scholar]
- Tommos C, Babcock G.T. Proton and hydrogen currents in photosynthetic water oxidation. Biochim. Biophys. Acta. 2000;1458:199–219. doi: 10.1016/s0005-2728(00)00069-4. doi:10.1016/S0005-2728(00)00069-4 [DOI] [PubMed] [Google Scholar]
- Turró C, Chang C.K, Leroi G.E, Cukier R.I, Nocera D.G. Photoinduced electron transfer mediated by a hydrogen-bonded interface. J. Am. Chem. Soc. 1992;114:4013–4015. doi:10.1021/ja00036a081 [Google Scholar]
- Uhlin U, Eklund H. Structure of ribonucleotide reductase protein R1. Nature. 1994;370:533–539. doi: 10.1038/370533a0. doi:10.1038/370533a0 [DOI] [PubMed] [Google Scholar]
- Vidakovic M, Sligar S.G, Li H, Poulos T.L. Understanding the role of the essential Asp251 in cytochrome p450cam using site-directed mutagenesis, crystallography, and kinetic solvent isotope effect. Biochemistry. 1998;37:9211–9219. doi: 10.1021/bi980189f. doi:10.1021/bi980189f [DOI] [PubMed] [Google Scholar]
- Volbeda A, Fontecilla-Camps J.C. Structure–function relationships of nickel–iron sites in hydrogenase and a comparison with the active sites of other nickel–iron enzymes. Coord. Chem. Rev. 2005;249:1609–1619. doi:10.1016/j.ccr.2004.12.009 [Google Scholar]
- Vrettos J.S, Brudvig G.W. Water oxidation chemistry of photosystem II. Phil. Trans. R. Soc. B. 2002;357:1395–1405. doi: 10.1098/rstb.2002.1136. doi:10.1098/rstb.2002.1136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward M.D. Photoinduced electron and energy transfer in non-covalently bonded supramolecular assemblies. Chem. Soc. Rev. 1997;26:365–376. doi:10.1039/cs9972600365 [Google Scholar]
- Whittaker J.W. The radical chemistry of galactose oxidase. Arch. Biochem. Biophys. 2005;433:227–239. doi: 10.1016/j.abb.2004.08.034. doi:10.1016/j.abb.2004.08.034 [DOI] [PubMed] [Google Scholar]
- Winkler J.R, Gray H.B. Electron transfer in ruthenium-modified proteins. Chem. Rev. 1992;92:369–379. doi: 10.1007/BF02110099. doi:10.1021/cr00011a001 [DOI] [PubMed] [Google Scholar]
- Yee C.S, Chang M.C.Y, Ge J, Nocera D.G, Stubbe J. 2,3-Difluorotyrosine at position 356 of ribonucleotide reductase R2: a probe of long-range proton-coupled electron transfer. J. Am. Chem. Soc. 2003a;125:10 506–10 507. doi: 10.1021/ja036242r. doi:10.1021/ja036242r [DOI] [PubMed] [Google Scholar]
- Yee C.S, Seyedsayamdost M.R, Chang M.C.Y, Nocera D.G, Stubbe J. Generation of the R2 subunit of ribonucleotide reductase by intein chemistry: insertion of 3-nitrotyrosine at residue 356 as a probe of the radical initiation process. Biochemistry. 2003b;42:14 541–14 552. doi: 10.1021/bi0352365. doi:10.1021/bi0352365 [DOI] [PubMed] [Google Scholar]
- Yeh C.Y, Chang C.J, Nocera D.G. “Hangman” porphyrins for the assembly of a model heme water channel. J. Am. Chem. Soc. 2001a;123:1513–1514. doi: 10.1021/ja003245k. doi:10.1021/ja003245k [DOI] [PubMed] [Google Scholar]
- Yeh C.Y, Miller S.E, Carpenter S.D, Nocera D.G. Structurally homologous β- and meso-amidinium porphyrins. Inorg. Chem. 2001b;40:3643–3646. doi: 10.1021/ic001387+. doi:10.1021/ic001387+ [DOI] [PubMed] [Google Scholar]
- Zhao X.G, Cukier R.I. Molecular dynamics and quantum chemistry study of a proton-coupled electron transfer reaction. J. Phys. Chem. 1995;99:945–954. doi:10.1021/j100003a017 [Google Scholar]